

Abstract
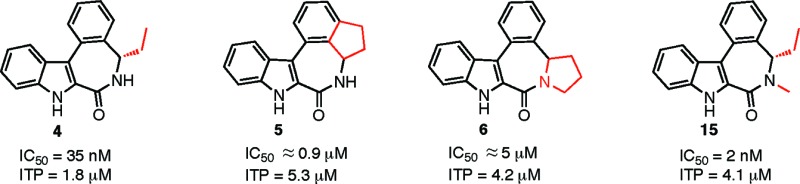
Two rigid analogues of 5-ethylindolobenzazepinone 4, a potent cytotoxic agent and inhibitor of tubulin polymerization, were prepared. The first was the indane derivative 5, in which the ethyl group is attached to the benzo moiety. The second was the pyrrolidine analogue 6, in which the ethyl chain was bound to the lactam nitrogen. While both compounds were considerably less active inhibitors of KB cell growth as compared to 4, inhibition of tubulin polymerization was only moderately reduced. Tubulin docking studies indicated that the aR and aS atropoisomers of 5 and 6 occupy different binding pockets at the colchicine binding site. Conversely, both aS-5 and aS-6 occupy the same binding pocket as aSS-4 but do not benefit from the favorable hydrophobic interactions provided by the C5 alkyl group of 4, thus possibly explaining their lower activities.
Keywords: Indolobenzazepinone, cytotoxicity, tubulin polymerization, atropoisomers, molecular docking
Tubulin remains one of the major therapeutic targets for clinically useful anticancer compounds,1 as reflected by the considerable success of the antimitotic vinca alkaloids (vincristin, vinblastin, and the synthetic analogue navelbine)2 and taxoid (taxol and its synthetic analogue taxotere)3 derivatives. The structural complexity of these families of compounds has spurred investigations to discover simpler structures having antimitotic activities. This has led, for example, to the development of N-acetyl-allocolchinol (NAC, 1),4 a structural analogue of one of the first described naturally occurring antimitotic agents, colchicine (2),5 as well as analogues of the combretastins (3) (see Figure 1).6 However, while some of these compounds or their analogues are in advanced clinical studies, none have yet been approved as drugs.
Figure 1.

In this regard, we recently described such a family of low molecular weight and synthetically accessible antimitotic agents, the C5-alkylated indolobenzazepinones, of which the most active member was the (S)-5-ethyl derivative 4.7 The latter showed potent antiproliferative activities in a variety of cancer cell lines with, for example, an IC50 value of 32 nM in KB cells. Compound 4 also inhibited the polymerization of tubulin with an IC50 of 1.8 μM, comparing favorably with the antimitotic activities of colchicine (2, IC50 = 2.2 μM) and NAC (1, IC50 = 3.0 μM) determined under the same test conditions. Compound 4 was, moreover, shown to be superior to colchicine and NAC in significantly reducing tumor size in the experimental model of glioma grafted on the chick chorio-allantoic membrane (CAM). As part of our ongoing structure–activity relationships development for this novel family of antimitotics, we prepared analogues of 4 in which the relatively flexible 7-membered benzazepinone ring was rendered more rigid by the presence of a supplementary ring. The first of these, the indane derivative 5, corresponds to active compound 4 in which the C5 ethyl group is linked to the benzo moiety, while in the second rigid target molecule, the same ethyl group is now connected to the nitrogen atom of the carboxamide to form the pyrrolidine structure 6.
The synthesis of compound 5 is depicted in Scheme 1. The bromoindanone 7(8) was treated with titanium tetra-isopropoxide and excess ammonia in ethanol to provide the corresponding imine, which was reduced with sodium borohydride to the amine. The latter was relatively unstable and was therefore transformed in situ to the N-Boc derivative 8, which was easily purified by chromatography. Reaction of compound 8 with pinacol borane in the presence of catalytic palladium diacetate and S-Phos in dioxane at 80 °C provided the pinocolindane derivative 9.9 Suzuki coupling of the latter with ethyl 1-benzenesulfonyl-3-iodoindole-2-carboxylate 10(7) using the combination palladium diacetate/dppf as the catalytic system and cesium fluoride as base provided compound 11 in approximately 50% yield from 8. Deprotection of the primary amine function of 11 using trifluoroacetic acid in dichloromethane gave compound 12. Finally, treatment of amine 12 with sodium in ethanol led to cyclization and concomitant deprotection of the indole nitrogen to afford the desired indane derivative 5.
Scheme 1.

Our second target molecule, the pyrrolidine derivative 6, corresponds to an N-alkylated version of compound 4. We have previously observed that N-methylation of the indole nitrogen as well as simultaneous methylation of the indole and lactam NH were detrimental to both the cytotoxic and the antimitotic activities of 4.7 Therefore, before undertaking the synthesis of the pyrrolidine derivative 6, it thus seemed appropriate to first investigate the effect of N-alkylation of only the lactam function of 4 on activity. Hence, for comparison with 6, we prepared the N-methyl derivative 15 (Scheme 2). To do so, the indole NH of compound 4 was first selectively protected by a Boc group to give 13. Methylation of the lactam NH of 13 was then effected with methyl iodide and sodium hydride in THF. Treatment of 14 with trifluoroacetic acid in dichloromethane then afforded monomethyl derivative 15.
Scheme 2.

Encouraged by the results obtained for 15 (vide infra), we then proceeded with the synthesis of the rigid pyrrolidine derivative 6 using the strategy shown in Scheme 3. N-Boc chloropropylamine 16 was treated with sodium hydride in THF, and the resulting sodium salt was reacted with 2-bromobenzyl bromide to provide 17 in 84% yield.10 Cyclization of the latter to the 2-bromophenylpyrrolidine 19 was effected in good yield (78%) by the action of LDA in THF. Applying the same coupling methodology used for the preparation of the indane derivative 5, compound 18 was first transformed into pinacol derivative 19, which was then coupled to 3-iodoindole derivative 10 under palladium catalysis to afford 20 in modest yield (28% for both steps). Removal of the N-Boc group of 20 with TFA followed by sodium ethanolate-mediated cyclization and N-benzenesulfonamide cleavage provided the desired pyrrolidino-benzazepine derivative 6.
Scheme 3.

Rigid analogues 5 and 6, as well as N-methyl derivative 15, were then evaluated for antiproliferative activities in KB cells (Table 1).7 Both 5 and 6 were considerably less active than the nonrigid derivative, compound 4, which possesses an IC50 of 35 nM. Thus, indane derivative 5 inhibited KB cell growth by 63% at 10–6 M (corresponding to an estimated IC50 of slightly less than 1 μM), while the pyrrolidine analogue 6 was even less cytotoxic, inhibiting KB cell growth by only 27% at 10–6 M. Interestingly, however, while the ability of compounds 5 and 6 to inhibit the polymerization of tubulin was also diminished (IC50 values of q5.3 and 4.2 μM, respectively) as compared to compound 4 (IC50 = 1.8 μM), this 2-fold decrease was modest as compared to the major loss of cytotoxic activity of both rigid molecules. These apparently paradoxical results may simply reflect poorer cell penetration by compounds 5 and 6 as compared to the less rigid analogue 4, a problem that does not come into consideration for evaluation of antimitotic activity. Gratifyingly, the N-methyl derivative of 4, compound 15, was observed to display an antiproliferative activity superior to that of 4 itself (IC50 values of 2 vs 34 nM, respectively), while inversely, the antimitotic activity of 15 (IC50 = 4.1 μM) was 2-fold less than that of 4 (IC50 = 1.8 μM) but equivalent to that of pyrrolidine derivative 6 (IC50 = 4.2 μM). This indicates that a free lactam NH function is not essential for activity and suggests that the loss of cytotoxic activity in the case of pyrrolidine derivative 6 is not due to the absence of the lactam NH.
Table 1. Antiproliferative Activities of Compounds 4–6 and 15 in KB Cells.
| % inhibition of KB cell growth |
|||
|---|---|---|---|
| compound no. | 10 μM | 1 μM | ITPc (μM) |
| 4 | 100 | 100a | 1.8 |
| 5 | 81 | 63 | 5.3 |
| 6 | 100 | 27 | 4.2 |
| 15 | 100 | 100b | 4.1 |
The IC50 of 4 was determined to be 35 nM.
The IC50 of 15 was determined to be 2 nM.
Inhibition of tubulin polymerization.
The antiproliferative activities of compounds 5 and 6 were also evaluated in three other cancer cell lines (HCT116, MCF7, and HL60), as well as in resistant strains of these same cells (HCT15, MCF7R, and HL60R). Interestingly, as shown in Table 2, while both compounds maintain micromolar activities in all cell lines, they were observed to be generally equally cytotoxic in resistant and nonresistant cells (HCT and HL60) and, in the case of MCF7 cells, more cytotoxic in the resistant strains. By comparison, compounds 4 and 15 were equally active in resistant and nonresistant cell lines except for HL60 where cytotoxic activity was slightly weaker in the resistant cells.
Table 2. Antiproliferative Activities of 5 and 6 as Compared to 4 and 15 in Resistant Cancer Cell Lines.
| 10/1 μM |
||||||
|---|---|---|---|---|---|---|
| compound no. | HCT116 (%)a | HCT15 (%) | MCF7 (%) | MCF7R (%) | HL60 (%) | HL60R (%) |
| 4 | 100/85 | 100/94 | 92/82 | 96/84 | 100/90 | 95/78 |
| 5 | 80/64 | 87/75 | 39/46 | 72/67 | 90/83 | 81/70 |
| 6 | 81/64 | 84/78 | 55/28 | 75/56 | 84/49 | 80/49 |
| 15 | 100/91 | 100/92 | 96/85 | 92/75 | 100/85 | 89/70 |
Percentage of inhibition of cell growth at 10 and 1 μM concentrations of test compound.
Joseph and colleagues have recently reported the antiproliferative and antimitotic activities of a series of 5-unsubstituted indolobenzazepinones (i.e., compounds of type 4 devoid of a C5 substituent).11,12 While these activities were generally less robust than in the case of our C5-alkylated derivatives, the authors were able to perform docking studies at the colchicine binding site of tubulin. Interestingly, no hydrogen-bonding interactions of the nitrogen amide with the protein were observed. The present study therefore corroborates this conclusion in that major structural modifications at this center, as in pyrrolidine derivative 6, have very little impact on the inhibition of tubulin polymerization.
Our own modeling studies did show some notable features that could explain the diminished antimitotic activities of the two rigid derivatives 5 and 6 versus more flexible compound 4. Specifically, we determined the conformational preferences of these three compounds using the Monte Carlo random search method with optimization by the PRCG molecular mechanics minimization technique using the Macromodel program (version 5.5)13 with the MM2 force field. Out of a search of 1000 conformers, only one atropoisomer aRR was found for both the indane and the pyrrolidine analogues 5 and 6, while both aRR and aSR atropoisomers were obtained for the indolobenzazepinone 4. In spite of differences in structural rigidity, all aRR atropoisomers adopt similar 3D structures (Figure S1 in the Supporting Information). The 3D structures of the missing 5-aSR and 6-aSR atropoisomers were constructed manually. Finally, the geometries of these six conformers, as well as those of the corresponding transition states, were optimized using the Gaussian 03 program14 at the HF/6-31G+(d,p) level (Figure 2). Subsequent vibrational frequency calculations confirmed that these conformations are local minima and maxima, respectively.
Figure 2.
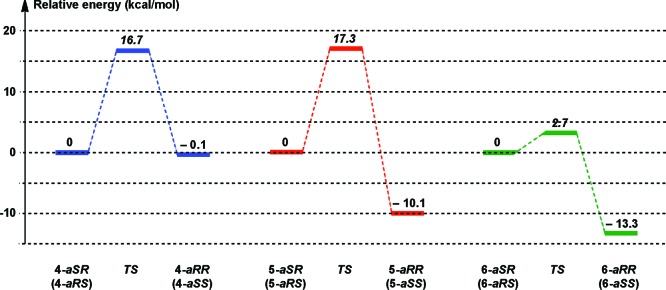
Transition state diagrams for atropoisomeric configuration inversion in the three systems studied.
Several conclusions can be drawn from these studies. First, in all three cases, the transition state energy of the atropoisomer inversion process allows the establishment, more or less rapidly, of a thermodynamic equilibrium. The similar energies calculated for the 4-aRR and 4-aSR atropoisomers are in good agreement with the diastereomeric mixture observed in solution,15 which is probably the consequence of atropoisomer interconversion at room temperature. In contrast, only one diastereoisomer is observed experimentally for compounds 5 and 6. In both cases, this can be formally predicted to be the more stable one (aRR), and its presence can be explained by the complete selectivity of the seven-membered ring closure reaction and/or by the easy conversion of aSR diastereoisomers into the aRR ones. Overall, these modeling studies predict that for compounds 5 and 6, the only species present in solution are the aRR diastereoisomers (and, of course, their aSS enantiomers). Thus, while hydrogen-bonding interactions of tubulin with the lactam function of these compounds may not be important, conformational considerations may affect binding to tubulin via unfavorable steric interactions.
Molecular docking studies16 were carried out to identify potential interactions during indolobenzazepinone 5 and 6 binding to tubulin. Thus, as mentioned above, all possible stereoisomers of compounds 5 and 6 (aRR, aSS, aSR, and aRS) were used for docking on tubulin, in one of the binding sites of DAMA-colchicine (Figure 3 and Figure S4 in the Supporting Information).17
Figure 3.

Detail of the tubulin binding site with (a) X-ray conformation of DAMA-colchicine (PDB code: 1SA0); (b) docking conformations of compounds 4-aRR (orange) and 4-aSS (green); (c) superposition of 6-aSS to the docking conformation of 4-aSR (green) showing a favorable fit for both molecules in the left-hand subpocket of the tubulin binding site; and (d) superposition of 6-aRR (magenta) to the docking conformation of 4-aRS (orange) showing potential steric clashes with the protein surface in the right-hand subpocket of the tubulin binding site.17
Previous molecular modeling studies with the C5-substituted indolobenzazepinone series, that is, of type 4, identified the existence of two distinct binding subpockets on the tubulin structure.7,15 These subpockets are partially overlapping (Figure 3b) and occupy approximately the same binding site as DAMA-colchicine (Figure 3a). The main criterion for ligand selectivity between the two subpockets is atropoisomerism; ligands with the aS configuration occupy principally the left subpocket, whereas those with aR configuration are positioned mainly in the right subpocket. It is noteworthy that the C5-alkyl substituents of compounds 4-aRR and 4-aSS occupy the same pocket as the C ring of colchicine (Figure 3a,b), and the favorable hydrophobic interactions with this pocket might explain the better biological activity of these compounds as compared with C5-unsubstituted derivatives.
Docking of compounds 5 and 6 in the colchicine binding site of tubulin followed the same trend, the compounds with aS configuration occupying mainly the left subpocket (Figure S2 in the Supporting Information) and those with aR configuration being positioned principally in the right subpocket (Figure S3 in the Supporting Information). In the first case, the docking conformations are very similar with the reference compound 4-aSR (Figure 3c and Figure S2a–d in the Supporting Information), and their superimposition does not show steric clashes with the protein surface (Figure S2e–h in the Supporting Information). This means that the binding of aS isomers of compounds 5 and 6 in the colchicine binding site of tubulin is favored but without the benefit of hydrophobic interactions observed for C5-alkyl indolobenzazepinones. This is in good agreement with the similar biological activities determined for the compounds 5 and 6 (IC50 = 4.2–5.3 μM, Table 1) and for the C5-unsubstituted indolobenzazepinone (IC50 = 5.3 μM).11,12
In the second case, the docking conformations are positioned quite differently as compared with the reference compound 4-aRS (Figure S3a–d in the Supporting Information), and their superimposition shows that the difference is due to important steric clashes between ligands and the protein surface, especially for 6-aRR and 6-aRS (Figure 3d and Figure S3e–h in the Supporting Information). Thus, it can be predicted that the binding of the aR isomers of compounds 5 and 6 in the colchicine site of tubulin is less favorable and that the biological activity of these compounds is most likely due to the binding of the aS isomers.
In tandem, the quantum chemistry calculations results, which indicate that the isomers aRR/aSS represent the only species present in solution, and the docking results, which provide evidence that binding of the aS isomers is favored, are strong indicators that 5-aSS and 6-aSS are the stereoisomers responsible for the biological activity of the compounds 5 and 6. A similar atropoisomer binding preference for biphenyl analogues of colchicine inhibitors has also been reported.18 The presented results should be very useful for the future, rational design of novel atropo- and enantioselective inhibitors of tubulin polymerization.
Acknowledgments
We thank Geneviève Aubert and Dr. Thierry Cresteil of the ICSN for the inhibition assays and Dr. Philippe Dauban for helpful discussions.
Supporting Information Available
Description of the tubulin docking studies with compounds 4–6, theoretical data, synthesis and characterization of compounds 5, 6, 8, 11, 15–18, and 20,and the cell proliferation inhibition assay. This material is available free of charge via the Internet at http://pubs.acs.org.
We thank the ICSN for fellowships (V.P. and S.B.).
Supplementary Material
References
- Jordan M. A.; Wilson L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [DOI] [PubMed] [Google Scholar]
- Guéritte F.; Fahy J.. The Vinca Alkaloids. In Anticancer Agents from Natural Products; Cragg G. M., Kingston D. G. I., Newman D. J., Eds.; CRC Press: Boca Raton, FL, 2005; pp 123–135. [Google Scholar]
- Kingston D. G. I.Taxol and Its Analogs. In Anticancer Agents from Natural Products; Cragg G. M., Kingston D. G. I., Newman D. J., Eds.; CRC Press: Boca Raton, FL, 2005; pp 89–122. [Google Scholar]
- Davis P. D.; Dougherty G. J.; Blakey D. C.; Galbraith S. M.; Tozer G. M.; Holder A. L.; Naylor M. A.; Nolan J.; Stratford M. R. L.; Chaplin D. J.; Hill S. A. ZD6126: A novel vascular-targeting agent that causes selective destruction of tumor vasculature. Cancer Res. 2002, 62, 7247–7253. [PubMed] [Google Scholar]
- Ravelli R. B. G.; Gigant B.; Curmi P. A.; Jourdain I.; Lachkar S.; Sobel A.; Knossow M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [DOI] [PubMed] [Google Scholar]
- Tron G. C.; Pirali T.; Sorba G.; Pagliai F.; Busacca S.; Genazzani A. A. Medicinal chemistry of combretastatin A4: Present and future directions. J. Med. Chem. 2006, 49, 3033–3044. [DOI] [PubMed] [Google Scholar]
- Keller L; Beaumont S; Liu J.-M.; Thoret S.; Bignon J. S.; Wdzieczak-Bakala J.; Dauban P.; Dodd R. H. New C5-alkylated indolobenzazepinones acting as inhibitors of tubulin polymerization: Cytotoxic and antitumor activities. J. Med. Chem. 2008, 51, 3414–3421. [DOI] [PubMed] [Google Scholar]
- Phong N.; Corpuz E.; Heidelbaugh T. M.; Chow K.; Garst M. E. A convenient synthesis of 7-halo-1-indanones and 8-halo-1-tetralones. J. Org. Chem. 2003, 68, 10195–10198. [DOI] [PubMed] [Google Scholar]
- Baudoin O.; Cesario M.; Guénard D.; Guéritte F. Application of the palladium-catalyzed borylation/Suzuki coupling (BSC) reaction to the synthesis of biologically active biaryl-lactams. J. Org. Chem. 2002, 67, 1199–1207. [DOI] [PubMed] [Google Scholar]
- Wu S.; Lee S.; Beak P. Asymmetric deprotonation by BuLi/(−)-sparteine: Convenient and highly enantioselective syntheses of (S)-2-aryl-Boc-pyrrolidines. J. Am. Chem. Soc. 1996, 118, 715–721. [Google Scholar]
- Putey A.; Joucla L.; Picot L.; Besson T.; Joseph B. Synthesis of latonduine derivatives via intramolecular Heck reaction. Tetrahedron 2007, 63, 867–879. [Google Scholar]
- Putey A.; Popowycz F.; Do Q.-T.; Bernard P.; Talapatra S. K.; Kozielski F.; Galmarini C. K.; Joseph B. Indolobenzazepino-7-ones and 6-, 8, and 9-membered ring derivatives as tubulin polymerization inhibitors: Synthesis and structure-activity relationship studies. J. Med. Chem. 2009, 52, 5916–5925. [DOI] [PubMed] [Google Scholar]
- http://www.schrodinger.com/.
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Montgomery J. A. Jr.; Vreven T.; Kudin K. N.; Burant J. C.; Millam J. M.; Iyengar S. S.; Tomasi J.; Barone V.; Mennucci B.; Cossi M.; Scalmani G.; Rega N.; Petersson G. A.; Nakatsuji H.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Klene M.; Li X.; Knox J. E.; Hratchian H. P.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Ayala P. Y.; Morokuma K.; Voth G. A.; Salvador P.; Dannenberg J. J.; Zakrzewski V. G.; Dapprich S.; Daniels A. D.; Strain M. C.; Farkas O.; Malick D. K.; Rabuck A. D.; Raghavachari K.; Foresman J. B.; Ortiz J. V.; Cui Q.; Baboul A. G.; Clifford S.; Cioslowski J.; Stefanov B. B.; Liu G.; Liashenko A.; Piskorz P.; Komaromi I.; Martin R. L.; Fox D. J.; Keith T.; Al-Laham M. A.; Peng C. Y.; Nanayakkara A.; Challacombe M.; Gill P. M. W.; Johnson B.; Chen W.; Wong M. W.; Gonzalez C.; Pople J. A.. Gaussian 03, revision E.01; Gaussian, Inc.: Wallingford, CT, 2003.
- Unpublished results.
- Docking studies were carried out using the GOLD 4.0 software (; Verdonk M. L.; Cole J. C.; Hartshorn M. J.; Murray C. W.; Taylor R. D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [DOI] [PubMed] [Google Scholar]; ) with the GoldScore scoring function and 100% search efficiency, all other parameters being used with default values. The binding site was defined as a 15 Å radius sphere around the SG atom of Cys241 from the chain D of the structure 1SA0 (; Ravelli R. B.; Gigant B.; Curmi P. A.; Jourdain I.; Lachkar S.; Sobel A.; Knossow M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202). The 3D structures of ligands were constructed with CORINA 3.44 (Molecular Networks GmbH). [DOI] [PubMed] [Google Scholar]
- Images were created with the Chimera software (Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E.. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612) and the PovRay module (http://www.povray.org/). [DOI] [PubMed] [Google Scholar]
- Herrbach A.; Marinetti A.; Baudoin O.; Guénard D.; Guéritte F. Asymmetric synthesis of an axially chiral antimitotic biaryl via an atropo-enantioselective Suzuki cross-coupling. J. Org. Chem. 2003, 68, 4897–4905. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.