

Abstract
Inhibition of dihydroorotate dehydrogenase (DHODH) for P. falciparum potentially represents a new treatment option for malaria, since DHODH catalyzes the rate-limiting step in the pyrimidine biosynthetic pathway and P. falciparum is unable to salvage pyrimidines and must rely on de novo biosynthesis for survival. We report herein the synthesis and structure–activity relationship of a series of 5-(2-methylbenzimidazol-1-yl)-N-alkylthiophene-2-carboxamides that are potent inhibitors against PfDHODH but do not inhibit the human enzyme. On the basis of efficacy observed in three mouse models of malaria, acceptable safety pharmacology risk assessment and safety toxicology profile in rodents, lack of potential drug–drug interactions, acceptable ADME/pharmacokinetic profile, and projected human dose, 5-(4-cyano-2-methyl-1H-benzo[d]imidazol-1-yl)-N-cyclopropylthiophene-2-carboxamide 2q was identified as a potential drug development candidate.
Keywords: DHODH, malaria, P. falciparum, drug candidate
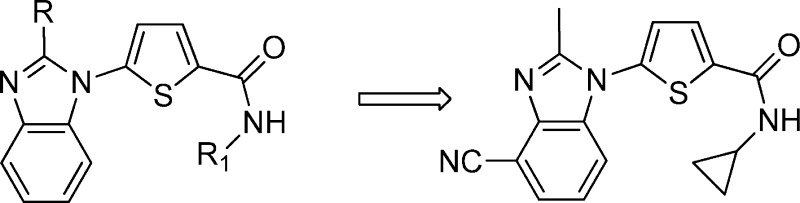
Malaria remains a major global health burden and is endemic in 87 countries where 2.5 billion people are at risk.1 While a number of highly effective chemotherapies have been developed over the years, widespread evolution of resistance has rendered most to be of limited value.2,3 Moreover, the reduced clinical efficacy of the frontline artemisinin derivatives emphasizes the urgent need for new antimalarial drugs.4,5 Given our interest in investigating the inhibition of novel biological targets, we collaborated with the Medicines for Malaria Venture on finding small molecule inhibitors of PfDHODH. Inhibition of human DHODH, the fourth step in the de novo pathway and the rate-limiting step for the synthesis of pyrimidines, has been shown to be effective in the clinic for the treatment of rheumatoid arthritis, resulting in the approval of leflunomide in 1998.6 Unlike the mammalian cell, the parasite is unable to salvage pyrimidines and depends on the de novo pathway for the supply of pyrimidines for survival. In recent years, there have been several reports on the synthesis of potent and selective inhibitors for PfDHODH as possible new targets for the treatment of malaria.7 Phillips recently reported the first example of a PfDHODH inhibitor that was efficacious in a mouse model for malaria.8 Previously, we reported that the benzimidazole analogue 1d had improved physicochemical properties and comparable activity as compared to the orginal hit identified from a high throughput screen (Figure 1).9 Recently, we disclosed that treatment of infected mice in the acute P. berghei efficacy model with 2e resulted in a sterile cure.10 We had also disclosed the in vitro activity data and ADME data for two further advanced analogues 2j and 2q.10 In this letter, we will describe our medicinal chemistry strategy, structure–activity relationship (SAR), and synthesis leading to compounds 2e, 2j, and 2q. Specifically, we will describe how we mitigated CYP and hERG inhibition liabilities of 2e and provide recently generated in vivo data that resulted in the selection of compound 2q as a potential drug development candidate (Figure 1).
Figure 1.

Structures of compounds 1 and 2.
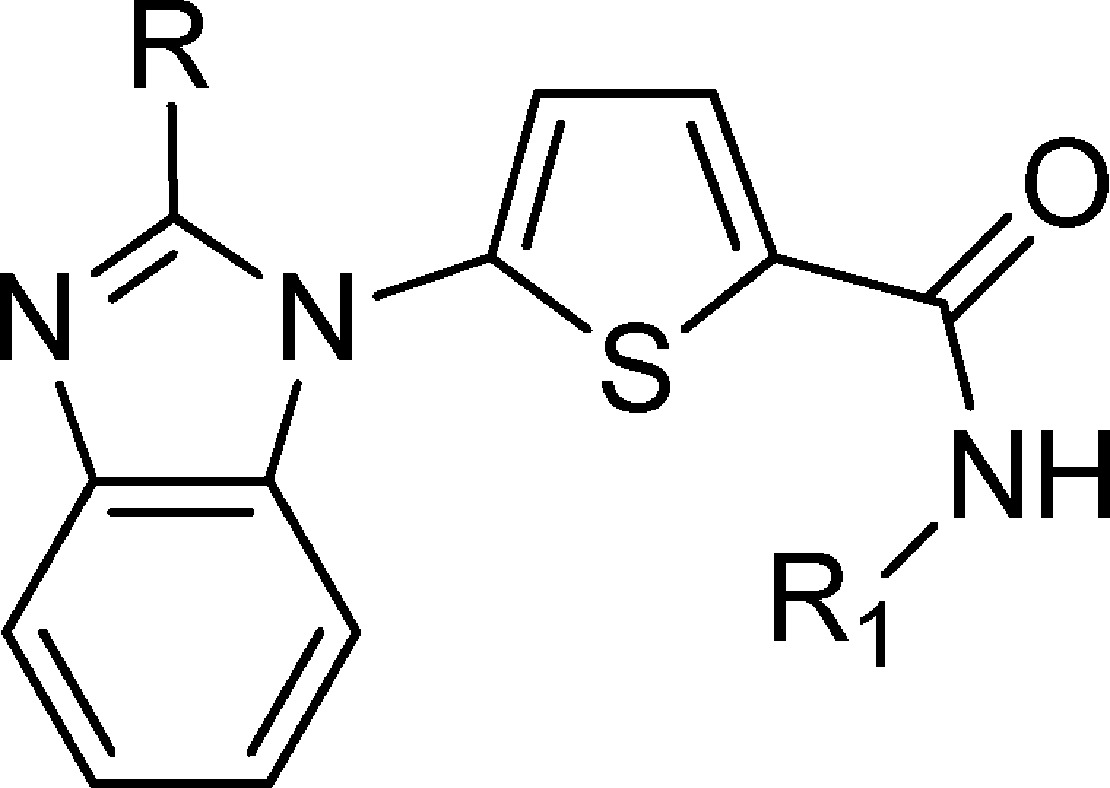
The SAR was based on the enzymatic PfDHODH activity and confirmed in a cell-based assay against the 3D7 and Dd2 drug-sensitive and drug-resistant strains of P. falciparum. In some cases (Table 2), the compounds appear to be more active in the cell-based assay than the enzymatic PfDHODH assay since the enzymatic assay was run at 12.5 nM, limiting our ability to measure inhibition in the very low nanomolar ranges. Our medicinal chemistry starting point was compound 1d, and the first region that we investigated was the effect of substitution at the secondary amide (Table 1). It was clear from the primary and tertiary amides 1a and 1j that a secondary amide was crucial for activity since both of these compounds were inactive in the PfDHODH enzymatic assay (IC50 > 30 μM). Another striking observation was the sensitivity of the PfDHODH activity and cell-based activity to subtle changes in the steric size of the amide. For instance, the PfDHODH IC50 values for compounds 1b (Me), 1c (Et), 1d (n-Pr), and 1h (n-Bu) were 1.08, 0.14, 0.70, and 28.6 μM, an approximate 200-fold decrease in activity between the Et and the n-Bu analogues. Although the Et analogue 1c was the most potent of this series, a moderate enhancement in potency was observed when the substituent was changed to a c-Pr ring 1f with an IC50 of 0.06 μM. This was verified by the 3D7 and Dd2 IC50 values, which were 0.08 and 0.09 μM, respectively. These data suggested that the cyclopropyl group was occupying a defined hydrophobic pocket, and this was confirmed by an X-ray crystal structure from the Philips laboratory10 of 2e bound to PfDHODH.10 Additional evidence that the c-Pr group was occupying a defined hydrophobic pocket was provided by the decrease in potency that was observed as the ring size was gradually increased. For example, in going from c-Pr 1f to c-pentyl 1i, the potency decreased 40-fold.
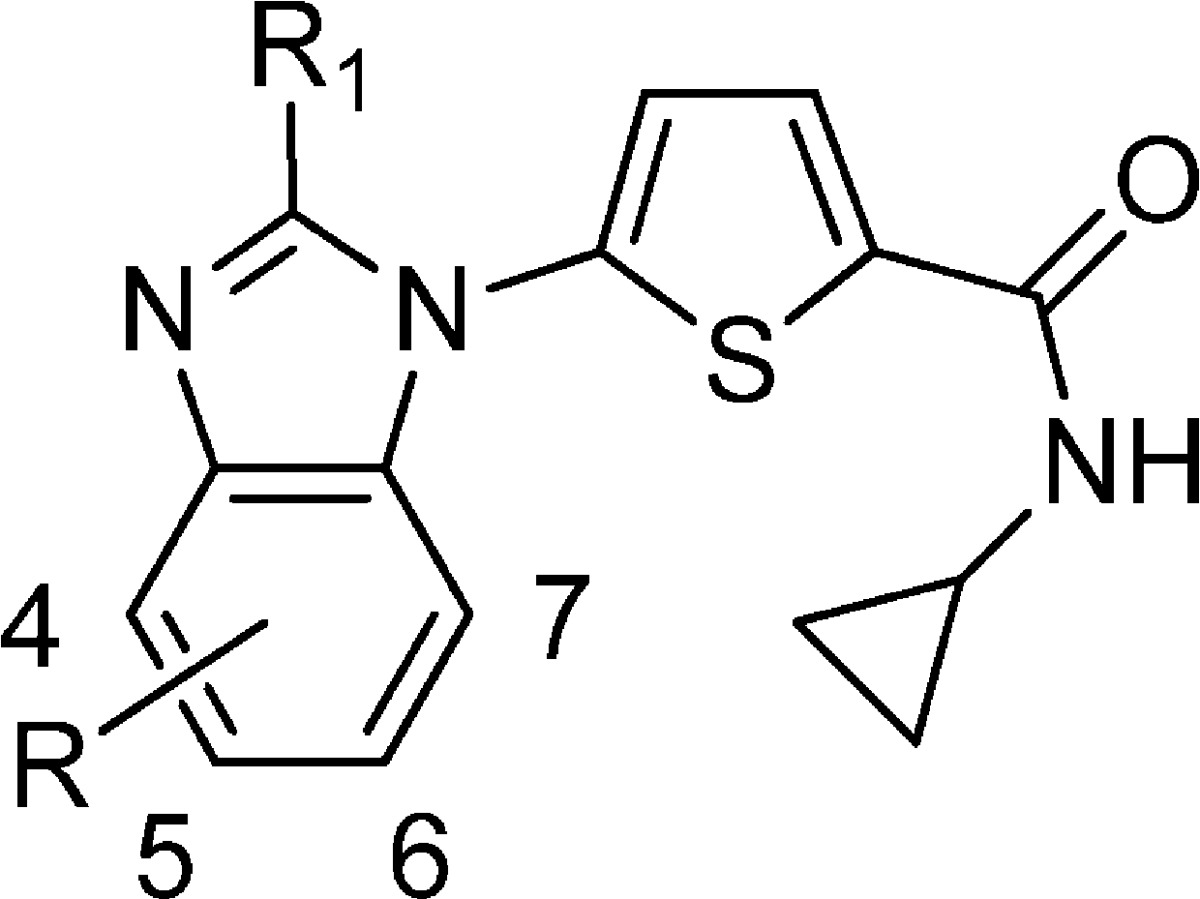
Table 2. Enzyme Inhibition, Parasite Viability, and Microsomal Stability of Compound 2a.
| IC50 (nM) |
predicted CLb (mL/min/kg) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | R | R1 | PfDHODH | HsDHODH | 3D7 | Dd2 | H | M | cLogP |
| 2a | 4-Me | Me | 39 (n = 4) | >30000 | 10 (n = 4) | 22 (n = 4) | 10 | 66 | 3.9 |
| 2b | 5-Me | 56 | 87 | 130 | 8 | >72 | 3.9 | ||
| 2c | 6-Me | 56 | 33 | 74 | 10 | >72 | 3.9 | ||
| 2d | 7-Me | 609 | 143 | 290 | 6 | >72 | 3.9 | ||
| 2e | 5-OCF3 | 22 (n = 4) | 7 (n = 4) | 9 (n = 4) | 7 | 43 | 4.7 | ||
| 2f | 6-OCF3 | 52 (n = 2) | 36 (n = 2) | 53 (n = 2) | 10 | 41 | 4.7 | ||
| 2g | 5-CF3 | 28 | 20 | 28 | 4 | >72 | 4.3 | ||
| 2h | 6-CF3 | 92 | 56 | 96 | 8 | 68 | 4.3 | ||
| 2i | 4-OMe | 162 | 49 | 109 | 4 | >72 | 3.6 | ||
| 2j | 4-OCHF2 | 44 (n = 6) | 19 (n = 6) | 26 (n = 6) | 4 | 33 | 4.1 | ||
| 2k | 4-OCH2CHF2 | 28 | 128 | 113 | na | na | 4.2 | ||
| 2l | 4-F | 34 (n = 2) | 8 (n = 2) | 6 (n = 2) | <4 | 37 | 3.5 | ||
| 2m | 6-F | 111 | 93 | 113 | <4 | >72 | 3.5 | ||
| 2n | 4-Cl | 21 (n = 2) | 5 (n = 2) | 6 (n = 2) | <4 | 19 | 4.1 | ||
| 2o | 5-Cl | 49 | 78 | 102 | na | na | 4.1 | ||
| 2p | 4-Br | 21 (n = 2) | 4 (n = 2) | 7 (n = 2) | 5 | 26 | 4.3 | ||
| 2q | 4-CN | 40 (n = 9) | 7 (n = 4) | 10 (n = 4) | <4 | 26 | 2.9 | ||
| 2r | 4-NMe2 | 181 | 75 | 159 | na | na | 3.9 | ||
| 2s | 4-F, 6-Me | 32 | 10 | 17 | 12 | >72 | 4.0 | ||
| 2t | 4-F, 6-Me | H | 17 | 3100 | 4 | 4 | 10 | >72 | 3.5 |
| 2u | 4-Cl, 6-F | Me | 25 | >30000 | 12 | 13 | 5 | 42 | 4.3 |
| 2v | 4-Cl, 6-F | H | 35 | 5600 | 8 | 18 | 7 | >72 | 3.8 |
| 2w | 5-Me, 7-F | H | 95 | 7300 | 36 | 60 | 11 | >72 | 3.5 |
| 2x | 5-CF3 | H | 19 | 17200 | 12 | 12 | 8 | >72 | 3.8 |
| 2y | 4-F | H | 62 | 14900 | 17 | 8 | 10 | >72 | 3.0 |
Assay conditions reported in ref (10).
Using microsomes and based on well-stirred model with no correction for microsomal or protein binding.
Table 1. Enzyme Inhibition and Parasite Viability of Compound 1a.
| compd | R | R1 | PfDHODH IC50 (μM) | HsDHODH IC50 (μM) | 3D7 IC50 (μM) | Dd2 IC50 (μM) | LC50 (μM)b |
|---|---|---|---|---|---|---|---|
| 1a | H | H | >30 | na | >40 | >30 | na |
| 1b | H | Me | 1.08 | >30 | 0.28 | 0.42 | >62 |
| 1c | H | Et | 0.14 | >30 | 0.10 | 0.15 | >62 |
| 1d | H | n-Pr | 0.70 | >30 | 0.47 | 1.10 | >62 |
| 1e | H | i-Pr | 0.68 | >30 | 0.56 | 0.81 | >62 |
| 1f | H | c-Pr | 0.06 | >30 | 0.08 | 0.09 | >62 |
| 1g | H | c-Bu | 0.42 | >30 | 0.41 | 0.44 | >62 |
| 1h | H | n-Bu | 28.6 | >30 | >20 | 7.30 | na |
| 1i | H | c-Pentyl | 2.51 | >30 | 4.40 | 5.50 | na |
| 1j | H | diMe | >30 | na | >40 | >40 | na |
| 1k | Me | c-Pr | 0.08 (n = 3) | >30 | 0.09 (n = 3) | 0.11 (n = 3) | >62 |
| 1l | Et | c-Pr | 0.11 | >30 | 0.12 | 0.18 | >62 |
| 1m | n-Pr | c-Pr | 0.18 | 15.1 | 0.39 | 0.63 | >62 |
| 1n | OH | c-Pr | >30 | na | >5 | >5 | na |
| 1o | CF3 | c-Pr | 0.57 | >30 | 0.83 | 1.71 | >62 |
| 1p | NMe2 | c-Pr | 5.6 | na | >1 | >1 | >62 |
Assay conditions reported in ref (10).
LC50 is the lethal concentration required to kill 50% of human renal proximal tubule cells.
Having determined that a secondary amide substituted with a c-Pr group afforded the optimal potency, we turned our attention to examine the effect of substitution at the 2-position of the benzimidazole ring (Table 1). Simple substitution of a methyl group 1k resulted in comparable potency to the unsubstituted analogue 1f, with an IC50 of 0.08 vs 0.06 μM, but more importantly, there was an improvement in in vitro hepatic metabolic stability based on data in human microsomes (data not shown). Increasing the steric bulk at the 2-position with an ethyl 1l or n-Pr 1m group was not beneficial from either a potency or an ADME perspective. Moreover, the addition of a polar hydroxyl group 1n or dimethyl amino group 1p was also detrimental to activity.
Compounds 1a–j were prepared as outlined in Scheme 1. The coupling of 5-bromothiophene-2-carboxylic acid 3a with benzimidazole in the presence of a catalytic amount of Cu2O and 2-amino-4,6-dihydroxypyrimidine afforded the key intermediate 3c, which underwent DCC coupling with a variety of commercially available amines to afford the secondary amides 1a–j. Conversely, compounds 1k–n and 1p were obtained by Cu(I)-catalyzed coupling of 5-bromo-N-cyclopropylthiophene-2-carboxamide 3b with the corresponding 2-substituted benzimidazoles in the presence of catalytic amounts of Cu2O and 4,7-dimethoxy-1,10-phenathroline.11
Scheme 1.

The substitution pattern of the thiophene ring was assessed. The 2,5-substitution pattern was found to be optimal since 2,4- and 3,4-substitution resulted in a significant loss in potency. Replacements of the thiophene ring were also examined and these included meta- and para-substituted phenyl, 2,5-substituted N-methyl pyrrole, pyrrole, thiazole, furan, and oxazole (data not shown). None of these replacements resulted in any appreciable activity other than the 2,5-substituted N-methyl pyrrole, which was approximately 3-fold less active than the corresponding thiophene. Although this series was not actively pursued, it was considered an important backup lead.
Next, we investigated the effect of substitution on the benzimidazole ring (Table 2). Simply walking a methyl group around the ring from the 4- to the 7-position (2a–d) resulted in an approximate 2-fold enhancement in activity for analogues 2a–c as compared to 1k but an approximate 8-fold reduction in potency for the 7-methyl analogue 2d, suggesting steric hindrance at the 7-position. This was confirmed by the 5-Me, 7-F analogue 2w (Table 2), which is isosteric to the 5-Me analogue 2b and had comparable enzymatic potency. Interestingly, substitution with electron-withdrawing groups such as CF3 (2g, 2h) and OCF3 (2e, 2f) at the 5- and 6-positions indicated that hydrophobic groups at the 5-position provided a 2–3-fold enhancement in potency as compared to 6-substitution. More pronounced was the difference in cellular activity, for instance, the 3D7 and Dd2 IC50 values were 7 and 9 nM vs 36 and 53 nM (5-OCF3 analogue 2e and 6-OCF3 analogue 2f). On the basis of the observed potency and reasonable ADME profile 2e was selected for efficacy studies.10 However, although 2e was curative when dosed orally in the acute P. berghei (ANKA strain) efficacy model,10 it exhibited significant ADME liabilities. Specifically, there was potential for drug–drug interactions with an observed IC50 of 0.1 μM for the CYP2D6 isozyme and a potential cardiovascular safety risk based on a hERG IC50 of <2.8 μM.
We determined that the ADME properties could be improved by reducing the lipohilicity as measured by cLogP. Our first strategy was to focus on 4-substituents since the 4-Me analogue 2a was approximately 3–6-fold more potent than the 5-Me and 6-Me analogues 2b−c (Table 2) in the cellular 3D7 and Dd2 assays. We were encouraged by the more hydrophilic 4-OMe analogue 2i (cLogP of 3.6), which was 9-fold less potent than 2e but did not inhibit hERG or CYP. In an attempt to improve the potency, the lipohilicity was slightly increased by preparing the 4-OCHF2 analogue 2j (cLogP of 4.1). Gratifingly, the potency was approximately 4-fold higher than 2i, PfDHODH IC50 of 44 vs 162 nM with a cellular activity in the range of 19–26 nM. More importantly, the P450 inhibition was >10 μM for all five primary drug-metabolizing isozymes, and the hERG IC50 was 28.8 μM. As a consequence, 2j was chosen for further in vivo evaluation.10
Although 2j had an acceptable profile, there was room for further potency enhancement. As a result, a series of substituents at the 4-position were prepared (Table 2), and it was found that electron-withdrawing groups such as F, Cl, Br, and CN (2l–q) afforded an approximate 5-fold gain in cellular potency as compared to 2j. For example, the IC50 of the 4-F compound 2l was 8 and 6 nM in 3D7 and Dd2, respectively. However, when considering physicochemical properties, potential drug–drug interactions mediated via CYPP450 inhibition and potential cardiovascular safety the 4-CN analogue 2q had the best overall profile.
The second strategy that was adopted to optimize the ADME/PK properties of 2j was to prepare the des 2-methyl benzimidazole analogues (2t, 2v–w, 2p) since the cLogP was reduced by approximately half a log unit (Table 2). These compounds were synthesized by the Cu(I)-catalyzed coupling of 3b with the corresponding substituted benzimidazoles in the presence of catalytic amounts of Cu2O and quinolin-8-ol in DMSO (Scheme 1). On the basis of the SAR from Table 1, we anticipated that the in vitro metabolic stability would be poorer for the des-methyl compounds, and indeed, this was the case. We were surprised, however, to observe that the selectivity for HsDHODH with the des-methyl compounds increased in the range of 3100–1700 vs >30000 nM for the 2-methyl analogues. Because of the poorer selectivity and low in vitro metabolic stability, these compounds were not further pursued.
The 5- and 6-substituted analogues were prepared by the Pd(0)-catalyzed coupling of the appropriately substituted 1-bromo-2-nitrobenzenes (4b–c, 4f–k) (Scheme 2) with 3d in the presence of Xantphos to afford intermediates 5 in good yield similar to a procedure reported by Hornberger et al.12 The intermediates 5 were readily transformed to the target compounds (2b–c, 2e–k, 2x) using the following four step sequence. The nitro group of 5 was reduced to the amine by hydrogenation in the presence of Pd(OH)2/C followed by cyclization with (EtO)3CMe to give the substituted benzimidazoles. To complete the synthesis, the ester was hydrolyzed to the acid using LiOH, which was then converted to the cyclopropyl amide via the acid chloride. It should be pointed out that during the cyclization reaction substituting (MeO)3CH for (EtO)3CMe afforded the des methyl compound 2x.
Scheme 2.

Compounds with electron-withdrawing groups (F, Cl, CN) at the 4-position (2l–n, 2q) were synthesized by the direct coupling of 3-fluoro-2-nitro(substituted)benzene (4l-o, 4q) with 3d in the presence of KOH and catalytic amounts of BnEt3NCl in DMF to afford the intermediates 5a. For the synthesis of the nitrile 2q, the nitro group of 5a was reduced with SnCl2 in HCl followed by cyclization to the benzimidazole and hydrolysis of the ester as described above. However, to avoid side reactions with the nitrile, the amide was formed via peptide coupling methodology using HCTU and cyclopropylamine to afford 5-(4-cyano-2-methyl-1H-benzo[d]imidazol-1-yl)-N-cyclopropylthiophene-2-carboxamide 2q (Scheme 2).
We examined the species selectivity for the series and found that the compounds exhibited equipotent activity against DHODH from P. falciparum, P. vivax, and P.berghei, an important attribute for developing a candidate with pan-parasite activity. Additionally, 2a–s and 2u lack activity against the human enzyme (HsDHODH IC50 > 30 μM). The series also showed an excellent correlation between enzymatic activity (DHODH from P. falciparum, P. berghei, and P. vivax) and cellular activity (3D7 and Dd2),10 suggesting that the species specificity exhibited for DHODH inhibition correlates with a specific effect on the parasites as compared with representative mammalian cells.
Additional in vivo and in vitro data for compound 2q including rodent 7 day repeat dose toxicology studies have been completed (Table 3). The pharmacokinetics of 2q in rat and dog were determined, and it was found to have moderate oral bioavailability (F %), 49 and 19%, respectively. The in vitro ADME profile was encouraging, particularly the predicted low hepatic metabolic clearance determined in human microsomes and heptocytes, <0.7 and <0.6 mL/min/kg, respectively. In a repeat dose toxicology study in rat at 0, 50, 150, 450, and 600 mg/kg PO QD for 7 days followed by a 7 day washout period, the NOAEL was >600 mg/kg (Cmax, 29 μg/mL; AUC, 328 μg*h/mL), and the predicted human therapeutic margin was >5.5. No off-target activities in a 54-receptor pharmacology panel (Cerep ExpresSProfile) at 10 μM were observed corroborating on target activity against pfDHODH. In addition, an Ames test using Salmonella typhimurium TA98, 100, and 1535, with and without S9 induction, revealed no evidence of activity at concentrations up to 100 μM.
Table 3. Pharmacokinetics, in Vitro ADMEa Properties, and in Vivo Efficacy of 2q.
| IV dose (1 mg/kg) |
PO dose (3 mg/kg) |
||||||
|---|---|---|---|---|---|---|---|
| species | AUC0-inf (ng*h/mL) | CL (mL/min/kg) | Vdss (mL/kg) | t1/2 (h) | AUC0-inf (ng*h/mL) | Cmax (ng/mL) | F (%) |
| rat | 703 | 24.0 | 995 | 0.85 | 1038 | 618 | 49 |
| dog | 1500 | 6.7 | 279 | 0.52 | 359 | 1110 | 19 |
| species | microsomes predicted CLb (mL/min/kg) | hepatocytes predicted CLc (mL/min/kg) | protein binding (%) | CYP450d IC50 (μM) | PAMPA permeability × 10−6 cm/s |
|---|---|---|---|---|---|
| mouse | 5.8 | 14 | 81.8 | na | 49 |
| rat | 5.3 | 16 | 78.0 | na | |
| dog | 3.8 | 12 | 68.8 | na | |
| human | <0.7 | <0.6 | 88.8 | all > 10 |
| in vivo efficacy model | ED50 (mg/kg/day) | ED90 (mg/kg/day) | ED99 (mg/kg/day) |
|---|---|---|---|
| P. berghei ANKA | 13 | 28 | 62 |
| P. falciparum 3D70087/N9 | 13 | 27 | 60 |
Assay conditions reported in ref (10).
Using well-stirred model with correction for microsomal and protein binding.
Using well-stirred model with correction for protein binding.
CYP1A2, CYP2C9, CYP2C19, CYP3A4, and CYP2D6 were evaluated.
Lastly, the efficacy of 2q was evaluated by PO dosing in two additional mouse models of malaria, the P. berghei ANKA and P. falciparum 3D70087/N9 models, and the ED50, ED90, and ED99 values ranged from 13 to 62 mg/kg/day, demonstrating the predicted potency. On the basis of these data and previously reported data10 [11% free fraction of drug in human plasma, no P450 inhibition (IC50 > 10 μM), hERG IC50 of 53 μM], 2q was considered for development.
In conclusion, we successfully optimized an early lead to generate a novel low molecular weight compound that is orally bioavailable in three species and efficacious in three mouse models of malaria. On the basis of these results and the favorable druglike properties, 2q has been chosen as a potential drug development candidate.
This work was supported by SPARC funds from the Broad Institute, with funds from Medicines for Malaria Venture, and with support from the Humanitarian Assistance for Neglected Diseases Initiative at Genzyme Corporation. Genzyme is committed to assisting in discovery of drugs for neglected diseases in collaboration with other partners and seeks no profit from drugs used for these applications.
Supporting Information Available
Experimental procedures and characterization data for the synthesis for compounds 1b–f,i,k,m,o, 2a–x, 3c–d, and 4a,q. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Greenwood B. M.; Fidock D. A.; Kyle D. E.; Kappe S. H.; Alonso P. L.; Collins F. H.; Duffy P. E. Malaria: Progress, perils, and prospects for eradication. J. Clin. Invest. 2008, 118, 1266–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava I. K.; Morrisey J. M.; Darrouzet E.; Daldal F.; Vaidya A. B. Resistance mutations reveal the atovaquone-binding domain of cytochrome b in malaria parasites. Mol. Microbiol. 1999, 33, 704–711. [DOI] [PubMed] [Google Scholar]
- Vleugls M. P.; Wetsteyn J. C.; Meuwissen J. H. Fansidar-resistant plasmodium falciparum infection from Tanzania. Trop. Geogr. Med. 1982, 34, 263–265. [PubMed] [Google Scholar]
- Yang H.; Liu D.; Yang Y.; Fan B.; Yang P.; Li X.; Li C.; Ying D.; Yang C. Changes in susceptibility of Plasmodium falciparum to artesunate in vitro in Yunnan Province, China. Trans. R. Soc. Trop. Med. Hyg. 2003, 97, 226–228. [DOI] [PubMed] [Google Scholar]
- Dondorp A. M.; Nosten F.; Yi P.; Das D.; Phyo A. P.; Tarning J.; Iwin K. M.; Ariey F.; Hanpithakpong W.; Lee S. J.; Ringwald P.; Silamut K.; Imwong M.; Chotivanich K.; Lim P.; Herdman T.; Sam An, S.; Yeung S.; Singhasivanon P.; Day N. P. J.; Lindergarth N.; Socheat D.; White N. J. Artemisinin resistance in plasmodium falciparum malaria. N. Engl. J. Med. 2009, 361, 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolen J. S.; Kalden J. R.; Scott D. L.; Rozman B.; Kvien T. K.; Larsen A.; Loew-Friedrich I.; Oed C.; Rosenburg R. Efficacy and safety of leflunomide compared with placebo and sulphasalazine in active rheumatoid arthritis: A double-blind, randomised, multicentre trial. European Leflunomide Study Group. Lancet 1999, 353, 259–266. [DOI] [PubMed] [Google Scholar]
- Phillips M. A.; Rathod P. K. Plasmodium dihydroorotate dehydrogenase: A promising target for novel anti-malarial chemotherapy. Infect. Disord.: Drug Targets 2010, 10, 226–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujjar R.; Mawaha A.; Mazouni F. E; White J.; White K. L.; Creason S.; Shackleford D. M.; Baldwing J.; Charman W. N.; Buckner F. S.; Charman S.; Rathod P. K.; Phillips M. A. Identification of a metabolically stable triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with antimalaria activity in mice. J. Med. Chem. 2009, 52, 1864–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel V.; Booker M.; Kramer M.; Ross L.; Celatka C.; Kennedy l. M.; Dvorin J. D.; Duraisingh M. T.; Sliz P.; Wirth D.; Clardy J. Identification and characterization of small molecule inhibitors of plasmodium falciparum dihydroorotate dehydrogenase. J. Biol. Chem. 2008, 283, 35078–35085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker M. L.; Bastos C. M.; Kramer M. L.; Barker R. H. Jr.; Skerlj R.; Sidhu A. B.; Deng X.; Celatka C.; Cortese J. F.; Guerrero Bravo J. E.; Crespo Llado K. N.; Serrano A. E.; Barturen I. A.; Jimenez Diaz M. B.; Viera S.; Garuti H.; Wittlin S.; Papastogiannidis P.; Lin J.; Janse C. J.; Khan S. M.; Duraisingh M.; Coleman B.; Goldsmith E. J.; Phillips M. A.; Munoz B.; Wirth D. F.; Klinger. J. D.; Weigand R.; Sybertz E. Novel inhibitors of plasmodium falciparum dihydroorotate dehydrogenase with antimalaria activity in the mouse model. J. Biol. Chem. 2010, 285, 33054–33064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman R. A.; Koval E. D.; Buchwald S. L. Copper-catalyzed N-arylation of imidazoles and benzimidazoles. J. Org. Chem. 2007, 72, 6190–6199. [DOI] [PubMed] [Google Scholar]
- Hornberger K. H.; Badiang J. G.; Salovich J. M.; Kuntz K. W.; Emmitte K. A.; Cheung M. Regioselective synthesis of benzimidazole thiophene inhibitors of polo-like kinase. Tetrahedron Lett. 2008, 49, 6348–6351. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.