

Abstract
MLN4924 is a selective inhibitor of the NEDD8-activating enzyme (NAE) and has advanced into clinical trials for the treatment of both solid and hematological malignancies. In contrast, the structurally similar compound 1 (developed by Millennium: The Takeda Oncology Company) is a pan inhibitor of the E1 enzymes NAE, ubiquitin activating enzyme (UAE), and SUMO-activating enzyme (SAE) and is currently viewed as unsuitable for clinical use given its broad spectrum of E1 inhibition. Here, we sought to understand the determinants of NAE selectivity. A series of compound 1 analogues were synthesized through iterative functionalization of the purine C6 position and evaluated for NAE specificity. Optimal NAE specificity was achieved through substitution with primary N-alkyl groups, while bulky or secondary N-alkyl substituents were poorly tolerated. When assessed in vitro, inhibitors reduced the growth and viability of malignant K562 leukemia cells. Through this study, we have successfully identified a series of sub-10 nM NAE-specific inhibitors and thereby highlighted the functionalities that promote NAE selectivity.
Keywords: NAE inhibitors, leukemia, anticancer drugs, proteasomal inhibitors
Ubiquitin (Ub) and ubiquitin-like (Ubl) proteins, such as neural precursor cell-expressed developmentally downregulated 8 (NEDD8) and small ubiquitin-related modifier (SUMO), are essential mediators of cellular function.1−3 Through a cascade of three enzymatically catalyzed events, Ub/Ubls are conjugated to target proteins, designating them for various fates such as degradation, translocation, signaling, and regulation of transcriptional activity.4,5 In an ATP-dependent step, Ub/Ubl binds to the Ub or Ubl activating enzyme (E1), resulting in Ub/Ubl adenylation. Subsequent conformational changes in E1 structure mediate intramolecular nucleophilic attack of the adenylated Ub/Ubl by a cysteine residue on E1, covalently attaching Ub/Ubl to E1 through a thioester linkage (Ub/Ubl-E1). A second Ub/Ubl is adenylated by E1, resulting in a doubly Ub/Ubl loaded E1. Next, Ub/Ubl is transferred to the Ub or Ubl conjugating enzyme (E2) via transthiolation, which, in concert with a Ub or Ubl ligase (E3), covalently attaches the Ub/Ubl to target proteins.6,7
NEDD8-labeled proteins play a crucial role in the assembly and function of the largest family of E3 Ub ligases, the cullin ring ligases (CRL).8 NEDDylated cullin proteins serve as a scaffold for the assembly of the CRL and enhance CRL efficiency by increasing the recruitment of Ub-E2 complexes and assisting in their most efficient positioning relative to the substrate to be labeled.9,10 These CRLs are therefore responsible for the ubiquitination and targeted degradation of a plethora of proteins within the cell. Other important NEDD8 targets include the mouse double minute 2 (MDM2) oncogene product,11 the p53 tumor suppressor protein,12 the von Hippel-Lindau protein (pVHL),13 the epidermal growth factor (EGF) receptor,14 the breast cancer-associated protein 3 (BCA3),15 and the estrogen receptor-α (ERα).16 Elevated levels of NEDDylation have been observed in aggressively proliferating malignant cells and have thus been identified as a promising target for cancer therapeutics.17
Recent studies by Millennium: The Takeda Oncology Company reported an AMP mimetic, compound 1 (Figure 1), composed of a purine base substituted at C6 and N9 with a hydrophobic 2,3-dihydroindene moiety and a ribose sulfamate substituent, respectively.18,19 Compound 1 strongly bound and equipotently inhibited NEDD8 activating enzyme (NAE), ubiquitin activating enzyme (UAE), and SUMO activating enzyme (SAE) with a low nanomolar potency (IC50 = 5 nM). Inhibition of NAE by compound 1 is achieved through a NEDD8–inhibitor adduct formed in situ that inhibits the normal progression of the cycle by preventing transthiolation to E2. The nucleophilic sulfamate attacks the thioester NEDD8-E1 intermediate, resulting in a covalently bound NEDD8–inhibitor adduct.19 This adduct effectively inhibited the activity of NAE, leading to increased levels of CRL proteins in cells treated with compound 1. MLN4924 (2, Figure 1),20 an analogous inhibitor also developed by the Millennium group, is a similarly potent NAE inhibitor but is significantly more specific for NAE than compound 1. The backbone of MLN4924 is composed of a pyrrolopyrimidine base, substituted at C6 and N9 with a hydrophobic 2,3-dihydroindene moiety and a (hydroxycyclopentyl)methylsulfamate appendage, respectively. In vitro, this compound reduces the growth and viability of malignant cells including HCT-116 (colorectal), Calu-6 (lung), SKOV-3 (ovarian), CWR22 (prostate), OCI-LY19 (lymphoma), and AML (acute myeloid leukemia).20−23 In vivo studies of MLN4924 with solid tumor xenografts (HCT-116, H522, and Calu-6) showed significant inhibition of tumor growth, and AML xenografts showed disease regression.20,23 Currently, this drug is being evaluated for the treatment of leukemia and lymphoma.
Figure 1.
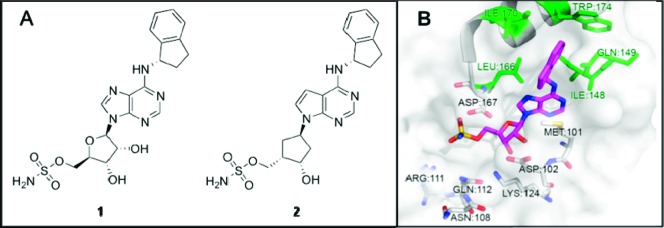
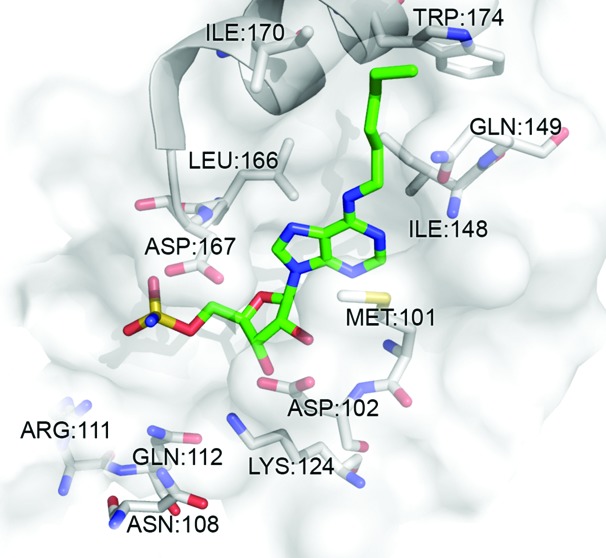
(A) Chemical structures of compound 1 and MLN4924 (2) and (B) GOLD docking of compound 1 in the ATP binding pocket of NAE (PDB: 3GZN).
The structures of compound 1 and MLN4924 are similar, but the molecules differ significantly in their specificity for NAE. Here, we explored the determinants of NAE selectivity. Virtual docking of compound 1 within the ATP pocket of NAE (PDB: 3GZN), using genetic optimization for ligand docking (GOLD) software,24 showed that the 2,3-dihydroindene moiety (present in both compound 1 and MLN4924) bound in the region above the ATP pocket formed by residues Ile148, Gln 149, Ile 170, and Trp174 in NAE (highlighted in green in Figure 1). This “gatekeeper” region of the ATP pocket appeared to be an attractive target for developing isoform-specific E1 inhibitors. Thus, with particular emphasis on understanding the determinants of NAE specificity, we prepared a series of 45 C6 N-alkylated adenosine-sulfamate analogues, incorporating substituents of varying size, flexibility, and functionality. From this focused library, we identified a series of NAE-specific inhibitors that mirror MLN4924's potency toward NAE but have reduced activity against other E1s, as compared with compound 1.
Briefly, C6 N-alkylated adenosine-sulfamate analogues, 8–51, were prepared via a facile synthetic route outlined in Scheme 1. First, the 2′,3′-ribose diol of 6-chloropurine riboside (3) was ketal protected using acetone to furnish 4 (95%). Next, microwave-assisted nucleophilic aromatic substitution of the 6-chloropurine with a series of primary and secondary amines produced 5(a–z, aa–as) in excellent yields (70–97%). Subsequent deprotonation of the 5′-hydroxyl group using NaH in THF and reaction with a preprepared sulfamoyl chloride reagent25 furnished the sulfamate 6(a–z, aa–as) in good yields (63–88%). Finally, removal of the ketal protecting group was achieved under acidic conditions using TFA:H2O to yield products 8–51 (50–89%). Compound 7 was synthesized in an analogous manner to that described in Scheme 1, starting from adenosine instead of 6-chloropurine riboside.
Scheme 1.

Inhibitors were principally assessed for effects on Ub/Ubl-E2 conjugate formation via a modified enzymatic assay, described previously26 and in the Supporting Information. Briefly, purified human recombinant E1 proteins were incubated with their respective Ub/Ubl and E2 proteins in a buffer containing 50 mM HEPES, pH 7.4, 5 mM MgCl2, 1–20 μM ATP, and increasing concentrations of compounds 7–51. The protein complexes generated were separated on a 10–20% gradient nondenaturing sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), followed by immunoblotting. The His6-tagged Ub–UbcH6 complex was detected using a mouse monoclonal primary antibody (1:2500) specific for His6. Rabbit polyclonal primary antibodies were used for the detection of other E2–Ubl complexes [anti-SUMO-1 (1:500) for SAE1/SAE2, anti-NEDD8 (1:500) for APPBP1-UBA3, and anti-UFM1 (1:1000) for UBA5]. Ub/Ubl–E2 complexes were revealed using enhanced chemiluminescent detection. As a control, we demonstrated that the NEDD8 linkage with the E1 and E2 loading was sensitive to reducing agents (Supporting Information). Densitometry was used to semiquantify the efficacy of the inhibitors in the gel-based assays.
The effect of substituting the C6 position of compound 1 with a variety of N-substituted amines on E1 enzymatic activity was examined (representative examples shown in Figure 2 and in the Supporting Information). In general, we found that the C6 analogues were selective for NAE over UAE, SAE, and UFM1 activating enzyme (UBA5) (Figure 2), regardless of the C6 amino substituent. The addition of sterically hindered substituents at C6 had detrimental effects on the potency of molecules against all E1 isoforms; however, these modifications were better tolerated by NAE (greater inhibition of activity) than UAE or SAE (Figure 2). We also noted that all molecules showed no effect against UBA5, even when tested at 10 μM. This suggests that the NAE specificity can be achieved with larger, more sterically hindered purine substrates.
Figure 2.

In vitro evaluation of compound 1 analogues against E1 enzymes. Inhibitors were evaluated in cell-free enzymatic assays to determine their effects on the activity of NAE (A), UAE (B), SAE (C), and UBA5 (D). Increasing concentrations of analogues were added to the appropriate E1, E2, and Ub/Ubl substrate along with ATP. After 1.5 h of incubation at room temperature, products were fractioned on SDS-PAGE gels followed by immunoblotting. Concentration of inhibitor: lane 0, 0 μM; lane 1, 0.001 μM; lane 2, 0.01 μM; lane 3, 0.1 μM; lane 4, 1 μM; and lane 5, 10 μM. Millennium's compound 1 and MLN4924 (2).
We were able to identify a number of interesting structure–activity relationship (SAR) trends, which help further explain the basis for NAE selectivity. Moreover, these SAR trends may be useful for the development of new E1-targeting scaffolds. First, the data revealed that compounds incorporating hydrophobic, straight chain, N-alkyl substituents at C6 were among the most potent NAE inhibitors, that is, 13, where R = n-hexyl, NAE IC50 < 10 nM (Figure 2). Interestingly, the length of the N-alkyl chain was shown to play an important role in NAE binding potency. For example, at 1 nM, when R = methyl (8) or n-propyl (9), there was no reduction in the formation of the UBC12–NEDD8 complex. However, when extended to either an n-butyl (11) or n-pentyl (12) chain, we observed <58 and <62% NAE inhibition, respectively, at 1 nM (Supporting Information). Moreover, further extension using an n-hexyl substituent (13) showed >70% inhibition of NAE activity at the same concentration. At this chain length, we believe that the alkyl group optimally occupies a hydrophobic region composed of residues Ile148, Gln149, Ile 170, and Trp174 (Figure 3). Further extension using an n-heptyl appendage (14) reduced NAE inhibition (no inhibition at 1 nM, Figure 2), suggesting that chain lengths >6 carbons are not well tolerated by NAE.
Figure 3.
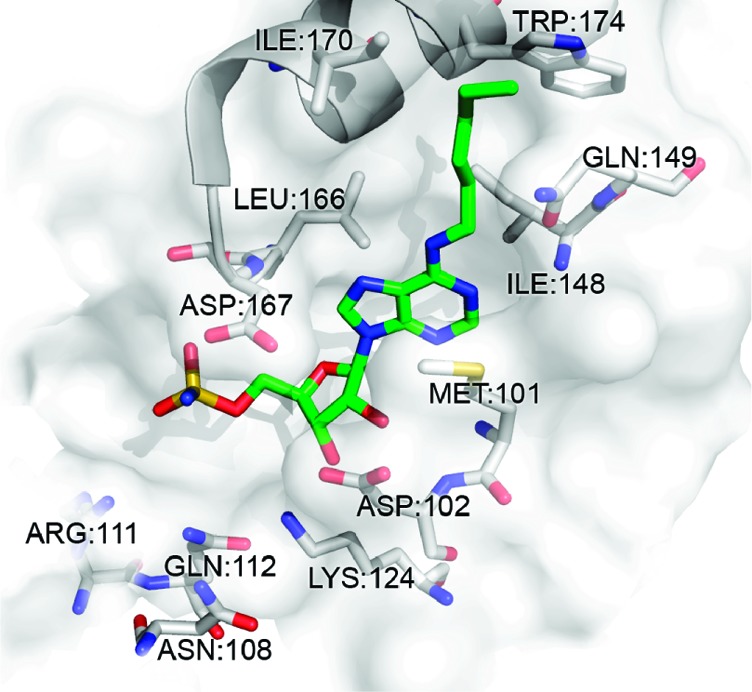
Low energy GOLD-docked 13 (green) in the ATP binding pocket of NAE (PDB: 3GZN).
As mentioned previously, incorporating steric bulk proximal to the C6 nitrogen negatively impacted inhibition of NAE activity. For example, in the straight chain alkyl series of compounds, decreased potency was observed when the carbon α to the C6 nitrogen was substituted with either a methyl (21, where R = 2-butyl, <25% inhibition of human UBC12–NEDD8 complex at 10 nM cf. 9, where R = n-propyl, <63% inhibition human UBC12–NEDD8 complex at 10 nM, Figure 2) or a gem-dimethyl group (20, R = 1,1,3,3-tetramethylbutylamine, no inhibition of UBC12–NEDD8 complex at 10 nM cf. 19, where R = 3,3-dimethylbutylamine, <29% inhibition of UBC12–NEDD8 complex at 10 nM, Figure 2). Interestingly, when 13 was substituted with a methyl group, as in 22, the compounds equipotently inhibited NAE (Figure 2). We believe the incorporation of the optimal alkyl chain overcomes the requirement for minimal steric bulk around the C6 nitrogen. When C6 was substituted with a series of tertiary amines, no inhibition of NAE activity was observed at concentrations <10 nM (i.e., 26–30, 34, 46, 47, and 50; see 27, 30, 34, and 50 in Figure 2; see 26 and 47 in the Supporting Information; 28, 29, and 46 data not shown). At higher concentrations, inhibitory effects were observed for tertiary amine analogues; however, the corresponding secondary amines all elicit higher potency than their more substituted counterparts (11 > 26, 15 > 28, and 33 > 34, data not shown). These data suggested that the peripheral region of the ATP-binding site is sterically constrained. In support of this theory, the unsubstituted 6-aminopurine (7) and N-methylated 6-aminopurine (8) both showed promising activity (7, <16% inhibition at 1 nM, and 8, <76% inhibition at 10 nM; Supporting Information). We also noted that cyclic aliphatic compounds, employed to reduce entropic binding costs and enhance binding efficiency, were less potent inhibitors of NAE than analogous straight chain alkyl groups (12 > 31, 13 > 38, 14 > 48, Supporting Information). Interestingly, we observed higher disruption of both NAE and UAE activity by increasing the ring size of the cyclic alkyl appendage (i.e., 5- < 6- < 7- < 8-membered rings) with compound 49 (an 8-membered ring) showing similar activity to 13 against NAE. However, increasing the ring size beyond eight members, using a 2-methyl-15-crown-5 appendage in 51, resulted in lower activity against both NAE and UAE, relative to 49.
We next evaluated a series of heterocyclic derivatives. The most potent inhibitors in this class of compounds were identified as the furan-3-ylmethanamine (33) and thiophen-3-ylmethanamine (35), which equipotently inhibited NAE activity (∼100% inhibition at 10 nM). However, both heterocyclic analogues were less potent than our N-hexyl derivative (13). Comparative inhibition of E1 activity by 13 and 35 is shown in Figure 2. Additionally, we confirmed that active compounds 1 and 13 inhibited loading of NEDD8 onto UBA3 and APPB1 (Supporting Information).
Our SAR identified determinants of selectivity for NAE as compared to UAE and SAE. However, AMP mimetics may exhibit promiscuity for other enzymes with ATP binding sites, such as kinases. Therefore, we evaluated the specificity of our lead NAE inhibitor, 13, against a panel of ATP-dependent kinases including JAK1, JAK3, ABL, and EGFR (Supporting Information). Encouragingly, 13 showed no activity against the selected kinases at a concentration of 100 nM.
Chemical and genetic inhibition of NAE reduces the proliferation of malignant cells in culture. Therefore, we evaluated the effects of our analogues on the growth and viability of leukemia cells. K562 leukemia cells were treated with increasing concentrations of selected analogues for 72 h. After incubation, cell growth and viability were measured by the CellTiter-Glo Luminescent Cell Viability assay (Table 1). Encouragingly, the ability of compounds to inhibit the proliferation of K562 matched their ability to inhibit NAE.
Table 1. Growth Inhibition (EC50) of K562 Leukemia Cells by Compound 1 Analogues (n = 2).
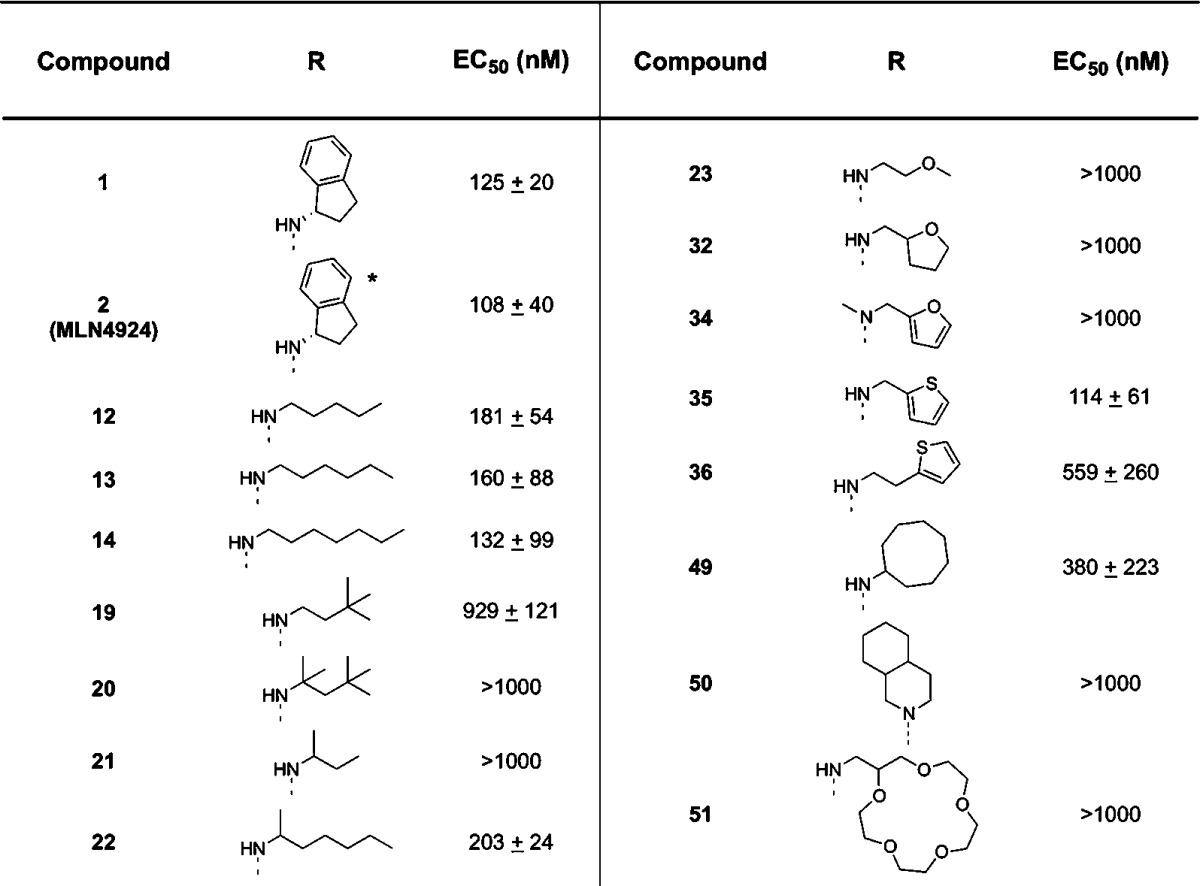
The backbone for compound 2 (MLN4924) differs from the other compounds listed. See Figure 1 for the complete structure.
We also determined the effects of 13 on the activity of NAE in vitro. K562 cells were treated with increasing concentrations of 13 for 24 h. After incubation, cells were harvested, and total cellular protein was isolated. The presence of NEDD8–cullin conjugates was evaluated by immunoblotting. Compound 13 inhibited the enzymatic activity of NAE as evidenced by reductions in NEDD8–cullin conjugates (Figure 4). Moreover, the concentration of 13 required to inhibit NAE matched the concentration required to inhibit cellular proliferation. Although 13 inhibited NEDD8 activity at concentrations associated with and times that preceded loss of cell growth and viability, we cannot exclude additional off-target effects that may contribute to cytotoxicity. Likewise, we cannot exclude additional targets for the other analogues tested in the cell growth and viability assays.
Figure 4.
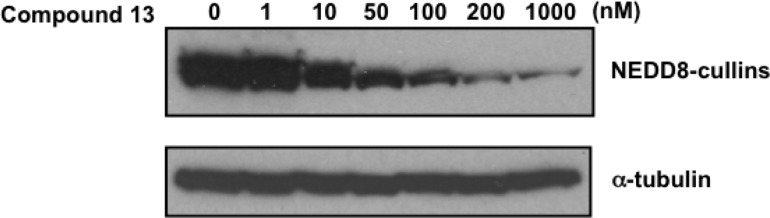
Inhibition of NEDD8 conjugation in K562 cells by 13.
Conclusions
Our preliminary SAR study of the purine C6 position identified selective and highly potent NAE inhibitors via substitution with N-aliphatic R groups. In particular, 13, which contains an N-hexyl R group, exhibited impressive inhibition with a potency that rivals MLN4924 (Figure 2). Finally, compounds containing furan and thiophene moieties (33 and 35, respectively), which could provide increased metabolic stability, showed minor reductions in potency, as compared to 13. Thus, by understanding the determinants of NAE selectivity, additional NAE inhibitors may be developed with different stability and metabolic profiles and offer insight into the design of selective inhibitors that target UAE and SAE.
This work was supported by the Ontario Ministry for Innovation (P.T.G.) and by an Ontario Postdoctoral Fellowship (J.L.L.).
Supporting Information Available
Full synthetic details, experimental procedures, and copies of 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ These three authors contributed equally to this work.
Supplementary Material
References
- Glotzer M.; Murray A. W.; Kirschner M. W. Cyclin is Degraded by the Ubiquitin Pathway. Nature 1991, 349, 132–138. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Yoshida Y.; Noda M. Cloning of a cDNA Which Encodes a Novel Ubiquitin-like Protein. Biochem. Biophys. Res. Commun. 1993, 195, 393–399. [DOI] [PubMed] [Google Scholar]
- Ihara M.; Koyama H.; Uchimura Y.; Saitoh H.; Kikuchi A. Noncovalent Binding of Small Ubiquitin-Related Modifier (SUMO) Protease to SUMO is Necessary for Enzymatic Activities and Cell Growth. J. Biol. Chem. 2007, 282, 16465–16475. [DOI] [PubMed] [Google Scholar]
- Kerscher O.; Felberbaum R.; Hochstrasser M. Modification of Proteins by Ubiquitin and Ubiquitin-Like Proteins. Annu. Rev. Cell Dev. Biol. 2006, 22, 159–180. [DOI] [PubMed] [Google Scholar]
- Hoeller D.; Hecker C. M.; Dikic I. Ubiquitin and Ubiquitin-Like Proteins in Cancer Pathogenesis. Nat. Rev. Cancer 2006, 6, 776–788. [DOI] [PubMed] [Google Scholar]
- Haas A. L.; Siepmann T. J. Pathways of Ubiquitin Conjugation. FASEB J. 1997, 11, 1257–1268. [DOI] [PubMed] [Google Scholar]
- Jentsch S. The Ubiquitin-Conjugation System. Annu. Rev. Genet. 1992, 26, 179–207. [DOI] [PubMed] [Google Scholar]
- Read M. A.; Brownell J. E.; Gladysheva T. B.; Hottelet M.; Parent L. A.; Coggins M. B.; Pierce J. W.; Podust V. N.; Luo R.; Chau V.; Palombella V. J. Nedd8Modification of Cul-1 Activates SCF Beta(TrCp)-Dependent Ubiquitination of I Kappa B Alpha. Mol. Cell. Biol. 2000, 20, 2326–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami T.; Chiba T.; Suzuki T.; Iwai K.; Yamanaka K.; Minato N.; Suzuki H.; Shimbara N.; Hidaka Y.; Osaka F.; Omata M.; Tanaka K. NEDD8 Recruits E2-Ubiquitin to SCF E3 Ligase. EMBO J. 2001, 20154003–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda D. M.; Borg L. A.; Scott D. C.; Hunt H. W.; Hammel M.; Schulman B. A. Structural Insights into NEDD8 Activation of Cullin-RING Ligases: Conformational Control of Conjugation. Cell 2008, 1346995–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xirodimas D. P.; Saville M. K.; Bourdon J.; Hay R. T.; Lane1 D. P. Mdm2-Mediated NEDD8 Conjugation of p53 Inhibits its Transcriptional Activity. Cell 2004, 118, 83–97. [DOI] [PubMed] [Google Scholar]
- Abida W. M.; Nikolaev A.; Zhao W.; Zhang W.; Gu W. FBXO11 Promotes the Neddylation of p53 and Inhibits its Transcriptional Activity. J. Biol. Chem. 2007, 282, 1797–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickle N. H.; Chung J.; Klco J. M.; Hill R. P.; Kaelin W. G. Jr.; Ohh M. PVHL Modification by NEDD8 is Required for Fibronectin Matrix Assembly and Suppression of Tumor Development. Mol. Cell. Biol. 2004, 2483251–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oved S.; Mosesson Y.; Zwang Y.; Santonico E.; Shtiegman K.; Marmor M. D.; Kochupurakkal B. S.; Katz M.; Lavi S.; Cesareni G.; Yarden Y. Conjugation to Nedd8 Instigates Ubiquitylation and Down-Regulation of Activated Receptor Tyrosine Kinases. J. Biol. Chem. 2006, 281, 21640–21651. [DOI] [PubMed] [Google Scholar]
- Gao F.; Cheng J.; Shi T.; Yeh E. T. H. Neddylation of a Breast Cancer-Associated Protein Recruits a Class III Histone Deacetylase that Represses NFκB-Dependent Transcription. Nat. Cell Biol. 2006, 8, 1171–1177. [DOI] [PubMed] [Google Scholar]
- Fan M.; Bigsby R. M.; Nephew K. P. The NEDD8 Pathway Is Required for Proteasome-Mediated Degradation of Human Estrogen Receptor (ER)-α and Essential for the Antiproliferative Activity of ICI 182,780 in ERα-Positive Breast Cancer Cells. Mol. Endocrinol. 2003, 17, 356–365. [DOI] [PubMed] [Google Scholar]
- Chairatvit K.; Ngamkitidechakul C. Control of Cell Proliferation Via Elevated NEDD8 Conjugation in Oral Squamous Cell Carcinoma. Mol. Cell. Biochem. 2007, 306, 163–169. [DOI] [PubMed] [Google Scholar]
- Critchley S.; Gant T. G.; Lagston S. P.; Olhava E. J.; Peluso S.. Preparation of nucleoside derivatives as inhibitors of E1 activating enzymes. PCT Int. Appl., WO 2006084281 Al, August 10, 2006.
- Brownell J. E.; Sintchak M. D.; Gavin J. M.; Liao H.; Bruzzese F. J.; Bump N. J.; Soucy T. A.; Milhollen M. A.; Yang X.; Burkhardt A. L.; Ma J.; Loke H.; Lingaraj T.; Wu D.; Hamman K. B.; Spelman J. J.; Cullis C. A.; Langston S. P.; Vyskocil S.; Sells T. B.; Mallender W. D.; Visiers I.; Li P.; Claiborne C. F.; Rolfe M.; Bolen J. B.; Dick L. R. Substrate-Assisted Inhibition of Ubiquitin-like Protein-Activating Enzymes: The NEDD8 E1 Inhibitor MLN4924 Forms a NEDD8-AMP Mimetic In Situ. Mol. Cell 2010, 37, 102–111. [DOI] [PubMed] [Google Scholar]
- Soucy T. A.; Smith P. G.; Milhollen M. A.; Berger A. J.; Gavin J. M.; Adhikari S.; Brownell1 J. E.; Burke K. E.; Cardin D. P.; Critchley1 S.; Cullis C. A.; Doucette A.; Garnsey J. J.; Gaulin J. L.; Gershman R. E.; Lublinsky1 A. R.; McDonald A.; Mizutani H.; Narayanan U.; Olhava E. J.; Peluso S.; Rezaei M.; Sintchak M. D.; Talreja T.; Thomas M. P.; Traore T.; Vyskocil S.; Weatherhead G. S.; Yu J.; Zhang J.; Dick L. R.; Claiborne C. F.; Rolfe M.; Bolen J. P.; Langston S. P. An Inhibitor of NEDD8-Activating Enzyme as a New Approach to Treat Cancer. Nature 2009, 458, 732–737. [DOI] [PubMed] [Google Scholar]
- Leck Y. C.; Choo Y. Y.; Tan C. Y.; Smith P. G.; Hagen T. Biochemical and Cellular Effects of Inhibiting Nedd8 Conjugation. Biochem. Biophys. Res. Commun. 2010, 398, 588–593. [DOI] [PubMed] [Google Scholar]
- Milhollen M. A.; Traore T.; Adams-Duffy J.; Thomas M. P.; Berger A. J.; Dang L.; Dick L. R.; Garnsey J. J.; Koenig E.; Langston S. P.; Manfredi M.; Narayanan U.; Rolfe M; Staudt L. M.; Soucy T. A.; Yu J.; Zhang J.; Bolen J. B.; Smith P. G. MLN4924, a NEDD8-Activating Enzyme Inhibitor, is Active in Diffuse Large B-cell Lymphoma Models: Rationale for Treatment of NF-kappa B-Dependent Lymphoma. Blood 2010, 116, 1515–1523. [DOI] [PubMed] [Google Scholar]
- Swords R. T.; Kelly K. R.; Smith P. G.; Garnsey J. J.; Mahalingam D.; Medina E.; Oberheu K.; Padmanabhan S.; O'Dwyer M.; Nawrocki S. T.; Giles F. J.; Carew J. S. Inhibition of NEDD8-Activating Enzyme: A Novel Approach for the Treatment of Acute Myeloid Leukemia. Blood 2010, 115, 3796–3800. [DOI] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. C.; Taylor R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. [DOI] [PubMed] [Google Scholar]
- Appel R.; Berger G. Hydrazinsulfonsaure-Amide. 1. Uber Das Hydrazodisulfamid. Chem. Ber. 1958, 91, 1339–1341. [Google Scholar]
- Xu G. W.; Ali M.; Wood T. E.; Maclean N.; Wang X.; Gronda M.; Skrtic M.; Li X.; Hurren R.; Mao X.; Venkatesan M.; Zavareh B.; Ketela T.; Reed J. C.; Rose D.; Moffat J.; Batey R. A.; Dhe-Paganon S.; Schimmer A. D. The Ubiquitin-Activating Enzyme E1 as a Therapeutic Target for the Treatment of Leukemia and Multiple Myeloma. Blood 2010, 115, 2251–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.