

Abstract

Short-acting oral calcilytics, calcium-sensing receptor (CaSR) antagonists, have been considered as alternatives for parathyroid hormone (PTH), an injectable bone anabolic drug used in the treatment of osteoporosis. Previously, we identified aminopropandiol 1, which transiently stimulated endogenous PTH secretion in rats. However, the inhibition of cytochrome P450 (CYP) 2D6 and the low bioavailability of 1 remain to be solved. Attempts to change the physicochemical properties of the highly lipophilic amine 1 by introduction of a carboxylic acid group as well as further structural modifications led to the discovery of the highly potent biphenylcarboxylic acid 15, with a markedly reduced CYP2D6 inhibition and a significantly improved bioavailability. Compound 15 evoked a rapid and transient elevation of endogenous PTH levels in rats after oral administration in a dose-dependent manner at a dose as low as 1 mg/kg. The PTH secretion pattern correlated with the pharmacokinetic profile and agreed well with that of the exogenous PTH injection which exerts a bone anabolic effect.
Keywords: Calcium-sensing receptor (CaSR) antagonist, calcilytics, PTH, short-acting, osteoporosis, cytochrome P450 inhibition
Osteoporosis is a bone disease marked by decreased bone strength and accompanied by an increased risk of bone fracture, affecting more than 75 million people in EU, U.S., and Japan.1 The current mainstream drugs for osteoporosis are antiresorptive agents such as bisphosphonates, estrogen, and selective estrogen receptor modulators (SERMs).2 These compounds suppress osteoclast cell function and prevent bone loss. However, an anabolic agent which stimulates osteoblast cells and leads to new bone formation is an attractive alternative.
The bone strength defined by bone density and quality is maintained through the balance of old bone resorption and new bone formation, known as bone remodeling. Parathyroid hormone (PTH) is a key intrinsic agent regulating bone remodeling in either the catabolic or the anabolic pathways, depending on the pattern of exposure to the hormone. Intermittent exposure to PTH is important to stimulate new bone formation and leads to anabolic effects, while prolonged exposure to elevated PTH levels increases bone turnover and results in bone loss.3 Since daily subcutaneous injection of teriparatide, the recombinant 1−34 amino acid fragment of human parathyroid hormone (PTH), improved bone mineral density (BMD) in the lumbar spine and reduced bone fracture rates in patients with postmenopausal osteoporosis,4 anabolic agents have received much attention. Currently, two injectable human PTH peptides, teriparatide and Preotact (hPTH 1−84), are on the market, but no orally administered anabolic drug is available.
Endogenous PTH is secreted from the parathyroid gland when lowered blood levels of ionized calcium are detected by the calcium-sensing receptor (CaSR), a G-protein coupled receptor expressed on the surface of parathyroid cells.5 Thereby, the antagonists, known as calcilytics, reduce the sensitivity of this receptor to extracellular calcium and stimulate PTH secretion. This outcome was first shown by an orally available calcilytic, NPS-2143 (Figure 1).6,7 At the same time, it was also reported that the long acting profile of this compound resulted in sustained high PTH levels and no net increase of bone mass. Therefore, research has been focused on the development of a CaSR antagonist with rapidly absorption and a short acting pharmacokinetic profile to achieve a PTH secretion pattern similar to that of the exogenously injected form.
Figure 1.

Structures of selected calcilytics.
Although many CaSR antagonists have been reported in the literature to date,8−16 the proof of concept in humans was recently shown by ronacarelet17 and JTT-30518 (MK-5442, 15 hemisulfate) (Figure 1). We have reported a new aminopropandiol class of calcilytics 1 (Figure 1) which demonstrated a rapid and transient stimulation of PTH secretion after oral administration in rats.12,19 However, compound 1 possessed a strong CYP2D6 inhibition (IC50 of 0.5 μM) and low bioavailability (BA, 13%). Here, we report our research efforts to improve the CYP inhibition and the low BA, which led to the discovery of short-acting oral calcilytic 15 with pulsatile secretion of PTH.
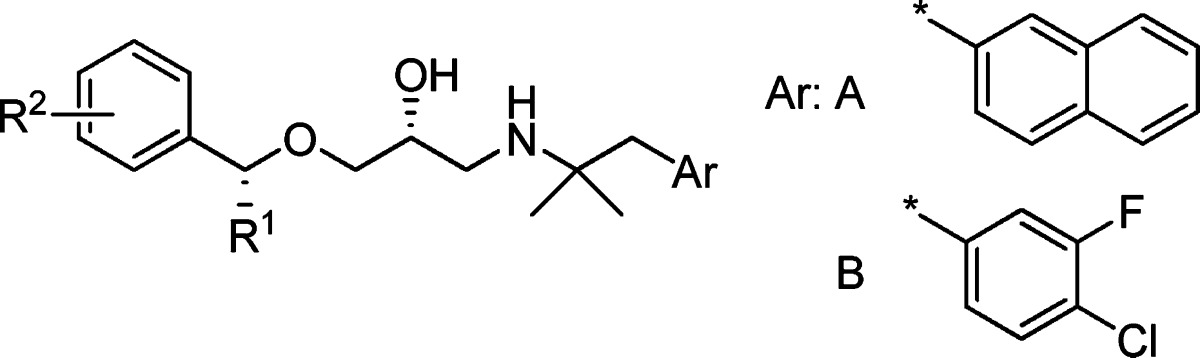
Molecules that are basic in character and highly lipophilic are conducive to CYP2D6 inhibition and also block the human ether-a-go-go related gene (hERG) ion channel.20 NPS-2143 and compound 1 exhibit both of these physicochemical characteristics. NPS-2143 has been reported to inhibit CYP2D6 and hERG strongly.10 In an examination of the metabolites of 1 in rats, two products in which the methyl group on the benzene ring was oxidized to either the alcohol or to the carboxylic acid were identified. Both these oxidative metabolites have reduced lipophilicity compared to that of parent compound 1. While the alcohol 2 still inhibited CYP2D6 (IC50 of 1.2 μM), the inhibitory potency of carboxylic acid 3 (diastereomixture) toward CYP2D6 activity was significantly weakened (IC50 > 10 μM). Although 3 exhibited significantly decreased CaSR antagonist activity, changing the position of the COOH group to the meta site (4) restored the antagonist activity (IC50 of 1.0 μM) and reduced CYP2D6 inhibition (IC50 > 10 μM). In light of this result, we attempted to introduce a carboxylic acid group into the molecule in an effort to develop an orally active aminopropandiol calcilytic without the unfavorable off-target activities. The compounds described in this paper were tested in the human CaSR antagonist assay (PC12h cells reporter gene assay) (Table 1) and by measurement of PTH secretion in rats (Table 2), as previously reported.19
Table 1. In Vitro hCaSR Antagonist Activity of the Aminopropandiol Derivatives.
 |
Table 2. In Vivo PTH Secretion Activity of the Selected Compounds in Rats.
| PTH secretiona | |||
|---|---|---|---|
| compd | 1 mg/kg | 3 mg/kg | 10 mg/kg |
| 8 | NT | + | NT |
| 12 | − | + | ++ |
| 13 | + | ++ | NT |
| 14 | − | ++ | NT |
| 15 | ++ | ++ | ++ |
Increases of serum PTH 1−34 levels (Δ) were compared with levels observed in control rats treated with vehicle. The peak levels, measured at 15 or 30 min after oral administration of the compounds in rats (n = 5), were compared with the levels observed in animals treated with compound 1 (30 mg/kg, po, ∼2−4-fold increases). ++, >80% of compound 1; +, 50−80% of compound 1; −, <50% of compound 1; NT, not tested.
The initial phase of the investigation focused on defining the site in the molecule where a COOH group could be introduced without affecting CaSR antagonist activity, as compared to 1. Benzoic acid analogue 5, having a COOH group at the para position of the left benzene ring, and 6, a methyl analogue of 3 with a benzylic substituent, exhibited significantly decreased antagonist activity, while the phenylacetic acid derivative 7 maintained the antagonist activity (IC50 of 2.2 μM). Further extension of the methylene chain to the phenylpropionic acid structure (8) increased inhibitory potency 100-fold (IC50 of 0.024 μM), which is comparable to that of compound 1, indicating that the COOH group in the area ortho to the site of attachment of the aminopropandiol chain could be accommodated. Compound 8 showed significantly reduced CYP2D6 inhibition (IC50 of 9.6 μM).
To define the optimum position for the COOH group, we prepared three carboxylic acids 9−11 in which the position of the COOH groups were fasten by the rigid biphenyl structure. Two of those, 10 and 11, with meta and ortho substituents, respectively, showed antagonist activities that were comparable to or more potent than those of 1. On the other hand, para substituent (9) displayed poor activity. Since these compounds are diastereomixtures at the benzylic Me substituent, we prepared an R,R-isomer of 11 (compound 12) with the same configuration as that of compound 1 . The R,R-enantiomer 12 showed the most potent antagonist activity of all tested analogues (IC50 of 0.004 μM) and a reduced CYP2D6 inhibition (IC50 > 10 μM).
We also examined the PTH secretion activity of 8 and 12 (Table 2). An oral dose of 12 at 10 mg/kg in rats elicited PTH 1−34 levels that were comparable to those obtained with a 30 mg/kg dose of 1. At the lower dose of 3 mg/kg, compounds 8 and 12 exhibited similar PTH secretion activities. These results confirmed that these carboxylic acid derivatives are orally active. However, their potencies in PTH secretion activity were not largely improved compared to that of 1, indicating that BA was not much improved in these compounds. In fact, a pharmacokinetic study of 12 in rats showed low BA (6.4%). We considered that the naphthyl structure on the right-hand side of the molecules might contribute to the low BA of this series. Among the compounds examined to improve BA, 4-chloro-3-fluorophenyl derivative 13 showed a better potency in vivo compared to 12 (PTH secretion levels at 3 mg/kg dose of 13 were similar to those at 10 mg/kg dose of compound 12), although the antagonist potency was lower (IC50 of 0.023 μM).
Another concern for the metabolism of these derivatives was the formation of conjugated metabolites (e.g., glucuronic acid conjugation). We hypothesized that the introduction of a Me group ortho to the COOH on the phenyl ring (compound 14) might cause steric hindrance and interfere with the glucuronate conjugate reaction. As a result, compound 14 exhibited a sustained antagonist activity but an improved potency in vivo compared to 12. Finally, all these modifications were successfully combined, as exemplified by compound 15, which showed a high antagonist activity (IC50 of 0.012 μM) and a potentiated PTH secretion activity in vivo (10-fold more potent than 12).
A PTH secretion time course study after oral administration of 15 in rats was conducted (Figure 2). Compound 15 afforded elevated endogenous PTH 1−34 levels rapidly, transiently, and in a dose-dependent manner at a dose as low as 1 mg/kg. A maximal level was observed at 15 min and was restored to baseline within2 h. The PTH secretion pattern is in good agreement with that observed with exogenous PTH injection,21,22 indicating that compound 15 could be a bone anabolic agent for osteoporosis. A bone anabolic effect of compound 15 on ovariectomized (OVX) rats has been observed (manuscript submitted).23 A pharmacokinetic study of 15 showed that the short-acting character of this compound was well correlated with the pharmacokinetic profile. It was rapidly absorbed after oral administration in rats (1 mg/kg, Tmax of 0.5 h, Cmax of 475 ng/mL) and disappeared shortly thereafter (T1/2 of 1.8 h). The BA was dramatically improved (64%). The strong CYP2D6 inhibition activity observed with compound 1 (IC50 of 0.5 μM) was significantly reduced in compound 15 (IC50 of 11 μM). Compound 15 showed a weak inhibition for β2-adrenergic receptor binding (IC50 of 1.7 μM) and did not show binding inhibition for α2-adrenergic receptor, dopamine transporter (D1), and serotonin receptors up to 10 μM.
Figure 2.
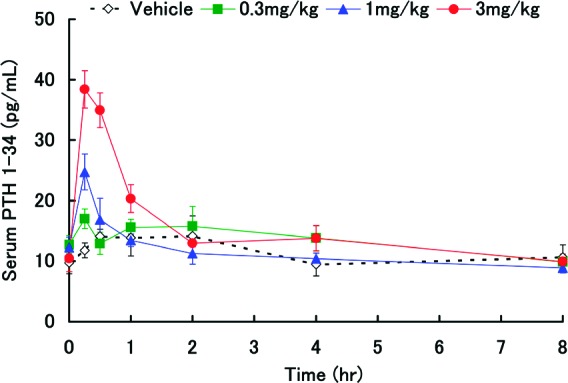
Time courses of PTH 1−34 levels after oral administration of 15 hemisulfate in rats. Data are expressed as mean values ± standard deviation, n = 5.
The compounds investigated in this study were synthesized as shown in Schemes 1 and 2. Alcohols 16(12) were converted to the glycidyl ethers 17−19 by alkylation with (R)-(−)-glycidyl 3-nitrobenzensulfonate in the presence of NaH in DMF or in THF−DMSO. Epoxy-opening reaction of 17 or 18 with 1,1-dimethyl-2-(2-naphthalenyl)ethylamine 20(19) gave the aminoalcohol 21 or 22, respectively. Hydrolysis of the nitrile group in 21 by KOH in ethylene glycol or the ester group in 22 by aq NaOH in MeOH afforded the carboxylic acids 3−6. Biphenyl carboxylic acids 9−11 were prepared from the glycidyl ether 19 in four steps as follows. Suzuki coupling of 19 with 4-formylphenylboronic acid and subsequent oxidation of the aldehyde by MnO2 in the presence of KCN in EtOH gave the esters 23, which were reacted with an amine 20 and hydrolyzed to the corresponding carboxylic acids 9−11. Optically active compounds 7, 8, and 12−15 were synthesized from chiral glycidyl ether 24 (prepared from the corresponding chiral alcohol by the same method described in Scheme 1). Either a coupling reaction of 24 with ethyl tributylstannyl acetate in the presence of Pd(PPh3)4, a Heck reaction of 24 with methyl acrylate followed by hydrogenation of the double bond in the presence of Rh/Al2O3, or Suzuki couplings with the corresponding boronic acids24,25 gave the epoxides 26−29, which were converted to 12−15 by epoxy-opening reaction with the corresponding amine 20 or 30(24) and subsequent hydrolysis of the ester, respectively.
Scheme 1.

Scheme 2.

In conclusion, we have discovered a potent and short-acting oral aminopropandiol calcilytic 15, bearing a biphenylcarboxylic acid structure. The strong CYP2D6 inhibition seen in the prototype compound 1 was significantly reduced by the introduction of a COOH group. Modification of the naphthylethylamine part and the incorporation of a biphenylcarboxylic acid moiety bearing a substituent on the position ortho to the COOH provided a significant improvement of BA in rats. Oral administration of 15 in rats led to a pulsatile secretion of endogenous PTH, which is a required profile for a bone anabolic agent. Compound 15 is now in a phase 2 clinical study.
Acknowledgments
The authors thank our Analytical Research and Development Laboratories for collecting analytical data and Dr. Jun-ichi Haruta for his continuous encouragement.
Author Contributions
† These three authors contributed equally to this work.
Supporting Information Available
Experimental details for the synthesis and characterization of the tested compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Report of a WHO Scientific Group. Prevention and management of osteoporosis; W. H. O. Tech. Rep. Ser.; World Health Organization: Geneva, 2003; Vol. 921, pp. 1−164. [PubMed]
- Deal C. Osteoporosis therapies Bisphosphonates, SERMs, PTH, and new therapies. Clin. Rev. Bone Miner. Metab. 2005, 3, 125–141. [Google Scholar]
- Kimmel D. B.; Bozzato R. P.; Kronis K. A.; Coble T.; Sindrey D.; Kwong P.; Recker R. R. The effect of recombinant human (1−84) or synthetic human (1−34) parathyroid hormone on the skeleton of adult osteopenic ovariectomized rats. Endoclinology 1993, 132, 1577–1584. [DOI] [PubMed] [Google Scholar]
- Neer R. M.; Arnaud C. D.; Zanchetta J. R.; Prince R.; Gaich G. A.; Reginster J. Y.; Hodsman A. B.; Eriksen E. F.; Ish-Shalom S.; Genant H. K.; Wang O.; Mitlak B. H. Effect of Parathyroid Hormone (1−34) on Fractures and Bone Mineral Density in Postmenopausal Women with Osteoporosis. N. Engl. J. Med. 2001, 344, 1434–1441. [DOI] [PubMed] [Google Scholar]
- Brown E. M.; MacLeod R. J. Extracellular Calcium Sensing and Extracellular Calcium Signaling. Physiol. Rev. 2001, 81, 239–297. [DOI] [PubMed] [Google Scholar]
- Nemeth E. F.; Delmar E. C.; Heaton W. L.; Miller M. A.; Lambert L. D.; Conklin R. L.; Gowen M.; Gleason J. G.; Bhatnagar P. K.; Fox J. Calcilytic Compounds: Potent and Selective Ca2+ Receptor Antagonists That Stimulate Secretion of Parathyroid Hormone. J. Pharm. Exp. Ther. 2001, 299, 323–331. [PubMed] [Google Scholar]
- Gowen M.; Stroup G. B.; Dodds R. A.; James I. E.; Votta B. J.; Smith B. R.; Bhatnagar P. K.; Lago A. M.; Callahan J. F.; Del Mar E. G.; Miller M. A.; Nemuth E. F.; Fox J. Antagonizing the parathyroid calcium receptor stimulates parathyroid hormone secretion and bone formation in osteopenic rats. J. Clin. Invest. 2000, 105, 1595–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W.; Ruan Z.; Wang Y.; Van Kirk K.; Ma Z.; Arey B. J.; Cooper C. B.; Seethala R.; Feyen J. H. M.; Dickson J. K. Jr. Discovery and Structure-Activity Relationships of Trisubstituted Pyrimidines/Pyridines as Novel Calcium-Sensing Receptor Antagonists. J. Med. Chem. 2009, 52, 1204–1208. [DOI] [PubMed] [Google Scholar]
- Balan G.; Bauman J.; Bhattacharya S.; Castrodad M.; Healy D. R.; Herr M.; Humphries P.; Jennings S.; Kalgutkar A. S.; Kapinos B.; Khot V.; Lazarra K.; Li M.; Li Y.; Neagu C.; Oliver R.; Piotrowski D. W.; Price D.; Qi H.; Simmons H. A.; Southers J.; Wei L.; Zhang Y.; Paralkar V. M. The Discovery of Novel Calcium Sensing Receptor Negative Allosteric Modulators. Bioorg. Med. Chem. Lett. 2009, 19, 3328–3332. [DOI] [PubMed] [Google Scholar]
- Marquis R. W.; Lago A. M.; Callahan J. F.; Rahman A.; Dong X.; Stroup G. B.; Hoffman S.; Gowen M.; DelMar E. G.; Van Wagenen B. C.; Logan S.; Shimizu S.; Fox J.; Nemeth E. F.; Roethke T.; Smith B. R.; Ward K. W.; Bhatnagar P. Antagonists of the Calcium Receptor. 2. Amino Alcohol-Based Parathyroid Hormone Secretagogues. J. Med. Chem. 2009, 52, 6599–6605. [DOI] [PubMed] [Google Scholar]
- Didiuk M. T.; Griffith D. A.; Benbow J. W.; Liu K. K.; Walker D. P.; Bi F. C.; Morris J.; Guzman-Perez A.; Gao H.; Bechle B. M.; Kelley R. M.; Yang X.; Dirico K.; Ahmed S.; Hungerford W.; DiBrinno J.; Zawistoski M. P.; Bagley S. W.; Li J.; Zeng Y.; Santucci S.; Oliver R.; Corbett M.; Olson T.; Chen C.; Li M.; Paralkar V. M.; Riccardi K. A.; Healy D. R.; Kalgutkar A. S.; Maurer T. S.; Nguyen H. T.; Frederick K. S. Short-Acting 5-(Trifluoromethyl)pyrido[4,3-d]pyrimidin-4(3H)-one Derivatives as Orally-Active Calcium-Sensing Receptor Antagonists. Bioorg. Med. Chem. Lett. 2009, 19, 4555–4559. [DOI] [PubMed] [Google Scholar]
- Shinagawa Y.; Inoue T.; Hirata K.; Katsushima T.; Nakagawa T.; Matsuo Y.; Shindo M.; Hashimoto H. New Aminopropandiol Derivatives as Orally Available and Short-Acting Calcium-Sensing Receptor Antagonists. Bioorg. Med. Chem. Lett. 2010, 20, 3809–3813. [DOI] [PubMed] [Google Scholar]
- Southers J. A.; Bauman J. N.; Price D. A.; Humphries P. S.; Balan G.; Sagal J. F.; Maurer T. S.; Zhang Y.; Oliver R.; Herr M.; Healy D. R.; Li M.; Kapinos B.; Fate G. D.; Riccardi K. A.; Paralkar V. M.; Brown T. M.; Kalgutkar A. S. Metabolism-Guided Design of Short-Acting Calcium-Sensing Receptor Antagonists. ACS Med. Chem. Lett. 2010, 1, 219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widler L.; Altmann E.; Beerli R.; Breitenstein W.; Bouhelal R.; Buhl T.; Gamse R.; Gerspacher R.; Halleux C.; John M. R.; Lehmann H.; Kalb O.; Kneissel M.; Missbach M.; Műller I. R.; Reidemeister S.; Renaud J.; Taillardat A.; Tommasi R.; Weiler S.; Wolf R. M.; Seuwen K. 1-Alkyl-4-phenyl-6-alkoxy-1H-quinazolin-2-ones: A Novel Series of Potent Calcium-Sensing Receptor Antagonists. J. Med. Chem. 2010, 53, 2250–2263. [DOI] [PubMed] [Google Scholar]
- Gerspacher M.; Altmann E.; Beerli R.; Buhl T.; Endres R.; Gamse R.; Kameni-Tcheudji J.; Kneissel M.; Krawinkler K. H.; Missbach M.; Schmidt A.; Seuwen K.; Weiler S.; Widler L. Penta-substituted benzimidazoles as potent antagonists of the calcium-sensing receptor (CaSR-antagonists). Bioorg. Med. Chem. Lett. 2010, 20, 5161–5164. [DOI] [PubMed] [Google Scholar]
- Yoshida M.; Mori A.; Inaba A.; Oka M.; Makino H.; Yamaguchi M.; Fujita H.; Kawamoto T.; Goto M.; Kimura H.; Baba A.; Yasuma T. Synthesis and structure−activity relationship of tetrahydropyrazolopyrimidine derivatives—A novel structural class of potent calcium-sensing receptor antagonists. Bioorg. Med. Chem. 2010, 18, 8501–8511. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick L. A.; Brennan E.; Kumar S.; Matheny C.; Yi B.; McLaughlin M.; Phillips J.; Skordos L.; Ethgen D.. Ronacaleret, a Novel Calcium-Sensing Receptor Antagonist, Demonstrates Potential as an Oral Bone-Forming Therapy in Healthy Postmenopausal Women. ASBMR 30th Annual Meeting_FINAL_Abstract_Book2008, 50, 1174.
- Fukumoto S.; Nakamura T.; Nishizawa Y.; Hayashi M.; Matsumoto T.. Randomized, Single-Blinded Placebo-Controlled Study of a Novel Calcilytic, JTT-305, in Patients with Postmenopausal Osteoporosis. J. Bone Miner. Res. 24 (Suppl. 1). Available at http://www.asbmr.org/Meetings/AnnualMeeting/AbstractDetail.aspx?aid=f7a434e5-ad26-4aa0-930d-5bca406c9317. Accessed July 1, 2009. [Google Scholar]
- Shinagawa Y.; Katsushima T.; Nakagawa T.. Calcium Receptor Antagonists. WO 2002/014259.
- Gleeson M. P. Generation of a Set of Simple, Interpretable ADMET Rules of Thumb. J. Med. Chem. 2008, 51, 817–834. [DOI] [PubMed] [Google Scholar]
- Teriparatide injection (rDNA origin). Data on file, Eli Lilly, USA, 2002. [online]. Available at http://www.accessdata.fda.gov/drugsatfda_docs/nda/2002/21-318_FORTEO_BioPharmr.pdf. Accessed July 1, 2010. [Google Scholar]
- Rubin M. R.; Bilezikian J. P. Parathyroid Hormone as an Anabolic Skeletal Therapy. Drugs 2005, 65, 2481–2491. [DOI] [PubMed] [Google Scholar]
- Kimura S. Manuscript submitted.
- Shinagawa Y.; Inoue T.; Kiguchi T.; Ikenogami T.; Ogawa N.; Fukuda K.; Nakagawa T.; Shindo M.; Soejima Y. CaSR Antagonist. WO 2004/094362.
- Shinagawa Y.; Inoue T.; Kiguchi T.; Ikenogami T.; Ogawa N.; Nakagawa T.; Shindo M.; Soejima Y. CaSR Antagonist. WO 2004/106280.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.