

Abstract

We have investigated a novel series of acid-derived γ-secretase modulators as a potential treatment of Alzheimer's disease. Optimization based on cellular potency and brain pharmacodynamics after oral dosing led to the discovery of 10a (BIIB042). Compound 10a is a potent γ-secretase modulator, which lowered Aβ42, increased Aβ38, but had little to no effect on Aβ40 levels both in vitro and in vivo. In addition, compound 10a did not affect Notch signaling in our in vitro assessment. Compound 10a demonstrated excellent pharmacokinetic parameters in multiple species. Oral administration of 10a significantly reduced brain Aβ42 levels in CF-1 mice and Fischer rats, as well as plasma Aβ42 levels in cynomolgus monkeys. Compound 10a was selected as a candidate for preclinical safety evaluation.
Keywords: Secretase, GSM, Alzheimer's, amyloid, Mannich reaction
Alzheimer's disease (AD) is a devastating neurodegenerative disorder for which there is no disease-modifying therapy currently available.1 Multiple lines of evidence suggest that amyloid-β (Aβ) peptide plays a pivotal role in the pathogenesis of AD. Preventing the accumulation and aggregation of Aβ peptide has been one of the major strategies in developing disease-modifying AD therapies.2−4 Aβ peptides are produced from sequential cleavage of amyloid precursor protein (APP) by β-secretase and γ-secretase in the brain.5 Cleavage by γ-secretase produces Aβ peptides that generally range from 37 to 42 amino acids in length. Aβ42 is more amyloidogenic as compared to the shorter Aβ isoforms and may serve as a key seed in the formation of senile plaques and may be at least partially responsible for mediating cognitive impairment in AD.6−8 γ-Secretase modulators (GSMs) preferentially shift the carboxy-terminal cleavage pattern on the β-secretase-processed APP (C99) to bias production away from longer isoforms such as Aβ42 in favor of shorter, less toxic Aβ isoforms, such as Aβ38. Because GSMs do not inhibit the enzymatic activity of γ-secretase, they are much less likely to interfere with processing and subsequent signaling activities of other important substrates, such as Notch, and could, therefore, potentially provide a better safety profile as compared to the γ-secretase inhibitors (GSIs).9,10
The first generation of GSMs originated from an epidemiological analysis, which showed that certain nonsteroidal anti-inflammatory drugs (NSAIDS) lowered the risk of developing AD, and the effect was independent of COX inhibition.11 NSAID-derived Tarenflurbil (1, EC50 = 300 μM, Figure 1)12 was progressed through phase III clinical trials for AD but failed to achieve efficacy, potentially due to weak GSM activity and poor brain penetration. Chiesi has recently advanced CHF5074 (2, EC50 = 40 μM, Figure 1) into a phase II clinical trial. Cognitive benefits and plaque load reduction in the brain were reported after chronic treatment of Tg2576 transgenic mice with CHF5074.13 This compound is about 7-fold more potent than Tarenflurbil and has improved brain exposure after prolonged dosing. There has been an intensive effort to further improve potency and brain exposure as exemplified by Merck's recent publications on a series of carboxylic acid-derived GSMs (e.g., 3, EC50 = 0.6 μM, Figure 1).14 Eisai's GSM (4, EC50 = 0.08 μM, Figure 1) represents a different class of GSMs that contain an aryl imidazole moiety instead of the carboxylic acid.15 Additional patent literature in this field has been extensively reviewed recently.15
Figure 1.

Structures of GSMs.
Our program aimed to identify potent GSMs with favorable brain exposure. As shown in Figure 2, our early lead 5 was designed to test the idea if an amino nitrogen atom was tolerated when introduced off the biaryl acetic acid scaffold that is similar to those of NSAIDs. Compound 5 was active in APP CHO cellular assay (EC50 = 1 μM). However, brain exposure was limited (0.6 μM, 4 h) in mice after 50 mg/kg oral dosing, even though the plasma concentration was excellent (14.9 μM). We then decided to constrain the side chain into a more rigid structure and incorporate a basic nitrogen atom, in an effort to improve brain penetration. Mannich reaction on 3-hydroxyphenyl acetic acid (35, Scheme 1) provided an interesting and facile route to construct a compact framework and incorporate amines such as piperidine. Multipoint optimization based on cellular potency and brain pharmacodynamic (PD) response in wild type mice after oral dosing lead to the discovery of the preclinical candidate BIIB042 (10a). Herein, we describe the syntheses, structure–activity relationships (SARs), and profiles of this novel series of carboxylic acid-derived GSMs.
Figure 2.

Structures of early lead 5 and BIIB042.
Scheme 1. Synthesis of Compound 10a.

(a) BnBr, K2CO3, acetonitrile, room temperature, 6 h, 87%. (b) LiHMDS, THF, CH3I, −78 °C to room temperature, 76%. (c) Pd/C, H2, MeOH, 3 h, 87%. (d) 4-Fluorobenzaldehyde, 4-methylpiperidine, trifluoro toluene, microwave, 120 °C, 1 h, 42%. (e) Tf2O, pyridine, CH2Cl2, 75%. (f) 4-(Trifluoromethyl)phenyl boronic acid, Pd(PPh3)4, 1,2-dimethoxyethane/ethanol/saturated Na2CO3, microwave, 120 °C, 0.5 h, 67%. (g) NaOH, MeOH/THF (1:1), microwave, 100 °C, 10 min, 86%. (h) SCF chiral separation, Chiralpak AD-H (2 cm × 15 cm), 10% isopropanol (0.1% DEA)/CO2, 100 bar, 60 mL/min, 220 nm.
Tables 1–3 summarize the SAR at various positions of the molecule. CHO cells overexpressing the APP V717F mutation were used to measure the effect of GSM compounds on Aβ peptide (Aβ42, Aβ40, and Aβ38) production using a multiplex Aβ ELISA assay to detect all isoforms simultaneously in a single well. In our investigation, compounds that lowered the levels of Aβ42 concomitantly elevated the levels of Aβ38, while leaving Aβ40 levels generally unchanged. The PD response of these compounds was measured in wild-type mice following oral administration. Animals were sacrificed 4 h after dosing, and brain and plasma samples were collected for analyses of both drug exposure and Aβ levels.
Table 1. SAR at the R1/R2, R3, and R5 Positions.
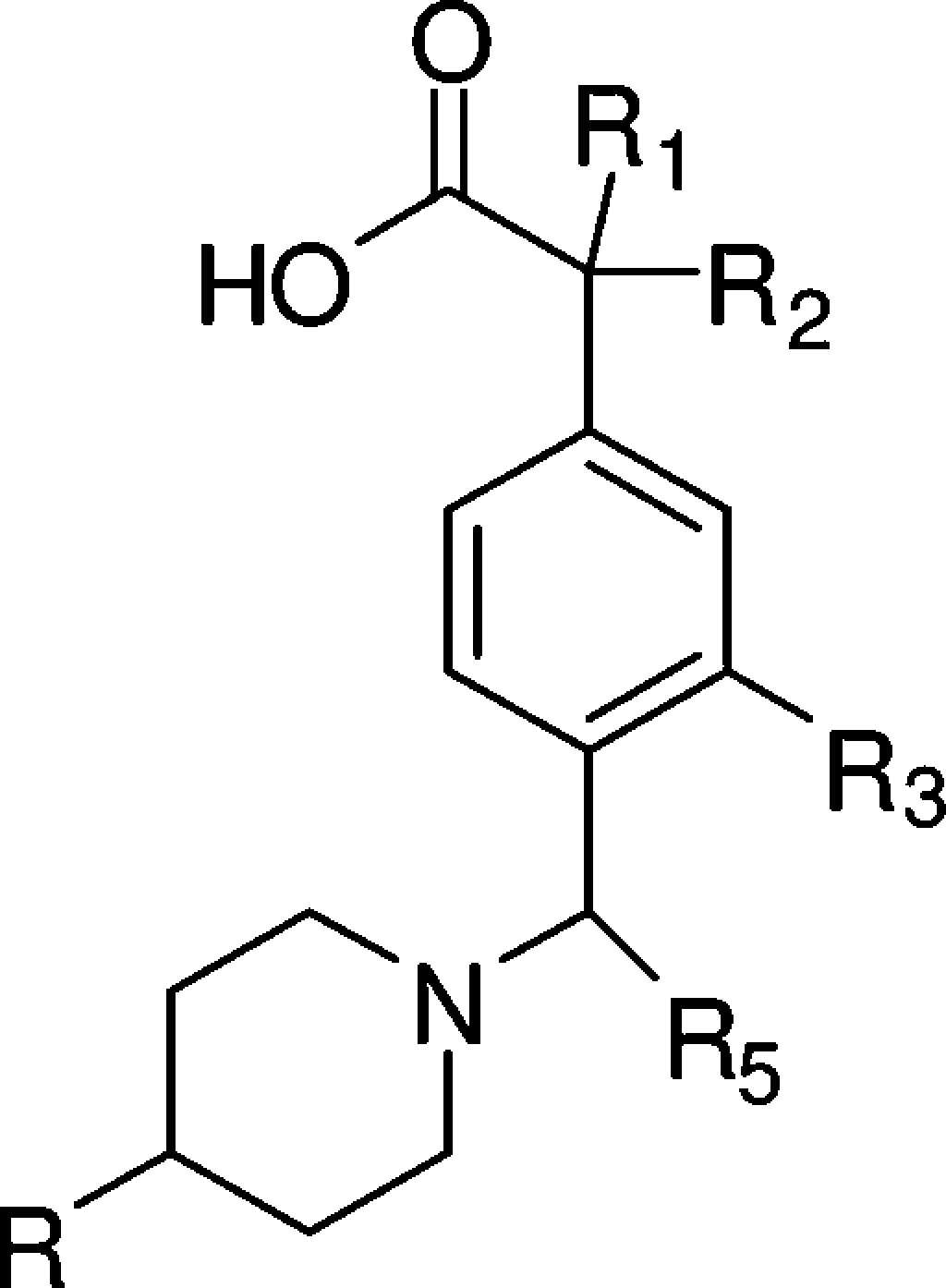
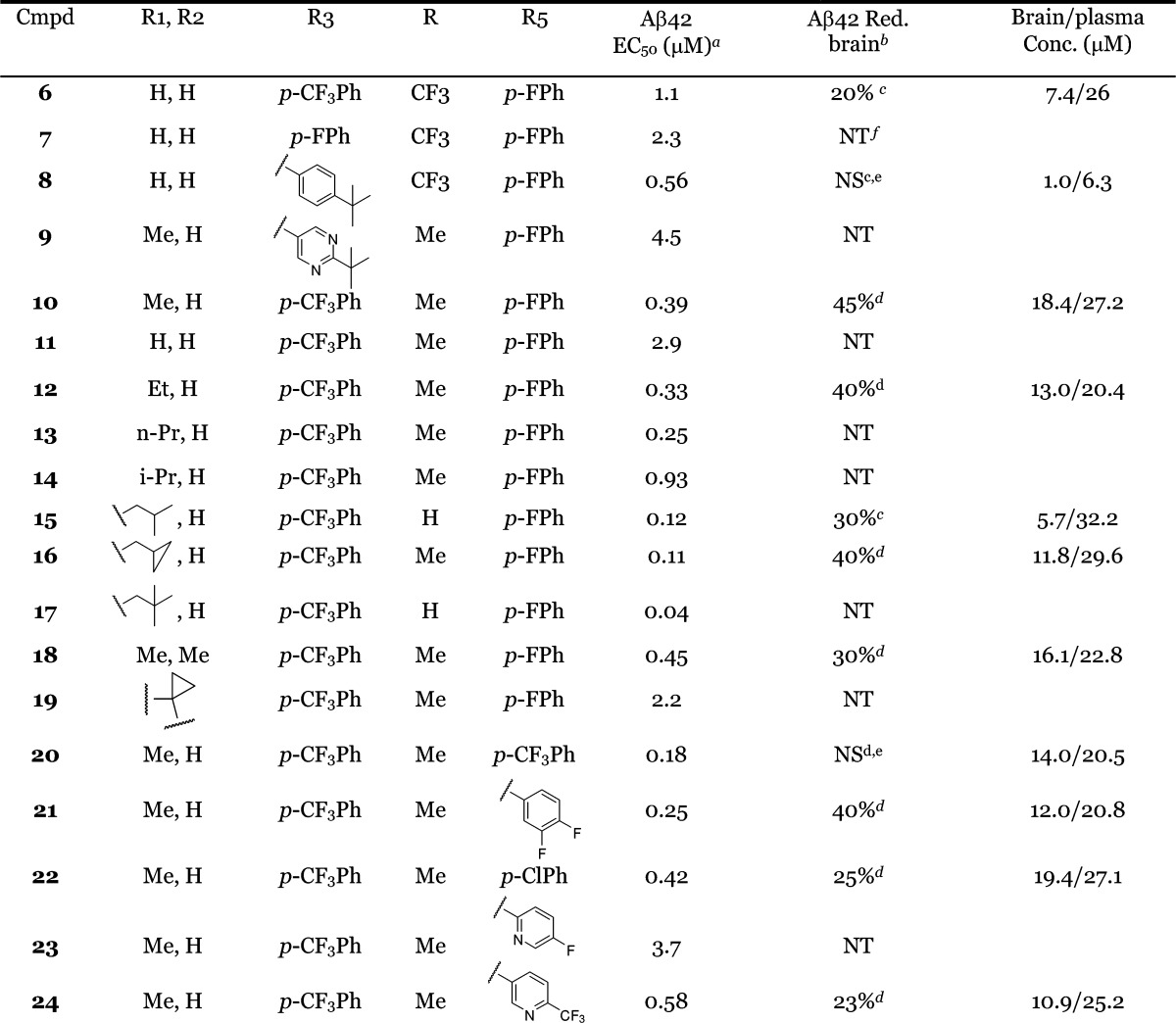
EC50 values were calculated as an average of at least two determinations.
PD responses were measure at 4 h after oral administration of compounds in a solution formulation (ethanol:propylene glycol:10% solutol = 1:1:8).
Oral dosing at 50 mg/kg.
Oral dosing at 30 mg/kg.
NS, not significant.
NT, not tested.
Table 3. Separated Single Isomers of Compound 10.
| compd | Aβ42 EC50 (μM) | Aβ42 red. brain (10 mpk) (%) | brain/plasma concn (μM) | hERG EC50 (μM) |
|---|---|---|---|---|
| 10a | 0.17 | 40 | 4.6/10 | 15 |
| 10b | 0.15 | 38 | 6.6/7.4 | 4.6 |
| 10c/10d | 0.89 | NT |
As shown in Table 1, with a much more rigid and compact structure and a basic amine as compared to the early lead 5, compound 6 maintained moderate potency (EC50 = 1.1 μM) for lowering Aβ42 in the APP CHO cellular assay. This was accompanied by a 10-fold improvement in brain exposure (brain concn = 7.4 μM, 4 h, po, 50 mg/kg) and a detectable PD response (20% Aβ42 reduction). At the R3 position, 4-CF3 phenyl was found to be optimal. As exemplified in Table 1, compound 7 (p-F-phenyl, EC50 = 2.3 μM) was less potent as compared to 6. Compound 8 (4-t-butyl phenyl, EC50 = 0.56 μM) demonstrated improved cellular potency; however, it had much lower brain exposure (1.0 μM, 4 h) as compared to 6 and did not reduce brain Aβ42 when dosed at 50 mg/kg orally. Heterocyclic moieties were not well tolerated at the R3 position as exemplified by compound 9 (EC50 = 4.5 μM).
Compounds 10–19 highlight the SAR at the R1/R2 position. Substitution at this position appeared to greatly improve cellular potency. As compared to compound 11, a simple methyl substitution at the R1/R2 position improved the cellular potency by 8-fold (Table 1, compound 10, EC50 = 0.39 μM). There was a trend that bulkier groups at the R1/R2 position provided more potent cellular activity as demonstrated by compounds 12, 13, and 15–17, with compound 17 achieving 40 nM EC50 in the APP CHO cellular assay. However, brain penetration tended to diminish as the number of rotatable bonds increased and the molecular weight passed 500. For example, compound 14 (EC50 = 0.12 μM) only achieved 30% Aβ42 reduction and 5.7 μM drug concentration in the brain when dosed at 50 mg/kg, while the methyl analogue 10 resulted in 45% Aβ42 lowering and 18.4 μM drug concentration in the brain when dosed at 30 mg/kg. Compounds 18 and 19 have an advantage of simplifying the stereochemistry. Unfortunately, the cyclopropyl analogue 19 was weaker in lowering Aβ42 in the cell-based assay (EC50 = 2.2 μM). The gem dimethyl analogue (18) showed reasonable cellular potency (EC50 = 0.45 μM), however, had only moderate brain PD response (30% Aβ42 reduction) in mice at 30 mg/kg and did not show PD response in the brain after oral dosing at 30 mg/kg in rat (data not shown).
The key SAR at the R5 position is highlighted by compounds 20–24. Various substitution patterns on the phenyl were explored; 4-F phenyl appeared to be the best as in compound 10. For reasons not clear to us, compound 20 did not achieve a PD response in the brain even though the cellular potency was better, and the brain exposure was significant. A heterocyclic moiety appeared to be not as well tolerated at the R5 position, as exemplified by compound 23. The best heterocyclic analogue (24, EC50 = 0.58 μM) in our collection achieved favorable brain exposure (brain concn = 10.9 μM), likely benefiting from a 4-CF3 substituted pyridyl group at the R5 position. However, the PD response of 24 was rather moderate (23% Aβ42 reduction, 30 mg/kg).
We then fixed R1/R2, R3, and R5 and explored the R4 position. As shown for compound 25 in Table 2, a piperazine moiety at this position led to much weaker cellular activity. However, hydrophobic substitution on the piperidine was beneficial in improving cellular potency. For example, the 4-CF3 substitution on piperidine rendered compound 26 2-fold more potent in the cell-based assay (EC50 = 0.18 μM) as compared to compound 10. Interestingly, the improved in vitro potency did not translate into a better PD response in the brain despite comparable brain exposures (Table 2). In the absence of a methyl substitution on the piperidine, compound 27 was about 4-fold less potent as compared to 10 in the cell-based assay. 4-Fluoro substitutions, as in 28 (EC50 = 2 μM), was not beneficial. The pyrrolidine analogue 29 was also weaker in the cell-based assay (EC50 = 2.3 μM). Compound 31 exhibited better cellular potency (EC50 = 0.17 μM); however, the brain exposure was much lower (2.5 μM), resulting in a weak PD response. As exemplified by compounds 32–34, spiro, bridged, and fused cyclic amines were well tolerated at the R4 position. The PD responses for these compounds were moderate (Table 2). Overall, compound 10 provided the most robust PD response in the brain.
Table 2. SAR at the R4 Position.
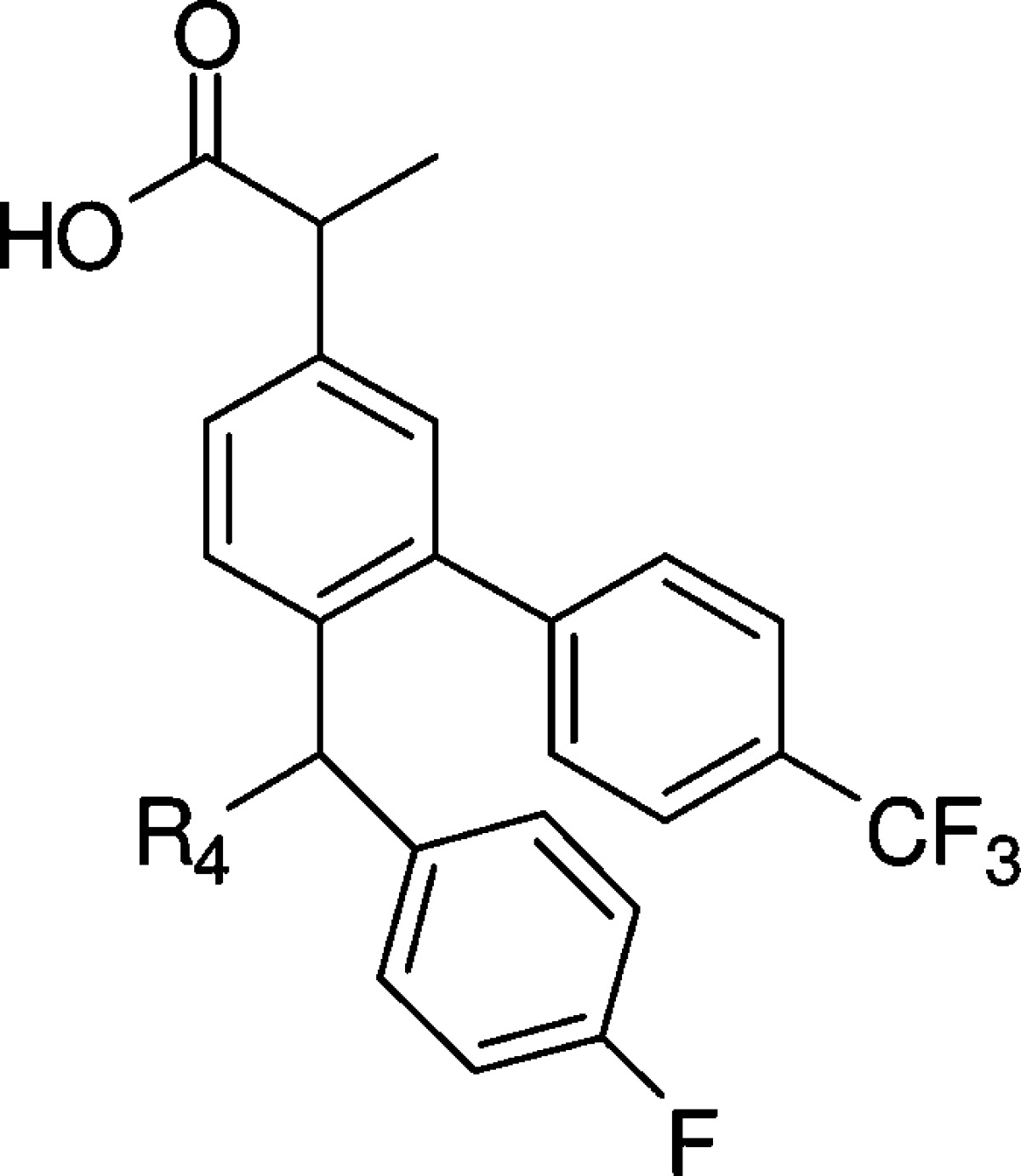
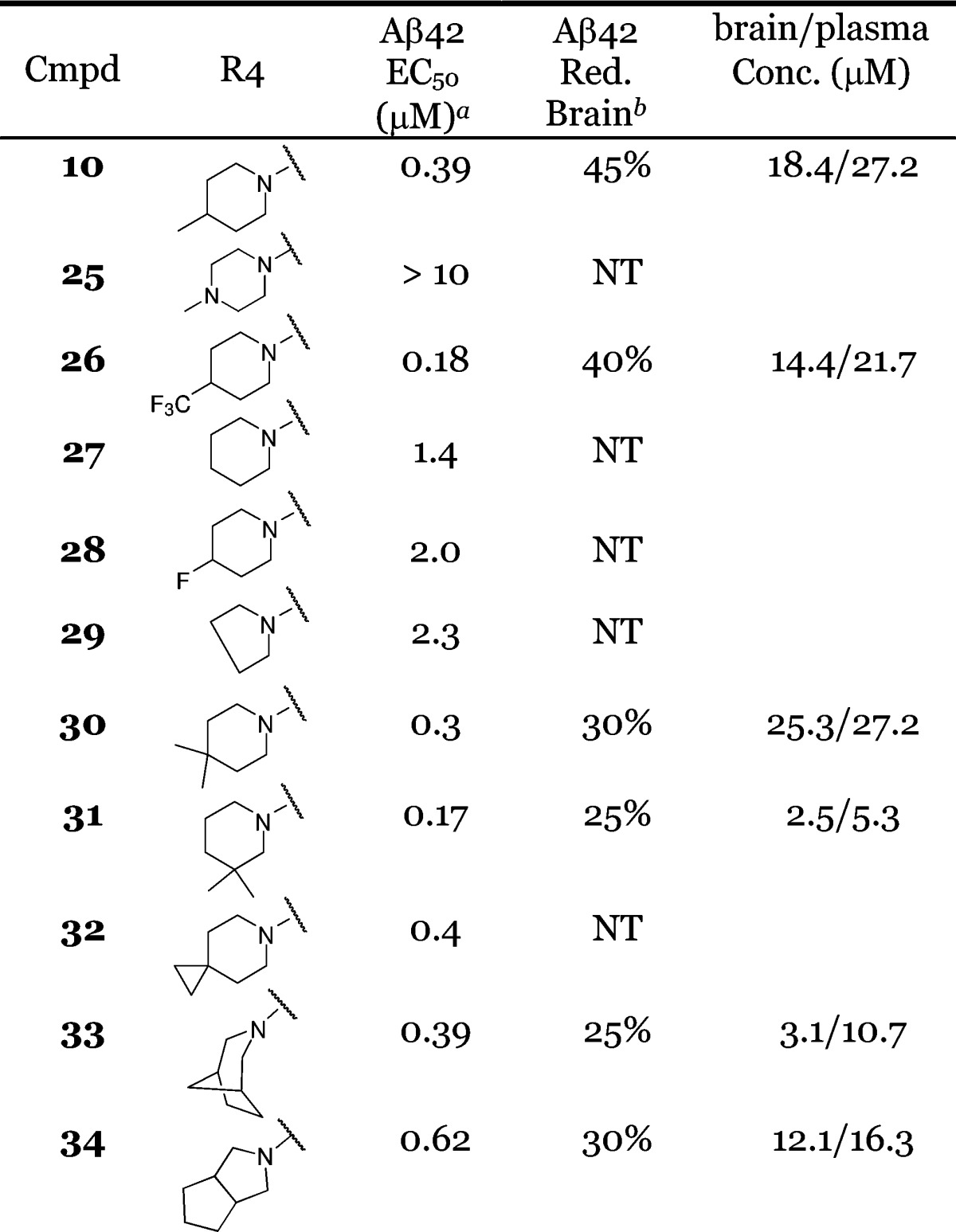
EC50 values were calculated as an average of at least two determinations.
PD responses were measure at 4 h after oral administration of compounds at 30 mg/kg in a solution formulation (ethanol:propylene glycol:10% solutol = 1:1:8).
The synthetic route to 10a is outlined in Scheme 1. The α-methyl group was installed by benzyl protection of methyl 2-(3-hydroxyphenyl)acetate (35) followed by methylation and deprotection to give intermediate 36. Then, 4-fluorobenzaldehyde and 4-methylpiperidine were condensed with 36 in trifluoro toluene via a three components Mannich reaction to afford intermediate 37. Triflation of 37 followed by Suzuki coupling with 4-(trifluoromethyl)phenyl boronic acid provided 39, which was hydrolyzed to compound 10. Using supercritical fluid (SCF) chiral method, racemate 10 was separated into two single isomers (10a and 10b). The absolute configuration of 10a was unambiguously established as R,R by a single crystal X-ray structure determination (Supporting Information). The other two isomers (10c + 10d) were very difficult to separate and were tested as a mixture in the assays.
As shown in Table 3, isomers 10a and 10b showed similar cellular potency in lowering Aβ42 (EC50 = 0.17 and 0.15 μM, respectively). The Aβ40 levels were unchanged, and Aβ38 levels were increased (EC50 = 0.15 and 0.11 μM, respectively). The mixture of the other two isomers was much less active (EC50 = 0.89 μM) and was not profiled further at the time. Both 10a and 10b demonstrated a very good PD response in the brain in wild-type mice after oral administration at 10 mg/kg. At 4 h postdose, these compounds achieved 4.6 and 6.6 μM brain concentrations, respectively, resulting in a 40 and 38% reduction in brain Aβ42 levels, as compared to the vehicle control. When tested in the FastPatch hERG assay, compound 10b had an EC50 of 4.6 μM, while isomer 10a had an EC50 of 15 μM. Therefore, we proceeded to profile 10a more thoroughly.
Compound 10a demonstrated excellent permeability in the Caco 2 assay [Papp(A – B) = 43.8 × 10–6 cm/s, p ratio (B – A/A – B) = 0.8] and was not a substrate for efflux transporters. It did not inhibit any of the five major human CYP isoforms assayed (3A4, 2C9, 2C19, 2D6, and 1A2; IC50 > 35 μM), and there was no evidence for time-dependent inhibition of human CYP3A4. The in vitro microsomal stability of 10a was good [Qh%: 27 (mouse), 24 (rat), 41 (dog), 25 (dog), and 34% (human)]. Compound 10a was highly protein-bound across species in mouse, rat, dog, monkey, and human plasma (fu% < 0.1%). The thermodynamic solubility was moderate and measured 20–30 μg/mL for a crystalline HCl salt. In addition to the low risk hERG readout, compound 10a was negative in the AMES mutagenicity assay.
In terms of selectivity, compound 10a inhibited COX1 or COX2 very weakly (IC50 = 35 and 27 μM, respectively). Compound 10a was tested in a cell-based assay in which the levels of HES1 protein were measured. HES1 expression is dependent upon an intact Notch signaling cascade and is mediated via cleavage of the Notch intracellular domain from the nascent transmembrane protein by γ-secretase.16 Compound 10a was found to have no effect on HES-1 after 24 h of incubation of the cell with 10 μM compound in this assay.
Compound 10a demonstrated very good pharmacokinetic characteristics. As summarized in Table 4, it achieved good oral bioavailability (44–106%) after a single oral dose at 10 mg/kg (crystalline HCl salt formulated in suspension) in rats, dogs, and cynomolgus monkeys. Compound 10a had low clearance in rats and cynomolgus monkeys with a t1/2 of 6 and 11 h, respectively. In dogs, the clearance was higher, and the t1/2 was approximately 2 h.
Table 4. Single Dose Pharmacokinetic Parameters of 10a in Rats, Dogs, and Cynomolgus Monkeysa.
| species | route | dose (mg/kg) | Clp (mL/min/kg) | Vss (L/kg) | t1/2 (h) | AUC0–24 h (μg/mL h) | Cmax (μg/mL) | Tmax (h) | F (%) |
|---|---|---|---|---|---|---|---|---|---|
| rat | ivb | 1 | 1.5 ± 0.1 | 0.8 ± 0.2 | 7.2 ± 1.7 | 10.4 ± 0.8 | |||
| poc | 10 | 6.6 ± 0.6 | 49.7 ± 8.8 | 6.0 ± 0.4 | 1.7 ± 0.6 | 47 ± 9 | |||
| dog | ivb | 1 | 14 ± 1 | 0.9 ± 0.1 | 1.3 ± 0.2 | 1.2 ± 0.1 | |||
| poc | 10 | 2.3 ± 0.2 | 5.2 ± 1.8 | 1.4 ± 0.5 | 1.3 ± 0.6 | 44 ± 16 | |||
| monkey | ivb | 1 | 3.7 ± 1.2 | 1.7 ± 0.3 | 11.2 ± 4.3 | 4.7 ± 1.4 | |||
| poc | 10 | 25 ± 6 | 48 ± 14 | 5.9 ± 2.3 | 2.0 ± 1.7 | 106 ± 33 |
Mean ± SD (n = 3).
Formulation iv EtOH:PEG400:water (10:40:50), 1 mL/kg.
Formulation po, 0.5% CMC, 0.2% Tween80, 5 mL/kg.
Compound 10a was also tested in human H4 cells that express human wild-type APP. In this assay, the compound displayed the same Aβ modulating profile as it had in the CHO-based cellular assay with an EC50 of 70 nM for the reduction of Aβ42. As discussed previously, compound 10a showed significant Aβ42 lowering (40%) in the brain after acute oral dosing at 10 mg/kg in wild type mice (CF-1). At the same oral dose, compound 10a also achieved 30% Aβ42 reduction in the brain in Fischer rats (4 h) and 30% Aβ42 reduction in the plasma in cynomolgus monkeys (5 h). The shifting of cleavage to shorter Aβ peptide in all three species appeared to match what was observed in the in vitro cellular assay: lowering Aβ42, increasing Aβ38, while having no effect on Aβ40 levels. The complete pharmacological data will be reported in due course.
In conclusion, we have designed a novel series of acid-based GSMs that are potent and have excellent brain exposure. On the basis of its potent lowering of brain Aβ, its lack of Notch activity, and its favorable pharmaceutical profiles, BIIB042 (10a) was selected as a candidate for preclinical safety evaluation.
Acknowledgments
We thank Drs. Alphonse Galdes, Wen-Cherng Lee, and William Harris for their strong support of the program, Harriet Rivera for compound management, DMPK group for the support on compound profiling and PK studies, and neuropharmacology group for the support on the in vivo studies.
Supporting Information Available
Experimental details for Aβ and Notch assays, synthetic procedures, and analytical data for compounds 6–34 and crystallographic data for compound 10a. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Citron M. Alzheimer's disease: Strategies for disease modification. Nature Rev. Drug Discovery 2010, 9, 387–398. [DOI] [PubMed] [Google Scholar]
- Hardy J.; Selkoe D. J. The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Tanzi R. E.; Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: A genetic perspective. Cell 2005, 120, 545–555. [DOI] [PubMed] [Google Scholar]
- Walsh D. M.; Selkoe D. J. Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron 2004, 44, 181–193. [DOI] [PubMed] [Google Scholar]
- De Strooper B.; Vassar R.; Golde T. The secretases: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett J. T.; Berger E. P.; Lansbury P. T. Jr. The carboxy terminus of β amyloid protein is critical for the seeding of amyloid formation: Implications for pathogenesis of Alzheimer's disease. Biochemistry 1993, 32, 4693–4697. [DOI] [PubMed] [Google Scholar]
- McGowan E.; Pickford F.; Kim J.; Onstead L.; Eriksen J.; Yu C.; Skipper L.; Murphy M. P.; Beard J.; Das P.; Jansen K.; DeLucia M.; Lin W.-L.; Dolios G.; Wang R.; Eckman C. B.; Dickson D. W.; Hutton M.; Hardy J.; Golde T. Aβ42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron 2005, 47, 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar G. M.; Li S.; Mehta T. H.; Garcia-Munoz A.; Shepardson N. E.; Smith I.; Brett F. M.; Farrell M. A.; Rowan M. J.; Lemere C. A.; Regan C. M.; Walsh D. M.; Sabatini B. L.; Selkoe D. J. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong G. T.; Manfra D.; Poulet F. M.; Zhang Q.; Josien H.; Bara T.; Engstrom L.; Pinzon-Ortiz M.; Fine J. S.; Lee H.-J. J.; Zhang L.; Higgins G. A.; Parker E. M. Chronic treatment with the γ-secretase inhibitor LY-411,575 inhibits β-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J. Biol. Chem. 2004, 279, 12876–12882. [DOI] [PubMed] [Google Scholar]
- Milano J.; McKay J.; Dagenais C.; Foster-Brown L.; Pognan F.; Gadient R.; Jacobs R. T.; Zacco A.; Greenberg B.; Ciaccio P. J. Modulation of Notch processing by γ-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol. Sci. 2004, 82, 341–358. [DOI] [PubMed] [Google Scholar]
- Weggen S.; Eriksen J. L.; Das P.; Sagi S. A.; Wang R.; Pietrizik C. U.; Findlay K. A.; Smith T. E.; Murphy M. P.; Bulter T.; Kang D. E.; Marquez-sterling N.; Golde T. E.; Koo E. H. A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 2001, 414, 212–216. [DOI] [PubMed] [Google Scholar]
- Eriksen J. L.; Sagi S. A.; Smith T. E.; Weggen S.; Das P.; McLendon D. C.; Ozols V. V.; Jessing K. W.; Zavitz K. H.; Koo E. H.; Golde T. E. NSAIDs and enantiomers of flurbiprofen target γ-secretase and lower Aβ42 in vivo. J. Clin. Invest. 2004, 112, 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbimbo B. P.; Hutter-Paier B.; Villetti G.; Facchinetti F.; Cenacchi V.; Volta R.; Lanzillotta A.; Pizzi M.; Windisch M. CHF5074, a novel γ-secretase modulator, attenuates brain β-amyloid pathology and learning deficit in a mouse model of Alzheimer's disease. Br. J. Pharmacol. 2009, 156, 982–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton M. G.; Hubbs J.; Sloman D.; Hamblett C.; Andrade P.; Angagaw M.; Bi G.; Black R. M.; Crispino J.; Cruz J. C.; Fan E.; Farris G.; Hughes B. L.; Kenific C. M.; Middleton R. E.; Nikov G.; Sajonz P.; Shah S.; Shomer N.; Szewczak A. A.; Tanga F.; Tudge M. T.; Shearman M.; Munoz B. Fluorinated piperidine acetic acids as γ-secretase modulators. Bioorg. Med. Chem. Lett. 2010, 20, 755–758. [DOI] [PubMed] [Google Scholar]
- Oehlrich D.; Berthelot D. J.-C.; Gijsen H. γ-Secretase modulators as potential disease modifying Anti-Alzheimer's drugs. J. Med. Chem. 2011, 54, 669–698. [DOI] [PubMed] [Google Scholar]
- Jarriault S.; Le Bail O.; Hirsinger E.; Pourquié O.; Logeat F.; Strong C. F.; Brou C.; Seidah N. G.; Isra I. A. Delta-1 activation of notch-1 signaling results in HES-1 transactivation. Mol. Cell. Biol. 1998, 18, 7423–7431. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.