

Abstract

Prostaglandin D2 (PGD2) plays a key role in mediating allergic reactions seen in asthma, allergic rhinitis, and atopic dermatitis. PGD2 exerts its activity through two G protein-coupled receptors (GPCRs), prostanoid D receptor (DP or DP1), and chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2 or DP2). We report the optimization of a series of phenylacetic acid derivatives in an effort to improve the dual activity of AMG 009 against DP and CRTH2. These efforts led to the discovery of AMG 853 (2-(4-(4-(tert-butylcarbamoyl)-2-(2-chloro-4-cyclopropylphenyl sulfonamido)phenoxy)-5-chloro-2-fluorophenyl)acetic acid), which is being evaluated in human clinical trials for asthma.
Keywords: AMG 853, CRTH2 receptor, DP receptor, prostaglandin D2, antagonist, asthma
Prostaglandin D2 (PGD2) is produced by mast cells in high concentrations during allergic responses and plays a key role in mediating allergic reactions seen in asthma, allergic rhinitis, atopic dermatitis, and allergic conjunctivitis.1,2 PGD2 exerts its activity through two G protein-coupled receptors (GPCRs), DP (prostanoid D receptor, DP1) and CRTH2 (chemoattractant receptor-homologous molecule expressed on Th2 cells, DP2). These two GPCRs act in concert to promote a number of biological effects associated with the development and maintenance of the allergic responses. Numerous studies using DP and CRTH2 antagonists, combined with genetic analysis, support the view that these receptors play a pivotal role in mediating allergic diseases.3−6 Therefore, there has been significant interest in the development of selective DP and CRTH2 antagonists.7−23 We have pursued the thesis that blockade of both receptors may prove more beneficial in alleviating allergic diseases triggered by PGD2 than inhibiting either one separately; therefore, we have been interested in identifying potent CRTH2/DP dual inhibitors. We previously reported on the discovery of AMG 009 (1; Figure 1) as a potent CRTH2 and DP dual antagonist.23 In this article, we report our continued optimization efforts, which led to the discovery of AMG 853 (2), which possesses increased DP and CRTH2 potency as compared to 1.
Figure 1.

AMG 009 and AMG 853.
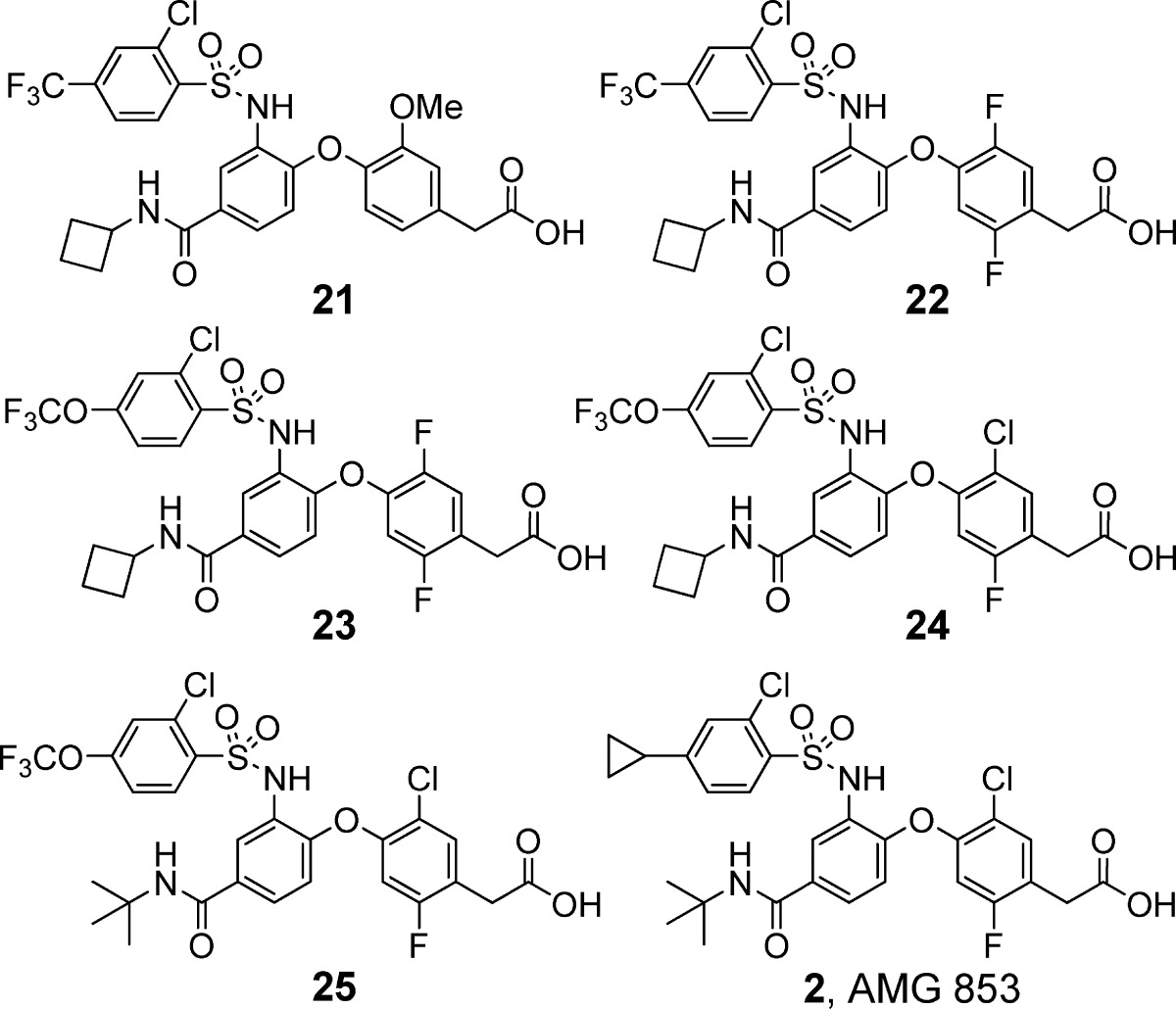
Compounds 1−25 were synthesized according to the chemistry depicted in Scheme 1. 4-Chloro-3-nitrobenzoyl chloride was reacted with amines to form the corresponding amides. Displacement of the chlorine adjacent to the nitro group with 4-hydroxyphenylacetic acids gave the bis-aryl ethers in good yields. Reduction of the nitro group followed by protection of the carboxylic acids as methyl esters provided the aniline methyl esters. The final two steps were carried out in one pot. The anilines were converted to the benzene sulfonamides by treating with sulfonyl chlorides, and then, hydrolysis of the methyl esters using sodium hydroxide afforded the final compounds (1−25).
Scheme 1.

Compound 1 had high affinity toward the CRTH2 receptor, even in the presence of human plasma. It inhibited the binding of 3H-PGD2 to the CRTH2 receptors on HEK-293 cells with an IC50 of 0.021 μM in the presence of human plasma. It also inhibited the binding of 3H-PGD2 to the DP receptors with a moderate IC50 of 0.28 μM in the presence of plasma. Therefore, we set out to find compounds with improved activity on DP while maintaining the excellent activity on CRTH2 displayed by 1. In addition, in vitro metabolism studies with 1 identified the methoxy, amide, and sulfonamide phenyl groups as potential metabolic sites. Thus, we focused our efforts on modifying these regions.
Compound 1 was one of the most potent dual CRTH2 and DP inhibitors identified from this series when it was selected for preclinical studies. Potency comparisons were mainly based on IC50 values in the presence of 50% human plasma, which correlated well with the functional activity in human whole blood. During earlier optimization efforts, it was found that single changes to the structure of 1 failed to yield significant improvements in potency. For example, a variety of chloro-substituted benzene sulfonamides were studied (Table 1). Two compounds (3 and 8) had similar potency as compared to 1; all other compounds suffered a loss in potency.
Table 1. Sulfonamide Modifications.
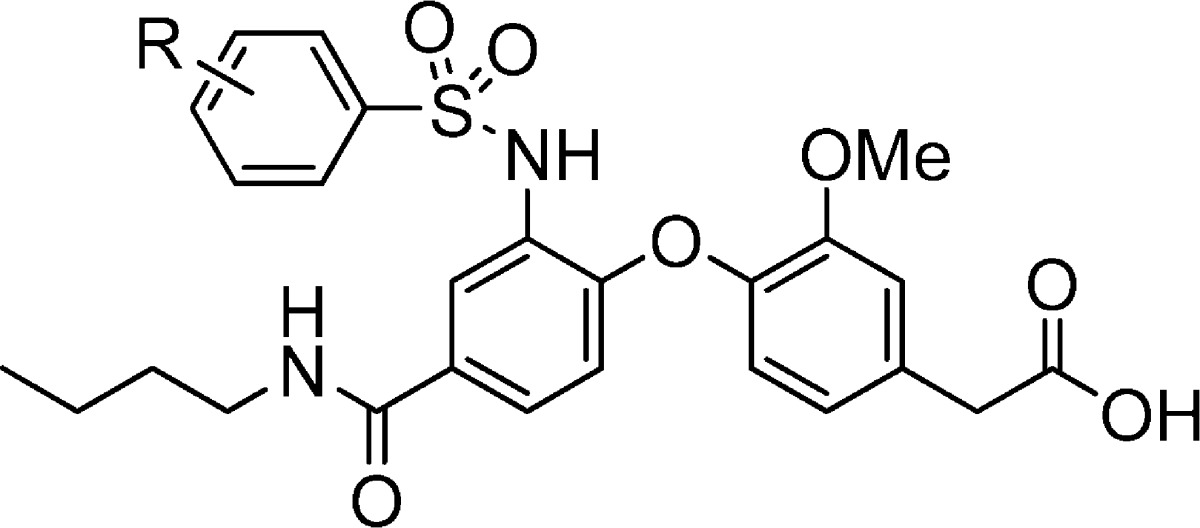
| IC50a in buffer/plasma (μM) |
|||
|---|---|---|---|
| compd | R | CRTH2 | DP |
| 1 | 2,4-Cl | 0.003/0.021 | 0.012/0.28 |
| 3 | 3,4-Cl | 0.005/0.023 | 0.021/0.12 |
| 4 | 2,5-Cl | 0.008/0.069 | 0.050/3.64 |
| 5 | 3,5-Cl | 0.68/4.38 | 0.93/>10 |
| 6 | 2,3-Cl | 0.032/0.30 | 0.52/2.34 |
| 7 | 2,6-Cl | 0.012/1.18 | >10/ND |
| 8 | 2-Cl-4-CF3 | 0.004/0.047 | 0.021/0.19 |
| 9 | 4-Cl | 0.004/0.10 | 0.013/ND |
Displacement of 3H-lPGD2 from the CRTH2 receptor expressed on HEK 293 cells. Assay run in buffer containing 0.5% bovine serum albumin. See ref (24) for assay protocol. Values are means of three experiments, and the standard deviation is ±30%.
Substitution of the phenyl ring of the phenylacetic acid moiety was also investigated (Table 2). A few substituents (methyl in 10 and difluoro in 14) provided similar CRTH2 and DP potencies as the methoxy group in 1. However, other substitutions, such as mono halogen (11 and 12) and cyano (13), afforded weaker potencies, especially in the presence of human plasma.
Table 2. Phenylacetic Acid Modifications.
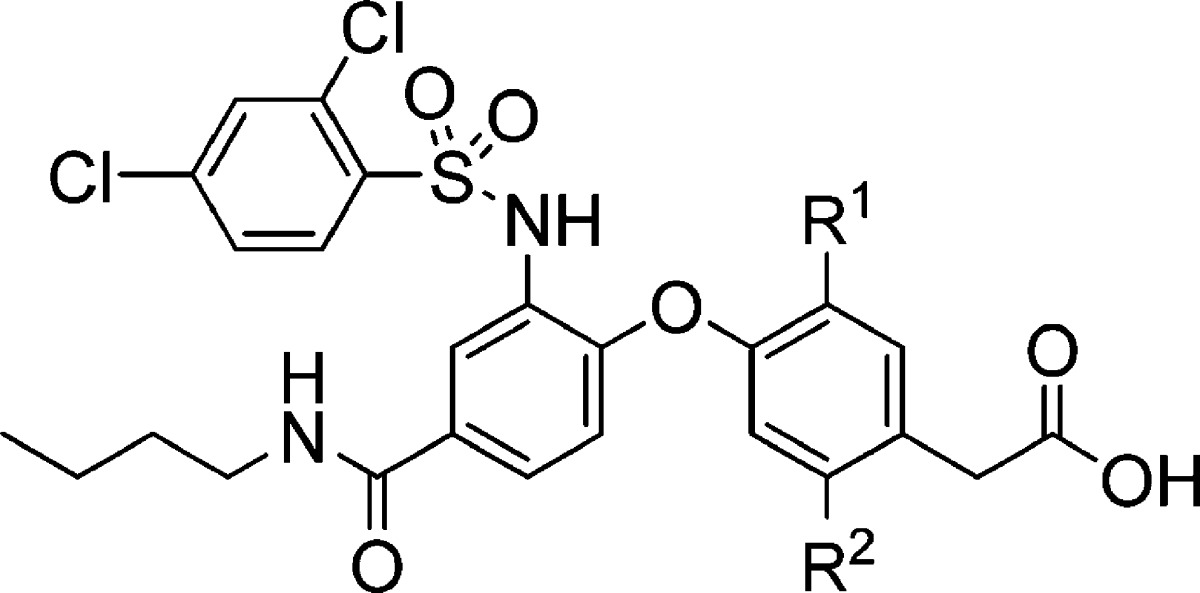
| IC50a in buffer/plasma (μM) |
||||
|---|---|---|---|---|
| compd | R1 | R2 | CRTH2 | DP |
| 1 | OMe | H | 0.003/0.021 | 0.012/0.28 |
| 10 | Me | H | 0.003/0.026 | 0.006/0.23 |
| 11 | Cl | H | 0.006/0.090 | 0.008/1.33 |
| 12 | F | H | 0.007/0.11 | 0.030/0.44 |
| 13 | CN | H | 0.018/0.21 | 0.31/ND |
| 14 | F | F | 0.004/0.032 | 0.007/0.60 |
Displacement of 3H-PGD2 from the CRTH2 or DP receptor expressed on HEK 293 cells. Assay run in buffer containing 0.5% bovine serum albumin or in 50% human plasma. See ref (24) for assay protocol. Values are means of three experiments, and the standard deviation is ±30%.
Furthermore, a previous study around the amide demonstrated that the n-butyl amide as in 1 was one of the most potent amides.23 A structure−activity relationship study also showed that the bis-aryl ether linker was preferred. It could be replaced by a methylene linker, and the potencies were maintained. All other linker modifications, such as sulfide, sulfoxide, sulfone, and amine linkers, afforded compounds exhibiting weaker potencies (data not shown).
When more than one change was made to the structure of 1, and interesting combination effects were seen. For example, substitution of the n-butyl amide by a cyclopropyl amide in combination with replacement of the methoxy moiety by chlorine afforded a compound (16) with similar potency to 1 in the displacement assays (Table 3). On the other hand, substitution of the n-butyl amide moiety of 1 by a cyclopropyl amide group provided a compound (15) with decreased DP potency (Table 3). Likewise, replacement of the methoxy group in the phenylacetic acid moiety of 1 by a chlorine atom also resulted in a compound (11) that was less potent than 1 in the presence of plasma. This suggested that the combination of modifications at different parts of the molecule needed to be taken into consideration when optimizing this series for potency in the presence of plasma.
Table 3. Combination of Amide and Phenylacetic Acid Modifications.
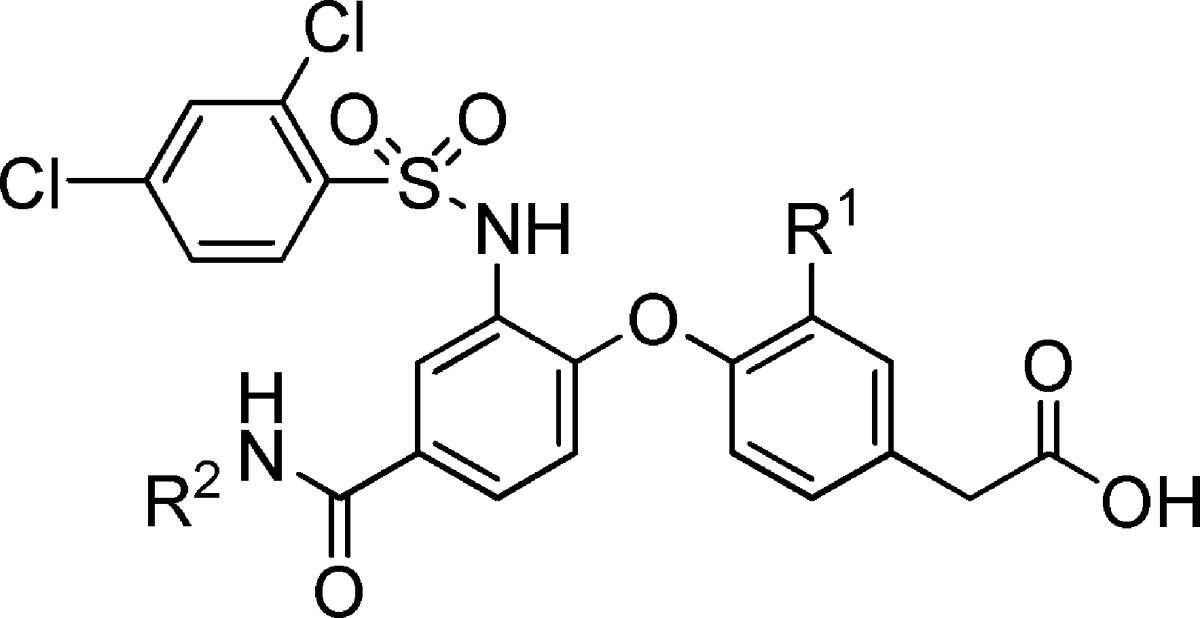
| IC50a in buffer/plasma (μM) |
||||
|---|---|---|---|---|
| compd | R1 | R2 | CRTH2 | DP |
| 1 | OMe | n-Bu | 0.003/0.021 | 0.012/0.28 |
| 15 | OMe | c-Pr | 0.004/0.028 | 0.041/1.03 |
| 11 | Cl | n-Bu | 0.006/0.090 | 0.008/1.33 |
| 16 | Cl | c-Pr | 0.007/0.025 | 0.014/0.22 |
Displacement of 3H-PGD2 from the CRTH2 or DP receptor expressed on HEK 293 cells. Assay run in buffer containing 0.5% bovine serum albumin or in 50% human plasma. See ref (24) for assay protocol. Values are means of three experiments, and the standard deviation is ±30%.
Table 4 further illustrates the effect that combination of different groups had on this series. The t-butyl amide functional group in combination with the 4-chlorobenzenesulfonamide moiety afforded a molecule (18) with decreased activity on the DP receptor, as compared to the analogous cyclo-butyl amide derivative (17). However, compound 20, which contained a t-butyl amide group in combination with the 2,4-dichlorobenzenesulfonamide moiety, had similar affinity for the DP receptor as the corresponding cyclo-butyl amide (19) in the presence of plasma.
Table 4. Combination of Sulfonamide and Amide Modifications.
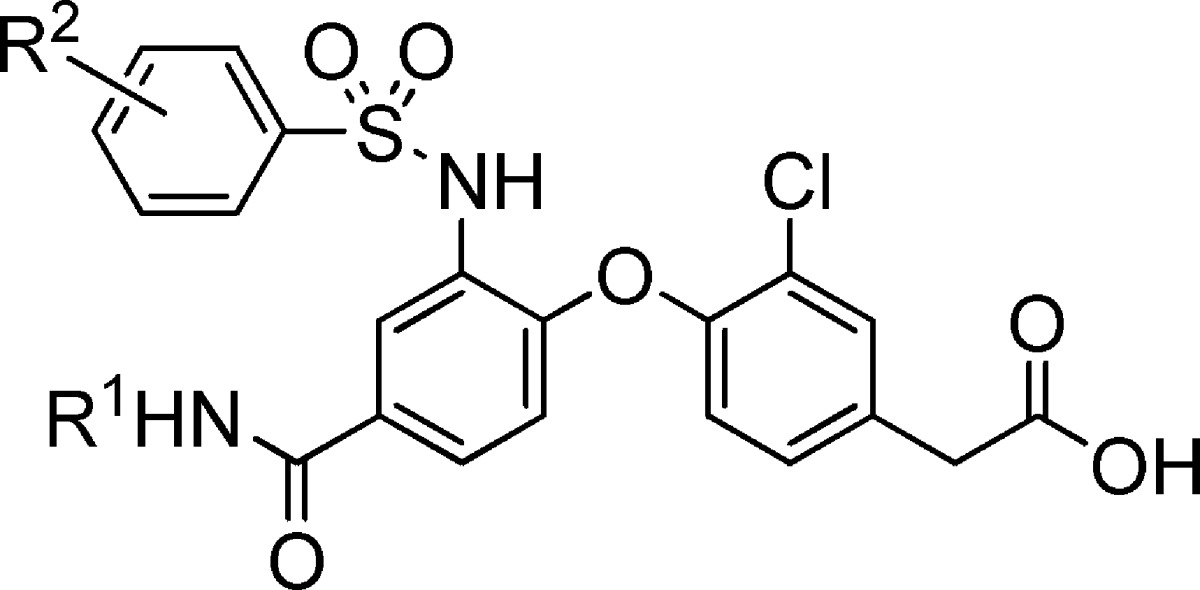
| IC50a in buffer/plasma (μM) |
||||
|---|---|---|---|---|
| compd | R1 | R2 | CRTH2 | DP |
| 17 | c-Bu | 4-Cl | 0.006/0.012 | 0.016/0.085 |
| 18 | t-Bu | 4-Cl | 0.009/0.015 | 0.046/0.45 |
| 19 | c-Bu | 2,4-Cl | 0.005/0.019 | 0.009/0.21 |
| 20 | t-Bu | 2,4-Cl | 0.005/0.012 | 0.015/0.23 |
Displacement of 3H-PGD2 from the CRTH2 or DP receptor expressed on HEK 293 cells. Assay run in buffer containing 0.5% bovine serum albumin or in 50% human plasma. See ref (24) for assay protocol. Values are means of three experiments, and the standard deviation is ±30%.
On the basis of the observations exemplified in Tables 3 and 4, we explored multiple combinations of preferred groups in the phenylacetic acid, amide, and sulfonamide regions. The combination produced dual CRTH2 and DP inhibitors with significantly improved DP potency as compared to 1. Table 5 shows a few examples of the potent dual antagonists. Compound 2 was among the potent dual antagonists discovered.
Table 5. Potent CRTH2 and DP Dual Antagonists.
| CRTH2 IC50a (μM) |
DP IC50a (μM) |
|||
|---|---|---|---|---|
| compd | in buffer | in plasma | in buffer | in plasma |
| 1 | 0.003 | 0.021 | 0.012 | 0.28 |
| 21 | 0.004 | 0.013 | 0.013 | 0.045 |
| 22 | 0.013 | 0.041 | 0.009 | 0.022 |
| 23 | 0.025 | 0.053 | 0.020 | 0.042 |
| 24 | 0.005 | 0.008 | 0.010 | 0.019 |
| 25 | 0.009 | 0.019 | 0.014 | 0.038 |
| 2 | 0.003 | 0.008 | 0.004 | 0.035 |
Displacement of 3H-PGD2 from the CRTH2 or DP receptor expressed on HEK 293 cells. Assay run in buffer containing 0.5% bovine serum albumin or in 50% human plasma. See ref (24) for assay protocol. Values are means of three experiments, and the standard deviation is ±30%.
Key compounds were also evaluated in CRTH2 and DP human whole blood functional assays. Consistent with the displacement assay data, 2 was also significantly more potent than 1 in the human whole blood functional assays. Compound 2 inhibited the PGD2-induced down-modulation of CRTH2 on CD16 negative granulocytes (eosinophils) in human whole blood with a Kb of 0.2 nM, while 1 had a Kb of 1 nM.25 Compound 2 also inhibited PGD2-induced cAMP response in platelets in 80% human whole blood with a Kb of 4.7 nM, which was significantly improved, as compared to the Kb of 148 nM of 1.26 In addition, 2 demonstrated similar antagonist activity in an aequorin assay using CRTH2-transfected HEK 293 cells and an eosinophil shape change assay, as compared to the CRTH2 receptor down-modulation human whole blood assay.25
The significant improvement of DP potency of 2 over 1 was also demonstrated in vivo in a guinea pig model of PGD2-induced airway constriction (Figure 2).27 In this model, airway resistance (Penh, enhanced pause) was measured in response to PGD2 challenge. The efficacy seen in this model was most likely due to the effect on the DP receptors, because CRTH2 selective antagonist had no effect in the model.23 The in vitro guinea pig DP potency was evaluated in guinea pig whole blood cAMP assay. Compound 2 had a Kb of 5 nM, while the Kb of 1 was 82 nM.
Figure 2.
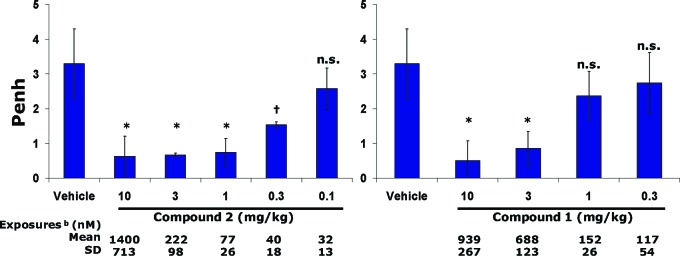
Evaluation of 2 and 1 in the guinea pig model of PGD2-induced airway constriction. Compounds 2 and 1 were dosed sc 4 h prior to aerosolized PGD2 exposure (0.6 mg/mL); n = 4 animals per group. bPlasma exposure at 4 h after dosing; *p < 0.02, †p < 0.05, and n.s., not significant relative to vehicle treatment.
The pharmacokinetic properties of 2 were evaluated in several species (Table 6) and were similar to those of 1.23 Compound 2 had low to moderate clearance across species, excellent oral absorption, and low potential of inducing drug−drug interactions as indicated by CYP inhibition and induction tests.
Table 6. Pharmacokinetic Properties of 2.
| species | clearance (L/h/kg)a | MRT (h) | Vdss (L/kg) | oral bioavailabilityb (%) |
|---|---|---|---|---|
| rat | 0.19 | 3.9 | 0.71 | 87 |
| dog | 0.9 | 3.0 | 2.72 | 100 |
| cynoc | 0.96 | 1.0 | 0.97 | 79 |
iv dose at 0.5 mg/kg.
Oral dose at 2 mg/kg.
Cynomolgus monkey.
In summary, further optimization of a series of phenylacetic acid derivatives led to the discovery of 2 (AMG 853), which displayed significantly improved potency, as compared to 1 (AMG 009), in CRTH2 and DP displacement and functional assays, including assays performed in human whole blood, and in vivo. Compound 2 was selected for evaluation in human clinical trials for asthma.
Supporting Information Available
Detailed synthetic experimental procedures and characterization for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Mitsumori S. Recent Progress in Work on PGD2 Antagonists for Drugs Targeting Allergic Diseases. Curr. Pharm. Des. 2004, 10, 3533–3538. [DOI] [PubMed] [Google Scholar]
- Nagai H. Prostaglandin as a Target Molecule for Pharmacotherapy of Allergic Inflammatory Diseases. Allergol. Int. 2008, 57, 187–196. [DOI] [PubMed] [Google Scholar]
- Schuligoi R.; Sturm E.; Luschnig P.; Konya V.; Philipose S.; Sedej M.; Waldhoer M.; Peskar B. A.; Heinemann A. CRTH2 and D-Type Prostanoid Receptor Antagonists as Novel Therapeutic Agents for Inflammatory Diseases. Pharmacology 2010, 85, 372–382. [DOI] [PubMed] [Google Scholar]
- Pettipher R. The roles of the prostaglandin D2 receptors DP1 and CRTH2 in promoting allergic responses. Br. J. Pharmacol. 2008, 153, s191–s199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettipher R.; Hansel T. T.; Armer R. Antagonism of the prostaglandin D2 receptors DP1 and CRTH2 as an approach to treat allergic diseases. Nat. Rev. Drug Discovery 2007, 6, 313–325. [DOI] [PubMed] [Google Scholar]
- Matsuoka T.; Hirata M.; Tanaka H.; Takahashi Y.; Murata T.; Kabashima K.; Sugimoto Y.; Kobayashi T.; Ushikubi F.; Aze Y.; Eguchi N.; Urade Y.; Yoshida N.; Kimura K.; Mizoguchi A.; Honda Y.; Nagai H.; Narumiya S. Prostaglandin D2 as a Mediator of Allergic Asthma. Science 2000, 287, 2013–2017. [DOI] [PubMed] [Google Scholar]
- Norman P. DP2 receptor antagonists in development. Expert Opin. Invest. Drugs 2010, 19, 947–961. [DOI] [PubMed] [Google Scholar]
- Medina J. C.; Liu J. PGD2 Antagonists. Annu. Rep. Med. Chem. 2006, 41, 221–235. [Google Scholar]
- Pettipher R.; Hansel T. T. Antagonists of the prostaglandin D2 receptor CRTH2. Drug News Perspect. 2008, 21, 317–322. [DOI] [PubMed] [Google Scholar]
- Philip G.; van Adelsberg J.; Loeys T.; Liu N.; Wong P.; Lai E.; Balachandra S.; Reiss T. F. Clinical studies of the DP1 antagonist laropiprant in asthma and allergic rhinitis. J. Allergy Clin. Pharmacol. 2009, 124, 942–948. [DOI] [PubMed] [Google Scholar]
- Sturino C. F.; O'Neill G.; Lachance N.; Boyd M.; Berthelette C.; Labelle M.; Li L.; Roy B.; Scheigetz J.; Tsou N.; Aubin Y.; Bateman K. P.; Chauret N.; Day S. H.; Lévesque J.; Seto C.; Silva J. H.; Trimble L. A.; Carriere M.; Denis D.; Greig G.; Kargman S.; Lamontagne S.; Mathieu M.; Sawyer N.; Slipetz D.; Abraham W. M.; Jones T.; McAuliffe M.; Piechuta H.; Griffith D. A. N.; Wang Z.; Zamboni R.; Young R. N.; Metters K. M. Discovery of a Potent and Selective Prostaglandin D2 Receptor Antagonist, [(3R)-4-(4-Chloro- benzyl)-7-fluoro-5-(methylsulfonyl)-1,2,3,4-tetrahydrocyclopenta[b]indol-3-yl]-acetic Acid (MK-0524). J. Med. Chem. 2007, 50, 794–806. [DOI] [PubMed] [Google Scholar]
- Beaulieu C.; Guay D.; Wang Z.; Leblanc Y.; Roy P.; Dufresne C.; Zamboni R.; Berthelette C.; Day S.; Tsou N.; Denis D.; Greig G.; Mathieu M. C.; O'Neill G. Identification of prostaglandin D2 receptor antagonists based on a tetrahydropyridoindole scaffold. Bioorg. Med. Chem. Lett. 2008, 18, 2696–2700. [DOI] [PubMed] [Google Scholar]
- Arimura A.Discovery of a First-in-Class Drug, a Prostaglandin D2 Antagonist, for the Treatment of Allergic Diseases. In Molecular Imaging for Integrated Medical Therapy and Drug Development; Tamaki N., Kuge Y., Eds.; Springer: New York, 2010; Part IV, pp 281−287. [Google Scholar]
- Mitsumori S.; Tsuri T.; Honma T.; Hiramatsu Y.; Okada T.; Hashizume H.; Kida S.; Inagaki M.; Arimura A.; Yasui K.; Asanuma F.; Kishino J.; Ohtani M. Synthesis and Biological Activity of Various Derivatives of a Novel Class of Potent, Selective, and Orally Active Prostaglandin D2 Receptor Antagonists. 2. 6,6-Dimethylbicyclo[3.1.1]heptane Derivatives. J. Med. Chem. 2003, 46, 2446–2456. [DOI] [PubMed] [Google Scholar]
- Oxagen Ltd. Novel CRTH2 Antagonist Shows Potential in Moderate Persistent Asthma: Presented at 19th Annual Congress of the European Respiratory Society (ERS), September 18, 2009; http://www.docguide.com/news/content.nsf/news/852576140048867A85257635005BC1C3.
- Tumey L. N.; Robarge M. J.; Gleason E.; Song J.; Murphy S. M.; Ekema G.; Doucette C.; Hanniford D.; Palmer M.; Pawlowski G.; Danzig J.; Loftus M.; Hunady K.; Sherf B.; Mays R. W.; Stricker-Krongrad A.; Brunden K. R.; Bennani Y. L.; Harrington J. J. 3-Indolyl sultams as selective CRTh2 antagonists. Bioorg. Med. Chem. Lett. 2010, 20, 3287–3290. [DOI] [PubMed] [Google Scholar]
- Grimstrup M.; Receveur J. M.; Rist O.; Frimurer T. M.; Nielsen P. A.; Mathiesen J. M.; Högberg T. Exploration of SAR features by modifications of thiazoleacetic acids as CRTH2 antagonists. Bioorg. Med. Chem. Lett. 2010, 20, 1638–1641. [DOI] [PubMed] [Google Scholar]
- Liu J.; Wang Y.; Sun Y.; Marshall D.; Miao S.; Tonn G.; Anders P.; Tocker J.; Tang H. L.; Medina J. Tetrahydroquinoline derivatives as CRTH2 antagonists. Bioorg. Med. Chem. Lett. 2009, 19, 6840–6844. [DOI] [PubMed] [Google Scholar]
- Stearns B. A.; Baccei C.; Bain G.; Broadhead A.; Clark R. C.; Coate H.; Evans J. F.; Fagan P.; Hutchinson J. H.; King C.; Lee C.; Lorrain D. S.; Prasit P.; Prodanovich P.; Santini A.; Scott J. M.; Stock N. S.; Truong Y. P. Novel tricyclic antagonists of the prostaglandin D2 receptor DP2 with efficacy in a murine model of allergic rhinitis. Bioorg. Med. Chem. Lett. 2009, 19, 4647–4651. [DOI] [PubMed] [Google Scholar]
- Ulven T.; Kostenis E. Minor Structural Modifications Convert the Dual TP/CRTH2 Antagonist Ramatroban into a Highly Selective and Potent CRTH2 Antagonist. J. Med. Chem. 2005, 48, 897–900. [DOI] [PubMed] [Google Scholar]
- Crosignani S.; Page P.; Missotten M.; Colovray V.; Cleva C.; Arrighi J. F.; Atherall J.; Macritchie J.; Martin T.; Humbert Y.; Gaudet M.; Pupowicz D.; Maio M.; Pittet P. A.; Golzio L.; Giachetti C.; Rocha C.; Bernardinelli G.; Filinchuk Y.; Scheer A.; Schwarz M. K.; Chollet A. Discovery of a New Class of Potent, Selective, and Orally Bioavailable CRTH2 (DP2) Receptor Antagonists for the Treatment of Allergic Inflammatory. Diseases. J. Med. Chem. 2008, 51, 2227–2243. [DOI] [PubMed] [Google Scholar]
- Armer R. E.; Ashton M. R.; Boyd E. A.; Brennan C. J.; Brookfield F. A.; Gazi L.; Gyles S. L.; Hay P. A.; Hunter M. G.; Middlemiss D.; Whittaker M.; Xue L.; Pettipher R. Indole-3-acetic Acid Antagonists of the Prostaglandin D2 Receptor CRTH2. J. Med. Chem. 2005, 48, 6174–6177. [DOI] [PubMed] [Google Scholar]
- Liu J.; Fu Z.; Wang Y.; Schmitt M.; Huang A.; Marshall D.; Tonn G.; Seitz L.; Sullivan T.; Tang H. L.; Collins T.; Medina J. Discovery and optimization of CRTH2 and DP dual antagonists. Bioorg. Med. Chem. Lett. 2009, 19, 6419–6423. [DOI] [PubMed] [Google Scholar]
- The CRTH2 or DP radioligand binding assay was performed on HEK-293 cells stably expressing human CRTH2 or DP. To measure binding, [3H]-PGD2 was incubated together with 293 (hCRTH2 or hDP) cells in the presence of increasing concentrations of compounds. After teh cells were washed, the amount of [3H]-PGD2 that remained bound to the cells was measured by scintillation counting, and the concentration of compounds required to achieve a 50% inhibition of [3H]-PGD2 binding (the IC50) was determined. The binding buffer contains either 0.5% BSA (buffer binding) or 50% human plasma (plasma binding).
- Human whole blood was drawn into acid-citrate-dextrose (ACD) anticoagulated tubes, treated with compounds or DMSO, and then stimulated with a dose response of PGD2. Fluorochrome conjugated antibodies were used to label CRTH2 positive granulocytes, and CRTH2 receptor internalization was monitored by flow cytometry. The Kb was determined using the Schild equation.
- Human whole blood was drawn into ACD vacutainer tubes, treated with compounds or DMSO, and then stimulated with a dose response of PGD2. Cells were lysed, and cAMP was measured using a competitive ELISA. Comparison of the dose response to PGD2 in samples containing DMSO only and samples containing compounds was used in determining Kb using the Schild equation.
- Hamelman E.; Schwarze J.; Takeda K.; Oshiba A.; Larsen G. L.; Irvin G.; Gelfand G. W. Am. J. Respir. Crit. Care Med. 1997, 156, 766–775. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.