

Abstract

S-Nitrosoglutathione reductase (GSNOR) regulates S-nitrosothiols (SNOs) and nitric oxide (NO) in vivo through catabolism of S-nitrosoglutathione (GSNO). GSNOR and the anti-inflammatory and smooth muscle relaxant activities of SNOs, GSNO, and NO play significant roles in pulmonary, cardiovascular, and gastrointestinal function. In GSNOR knockout mice, basal airway tone is reduced and the response to challenge with bronchoconstrictors or airway allergens is attenuated. Consequently, GSNOR has emerged as an attractive therapeutic target for several clinically important human diseases. As such, small molecule inhibitors of GSNOR were developed. These GSNOR inhibitors were potent, selective, and efficacious in animal models of inflammatory disease characterized by reduced levels of GSNO and bioavailable NO. N6022, a potent and reversible GSNOR inhibitor, reduced bronchoconstriction and pulmonary inflammation in a mouse model of asthma and demonstrated an acceptable safety profile. N6022 is currently in clinical development as a potential agent for the treatment of acute asthma.
Keywords: GSNOR, GSNO, N6022, asthma, pyrrole, nitric oxide
The reaction of glutathione with reactive nitrogen species gives S-nitrosoglutathione (GSNO) as a major nitric oxide (NO) metabolite.1S-Nitrosoglutathione reductase (GSNOR), a member of the alcohol dehydrogenase (ADH) family,2 has been reported to regulate intracellular S-nitrosothiols (SNOs), a biologically important class of stable NO adducts, by reducing GSNO.3 GSNO has been shown to elicit many biological functions of NO and also serves as a durable depot for NO, which has a short biological half-life.4 Increases in bioavailable NO are associated with anti-inflammatory and smooth muscle relaxant effects, especially in organ systems characterized by smooth muscle and endothelial/epithelial layers such as the respiratory, cardiovascular, and gastrointestinal systems.5−7
GSNOR dysregulation has recently been implicated in respiratory diseases.8,9 Under normal conditions, NO and GSNO can maintain lung function through their influence on bronchial smooth muscle tone and anti-inflammatory activities.9,10 In human asthma, there are lowered SNO concentrations in the lungs, likely attributable to up-regulated GSNOR activity.8 Mice with genetic deletion of GSNOR exhibit increases in lung SNOs and are protected from airway hyperresponsivity.11 In these mice, GSNOR has been shown to have an important influence on NO containing species, regulation of smooth muscle tone in the airways, and function of adrenergic receptors in lungs and heart.5,8,12 GSNO also plays an important role in inflammatory bowel disease (IBD). NO and GSNO maintain normal intestinal physiology via anti-inflammatory actions and maintenance of the intestinal epithelial cell barrier. In IBD, reduced levels of GSNO and NO are evident and may also occur via up-regulation of GSNOR activity.13
Given such findings, GSNOR has emerged as a potentially important target for the treatment of respiratory, cardiovascular, and gastro-intestinal diseases, many of which have lowered levels of NO as a basis to their pathophysiology.6,13−15 N30 Pharmaceuticals initiated a discovery effort to identify small molecule inhibitors of GSNOR through high throughput screening of commercially available compounds followed by structure based lead optimization. The goal of the effort was to identify potent GSNOR inhibitors that can be administered either intravenously (IV) or orally (PO) to treat these diseases. In this paper, we report potent GSNOR inhibitors with low nanomolar IC50, which demonstrated in vivo efficacy and safety in preclinical models and have advanced into clinical development. We also describe the structure−activity relationships of GSNOR inhibitors and the crystal structure of an enzyme−NAD+−inhibitor complex, and we report some initial biological and pharmacological properties of these inhibitors.
The majority of GSNOR inhibitors reported in this study were synthesized according to Scheme 1 or 2. Other compounds were synthesized using methods described in the Supporting Information.
Scheme 1. Synthetic Route of GSNOR Inhibitors.

Conditions: (a) 2-furanaldehyde, NaOMe/MeOH. (b) conc HCl/EtOH, reflux. (c) conc HBr/EtOH, reflux. (d) aniline, TsOH/EtOH, reflux. (e) aniline, AcOH/EtOH, reflux. (f) 1 N NaOH/EtOH, 45 °C. (g) 1 N NaOH/MeOH, 45 °C.
Scheme 2. Alternate Synthesis of GSNOR Inhibitors.

Conditions: (a) 2,5-dimethoxytetrahydrofuran/AcOH. (b) POCl3/DMF. (c) Ph3P = CHCO2Et, toluene, reflux. (d) potassium 3-ethoxy-3-oxopropanoate, DMAP/piperidine/AcOH/DMF, 80 °C. (e) H2, 10% Pd/C in EtOH. (f) NBS. (g) boronic acid, Pd(PPh3)4, Na2CO3. (h) K2CO3, H2O2/DMSO, room temperature. (i) LiOH.
In Scheme 1, condensation of ketone 1 and 2-furanaldehyde afforded intermediate 2 in high yield.16 Furan ring-opening of intermediate 2 by HCl or HBr in alcohol under reflux conditions provided diketone 3.17 Pyrrole formation was achieved by condensation of diketone 3 and anilines under acidic conditions.18 Hydrolysis of intermediate 4 by NaOH in alcohol afforded final products 5a−5j in 5−15% overall yield.
Compounds with structural variations on the phenyl ring at the C-5 position of the pyrrole ring were synthesized according to Scheme 2. Condensation of 4-amino-3-methylbenzamide with 2,5-dimethoxytetrahydrofuran in AcOH led to the formation of a key intermediate pyrrole 6.19 Formylation of the pyrrole ring of 6 under the Vilsmeier conditions produced 7 in good yield. The acrylate 8 was obtained through a Wittig reaction of 7 with (carbethoxymethylene)triphenylphosphorane20 or reaction of aldehyde 7 with potassium monoethyl malonate and DMAP in DMF followed by addition of AcOH and piperidine.21 Hydrogenation of the acrylate 8 using Pd/C afforded the propionic ester 9, bromination of which resulted in the key building block 10.22 Derivatization of 10 with a variety of boronic acids or esters under Suzuki conditions provided intermediate 11.23 The nitrile 11 was converted to amide 12 by K2CO3 in the presence of H2O2 in DMSO. Final hydrolysis under basic conditions afforded 13a−13i, with overall yields of 5−20%.
The GSNOR inhibitors reported here bind reversibly to the HMGSH binding pocket.24 Although the structures of apo, binary, and ternary complexes of GSNOR have been published,25 no crystal structure of the GSNO−GSNOR complex has been reported. To guide design and optimization of GSNOR inhibitors, the structures of inhibitors bound to GSNOR and NAD+ were determined by X-ray crystallography. The crystal structure of the ternary complex of GSNOR, NAD+, and N6022 was solved to 1.9 Å (Figure 1) (pdb code: 3QJ5). The enzyme−ligand complex crystallized as a homodimer with N6022 binding to each GSNOR monomer through the following key interactions: (1) the imidazole in the inhibitor interacts with one of the structural zincs, which is also coordinated to histidine 66, cysteine 44, and cysteine 173 to form the zinc coordination tetrahedral; (2) the carboxylic acid of N6022 hydrogen bonds to the glutamine 111 and forms a salt bridge with arginine 114 and lysine 283 from the second monomer; (3) the N6022 carboxamide hydrogen bonds to glutamine 117; (4) the N6022 imidazole ring forms a π−π interaction with the nicotinamide ring of NAD+.
Figure 1.
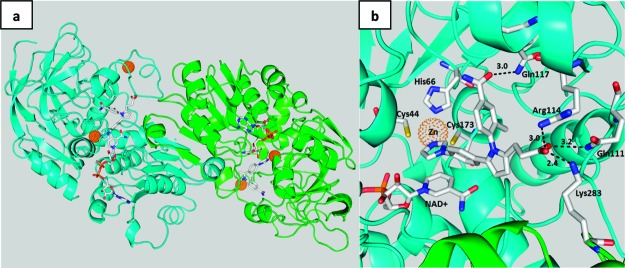
Dimeric structure of N6022 bound to the GSNOR enzyme−NAD+ complex (a) and the GSNOR binding pocket of N6022 (b).
Compound 5a (GSNOR IC50 = 570 nM), which was identified by screening a library of commercially available compounds, was the starting point for lead optimization based on ability to inhibit GSNOR activity in vitro (Table 1). Introduction of a methyl group at the R3-position of the N-phenyl ring as shown in Scheme 1 resulted in slight improvement of IC50 from 570 nM to 360 nM (5b). Substitution at the 4-position of the C5-phenyl ring (R1) with a methoxy group also improved IC50 to 460 nM (5c). IC50 = 210 nM was achieved by combining both functionalities (5f). Structure−activity relationships (SAR) involving substituents at the R3 position were further explored by replacing the methyl group with halogens (5d and 5e) or a trifluoromethyl group (14). Slightly improved activity was observed for the chloro analogue 5e. The R1 position was further examined by substitution with a variety of groups. Subtle increases in potency were obtained for the chloro 13b, bromo 13c, and hydroxyl 5h analogues. Significant improvement was realized by substituting the R1 position with imidazole, leading to N6022 with IC50 of 20 nM. The increased potency of N6022 can be explained by the fact that the imidazole interacts with one of the structural Zn in GSNOR (Figure 1). Other potential Zn binding moieties including heterocycles were explored, but imidazole appeared to be the best.
Table 1. SAR and PK Properties of GSNOR Inhibitors.
| compd | R1 | R2 | R3 | GSNOR IC50 (nM) | % F | CL (mL/min/kg) |
|---|---|---|---|---|---|---|
| 5a | H | H | H | 570 | 86.5 | 8.9 |
| 5b | H | H | Me | 360 | 46.8 | 11.8 |
| 5c | OMe | H | H | 460 | ||
| 5d | OMe | H | F | 240 | ||
| 5e | OMe | H | Cl | 190 | ||
| 14 | OMe | H | CF3 | 450 | ||
| 5f | OMe | H | Me | 210 | 16.2 | 35.4 |
| 13a | F | H | Me | 500 | ||
| 13b | Cl | H | Me | 120 | 20.7 | 16.0 |
| 13c | Br | H | Me | 160 | 18.0 | 21.4 |
| 5g | CN | H | Me | 563 | 16.3 | 25.5 |
| 5h | OH | H | Me | 160 | ||
| 13d | CF3 | H | Me | 3990 | ||
| 15 | CONH2 | H | Me | 3920 | ||
| N6022 | 1H-imidazol-1-yl | H | Me | 20 | 4.4 | 37.8 |
| 16 | Cl | MeO | Me | 56 | 22.4 | 20.2 |
| 13e | MeO | Cl | Me | 200 | ||
| 17 | Br | MeO | Me | 96 | 21.8 | 22.4 |
| 5j | Cl | OH | Me | 55 | 2.72 | 33.4 |
| 13f | Cl | CONH2 | Me | 160 | ||
| 18 | Cl | EtO | Me | 170 | ||
| 19 | Cl | PrO | Me | 110 | ||
| 20 | Cl | NMe2 | Me | 350 | ||
| 21 | Cl | NHCHO | Me | 580 | ||
| 13g | Cl | Cl | Me | 160 | ||
| 13h | Cl | F | Me | 120 | ||
| 13i | Cl | CF3 | Me | 330 |
SAR involving substituents of the C5-phenyl ring were further explored by substituting at both the ortho and para positions (Table 1). In most cases, the R1 position was substituted with chlorine for comparison and R2 was explored for its effect on IC50 and pharmacokinetic properties. When R2 was a methoxy (16), IC50 = 56 nM was achieved. Interchanging the position of the two groups resulted in a significant loss of potency (IC50 = 200 nM, 13e). The bromo analogue (17) of compound 16 also has a low IC50 (96 nM). The hydroxyl analogue 5j has similar activity to its methoxy comparator 16. Increasing the size of the alkoxy group at the R2 position resulted in a 2−3-fold reduction in potency (18 and 19). Simple nitrogen containing groups did not improve the inhibitory activity (13f, 20, and 21). Neither disubstitution with halogens (13g and 13h) nor the CF3 (13i) improved inhibitory activity.
Due to the potential metabolic liability of carboxylic acids, this group was replaced with a tetrazole moiety (Figure 2, Table 2), a commonly used bioisostere for carboxylic acid,26 connecting through the carbon of the tetrazole ring. The data in Table 2 demonstrate that the carboxylic acid can be replaced without a huge loss in potency, particularly when R1 was bromo (24) as compared to its carboxylic acid counterpart 13c (IC50 = 160 nM) or R1 was the N-imidazole group (28 and 29). The length of the chain linking pyrrole and tetrazole (Figure 2) played a critical role. When R1 was a relatively small group (e.g., methoxy and bromo), better inhibition was achieved with longer alkyl chains (22, 24, and 26). However, when R1 was a relatively larger group such as imidazole, the shorter chain length was preferred (29), due to size restriction in the binding pocket.
Figure 2.

Structures of tetrazole containing GSNOR inhibitors.
Table 2. SAR of Tetrazole Containing GSNOR Inhibitors.
| compd | R1 | R3 | R4 | n | GSNOR IC50 (nM) |
|---|---|---|---|---|---|
| 22 | OMe | Me | CONH2 | 1 | 1530 |
| 23 | OMe | Me | CONH2 | 0 | 2910 |
| 24 | Br | Me | CONH2 | 1 | 190 |
| 25 | Br | Me | CONH2 | 0 | 560 |
| 26 | Br | H | OH | 1 | 450 |
| 27 | Br | H | OH | 0 | 4350 |
| 28 | 1H-imidazol-1-yl | H | OH | 1 | 300 |
| 29 | 1H-imidazol-1-yl | H | OH | 0 | 18 |
Several compounds were selected for pharmacokinetic studies in mice. The oral bioavailability of the compounds tested ranged from 2.72% to 86.5% (Table 1), and the plasma clearance (CL) after IV administration ranged from 8.9 to 37.8 mL/min/kg. Compound 5a demonstrated good oral bioavailability (86.5%), although potency was poor. Despite the high potency of N6022, its oral bioavailability was very low (4.4%), showing high clearance (37.8 mL/min/kg), potentially due to the high polarity of the imidazole. Compound 16 was the most potent non-imidazole-containing analogue, with an oral bioavailability of 22%.
A battery of in vitro assays was employed to assess the potential off-target activity of the GSNOR inhibitors on 54 transmembrane and soluble receptors, ion channels, and monoamine transporters. Off-target effects were estimated from the percent inhibition of receptor radioligand binding at a 10 μM concentration of test compound. Limited off-target activity was observed toward the δ2 opiate receptor for N6022 (66% inhibition); no other potential off-target activity was detected. Selected compounds were also screened for cytotoxicity toward A549 epithelial lung cells. Minimal cytotoxicity (IC50's > 100 μM) was observed using this assay. Selected compounds were also screened in cytochrome P450 assays. All compounds screened in these assays exhibited minimal inhibition at 10 μM compound concentration except N6022, which had an IC50 = 0.77 μM for CYP 2C19. N6022 was negative in a hERG in vitro screen (>100 μM) and showed no evidence of mutagenicity in a bacterial mutagen screen.
Selected GSNOR inhibitors were tested in 5-day toxicology studies in mice. Compounds were administered to mice by IV or PO routes. The selected inhibitors were well-tolerated. The no observable adverse effect levels (NOAELs) for IV administration of N6022, 5f, and 13c were 30, 50, and 50 mg/kg/day, respectively. The NOAEL for PO administration of N6022 for 5 days was 100 mg/(kg day).27
The efficacy of GSNOR inhibitors was assessed in animal models of asthma and other inflammatory diseases influenced by dysregulated GSNOR. In a mouse model of ovalbumin-induced asthma,28N6022 attenuated methacholine-induced bronchoconstriction (airway hyperresponsiveness) in a dose- and time-dependent manner, with significant efficacy achieved with a single IV dose ≥0.01 mg/kg and when administered from 30 min to 48 h prior to the methacholine challenge. N6022 also significantly attenuated eosinophil infiltration into the lungs with a single IV dose ≥0.0005 mg/kg.29 In a mouse model of DSS induced IBD, N6022 demonstrated significant efficacy with either oral or IV dosing at 1 and 10 mg/kg/day for 10 days. The efficacy of inhibitors 5j, 5f, 13c, and 16 was assessed using one or more of the mouse models of ovalbumin-induced asthma, porcine pancreatic elastase (PPE)-induced COPD, DSS-induced IBD, and a rat model of high dietary salt-induced hypertension (DAHL/S). Significant efficacy was observed across all models.30−32
These data suggest that GSNOR inhibitors have potential as therapeutic agents to modulate GSNOR activity and mitigate GSNOR-mediated pathologies in vivo. Thus, N6022 is being advanced in preclinical studies and is currently in phase I clinical trials for the treatment of acute asthma. Other compounds described in this report are under evaluation for treating other types of GSNOR-mediated diseases.
In summary, structure based lead optimization led to a number of potent GSNOR inhibitors with favorable pharmacological properties in multiple disease models. We have identified a potent and reversible GSNOR inhibitor N6022, which demonstrated excellent in vivo activity in the ovalbumin induced asthma animal model. This compound also demonstrated an acceptable safety profile in the nonclinical toxicology evaluations. N6022 is currently under further development for its clinical potential to treat acute asthma.
Acknowledgments
The authors are grateful to Dr. J. Singh and his team at deCode chemistry for the synthetic chemistry support. We also thank Dr. Zhaojun Zhang and his colleagues at Chempartner for the synthesis of some of the inhibitors and Larry Ross at SRI for screening GSNOR inhibitors in the enzyme assay.
Glossary
Abbreviations
- GSNOR
S-nitrosoglutathione reductase
- GSNO
S-nitrosoglutathione
- NO
nitric oxide
- SNO
nitrosothiol
Supporting Information Available
Experimental details and characterization of selected compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Hess D. T.; Matsumoto A.; Kim S.-O.; Marshall H. E.; Stamler J. S. Protein S-nitrosylation: purview and parameters. Nature Rev., Mol. Cell Biol. 2005, 6, 150–166. [DOI] [PubMed] [Google Scholar]
- Eklund H.; Muller-Wille P.; Horjales E.; Futer O.; Holmquist B.; Vallee B. L.; Hoog J. O.; Kaiser R.; Jornvall H. Comparison of three classes of human liver alcohol dehydrogenase: Emphasis on different substrate binding pockets. Eur. J. Biochem. 1990, 193, 303–310. [DOI] [PubMed] [Google Scholar]
- Liu L.; Hausladen A.; Zeng M.; Que L.; Heltman J.; Stamler J. S. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature 2001, 410, 490–494. [DOI] [PubMed] [Google Scholar]
- Myers P. R.; Minor R. L. Jr.; Guerra R. Jr.; Bates J. N.; Harrison D. G. Vasorelaxant properties of the endothelium-derived relaxing factor more closely resemble S-nitrosocysteine than nitric oxide. Nature 1990, 345, 161–163. [DOI] [PubMed] [Google Scholar]
- Whalen E. J.; Foster M. W.; Matsumoto A.; Ozawa K.; Violin J. D.; Que L. G.; Nelson C. D.; Benhar M.; Keys J. R.; Rockman H. A.; Koch W. J.; Daaka Y.; Lefkowitz R. J.; Stamler J. S. Regulation of β-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell 2007, 129, 511–522. [DOI] [PubMed] [Google Scholar]
- Foster M. W.; Hess D. T.; Stamler J. S. Protein S-nitrosylation in health and disease: a current perspective. Trends Mol. Med. 2009, 15, 391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P.; Beckman J. S.; Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que L. G.; Yang Z.; Stamler J. S.; Lugogo N. L.; Kraft M. S-Nitrosoglutathione reductase: an important regulator in human asthma. Am. J. Respir. Crit. Care Med. 2009, 180, 226–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson E. M.; Gaston B. SNOR and wheeze: the asthma enzyme?. Trends Mol. Med. 2005, 11, 481–484. [DOI] [PubMed] [Google Scholar]
- Fortenberry J. D.; Owens M. L.; Chen N. X.; Brown L. A. S-nitrosoglutathione inhibits TNF-alpha-induced NF-κB activation in neutrophils. Inflamm. Res. 2001, 50, 89–95. [DOI] [PubMed] [Google Scholar]
- Que L. G.; Liu L.; Yan Y.; Whitehead G. S.; Gavett S. H.; Schwartz D. A.; Stamler J. S. Protection from experimental asthma by an endogenous bronchodilator. Science 2005, 308, 1618–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L.; Yan Y.; Zeng M.; Zhang J.; Hanes M. A.; Ahearn G.; McMahon T. J.; Dickfeld T.; Marshall H. E.; Que L. G.; Stamler J. S. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell 2004, 116, 617–628. [DOI] [PubMed] [Google Scholar]
- Savidge T. C.; Newman P.; Pothoulakis C.; Ruhl A.; Neunlist M.; Bourreille A.; Hurst R.; Sofroniew M. V. Enteric glia regulate intestinal barrier function and inflammation via release of S-nitrosoglutathione. Gastroenterology 2007, 132, 1344–1358. [DOI] [PubMed] [Google Scholar]
- Sanghani P. C.; Davis W. I.; Fears S. L.; Green S. L.; Zhai L.; Tang Y.; Martin E.; Bryan N. S.; Sanghani S. P. Kinetic and cellular characterization of novel inhibitors of S-nitrosoglutathione reductase. J. Biol. Chem. 2009, 284, 24354–24362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal G. J.; Blonder J.; Damaj B.; Richards J.; Elia M.; Scoggin C. A Novel compound that inhibits S-nitrosoglutathione reductase protects against experimental asthma. Am. Respir. Crit. Care Med. 2009, 179, A4151. [Google Scholar]
- Chong J. M.; Shen L.; Taylor N. J. Asymmetric conjugate addition of alkynylboronates to enones. J. Am. Chem. Soc. 2000, 122, 1822–1823. [Google Scholar]
- Nasipuri D.; Konar S. K. Polycyclic systems. Part XVIII. Synthesis of methyl 2-(1-naphthyl)-5-oxocyclopent-1-enylacetate, methyl β-5-(1-naphthyl)-2-furylpropionate, and a few related compounds. J. Indian Chem. Soc. 1978, 55, 580–583. [Google Scholar]
- Blicke F. F.; Warzynski R. J.; Faust J. A.; Gearien J. E. Preparation of certain acids and esters which contain phenylpyrryl nuclei. J. Am. Chem. Soc. 1944, 66, 1675–1677. [Google Scholar]
- Paillet-Loilier M.; Fabis F.; Lepailleur A.; Bureau R.; Butt-Gueulle S.; Dauphin F.; Delarue C.; Vaudry H.; Rault S. Phenylpyrroles, a new chemolibrary virtual screening class of 5-HT7 receptor ligands. Bioorg. Med. Chem. Lett. 2005, 15, 3753–3757. [DOI] [PubMed] [Google Scholar]
- Martinez G. R.; Hirschfeld D. R.; Maloney P. J.; Yang D. S.; Rosenkranz R. P.; Walker K. A. M. [(1H-Imidazol-1-yl)methyl]- and [(3-pyridinyl)methyl]pyrroles as thromboxane synthetase inhibitors. J. Med. Chem. 1989, 32, 890–897. [DOI] [PubMed] [Google Scholar]
- New J. S.; Christopher W. L.; Yevich J. P.; Butler R.; Schlemmer R. F. Jr.; VanderMaelen C. P.; Cipollina J. A. The thieno[3,2-c]pyridine and furo[3,2-c]pyridine rings: new pharmacophores with potential antipsychotic activity. J. Med. Chem. 1989, 32, 1147–1156. [DOI] [PubMed] [Google Scholar]
- He H.; Jiang X. Preparation of twelve 1-methyl-2-formyl-5-substituted pyrroles and their hydrazones. Chin. J. Chem. 1999, 17, 171–183. [Google Scholar]
- Vachal P.; Toth L. M. General facile synthesis of 2,5-diarylheteropentalenes. Tetrahedron Lett. 2004, 45, 7157–7161. [Google Scholar]
- Richards J.; et al. Kinetic characterization of N6022, a potent first-in-class drug inhibiting GSNOR. Unpublished results.
- Sanghani P. C.; Robinson H.; Bosron W. F.; Hurley T. D. Human glutathione-dependent formaldehyde dehydrogenase. Structures of apo, binary, and inhibitory ternary complexes. Biochemistry 2002, 41, 10778–10786. [DOI] [PubMed] [Google Scholar]
- Herr R. J. 5-Substituted-1H-tetrazoles as carboxylic acid isosteres: medicinal chemistry and synthetic methods. Bioorg. Med. Chem. 2002, 10, 3379–3393. [DOI] [PubMed] [Google Scholar]
- Colagiovanni D. B.; et al. Inhibition of S-nitrosoglutathione reductase does not cause mechanism-based toxicity. The Toxicologist 2011, 120S-2172. [Google Scholar]
- Zosky G. R.; Sly P. D. Animal models of asthma. Clin. Exp. Allergy: J. Br. Soc. Allergy Clin. Immunol. 2007, 37, 973–988. [DOI] [PubMed] [Google Scholar]
- Blonder J.;. et al. A GSNOR inhibitor protects against experimental asthma via bronchodilatory and anti-inflammatory activities. Unpublished results.
- Blonder J.;. et al. GSNOR inhibitors protect against experimental IBD. Unpublished results.
- Blonder J.;. et al. Oral GSNOR inhibitors attenuate pulmonary inflammation and decrease airspace enlargement in experimental models of chronic obstructive pulmonary disease. Unpublished results.
- Chen Q.;. et al. Pharmacological inhibition of GSNOR improves endothelial vasodilatory function in living rats. Unpublished results.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.