

Abstract

Phospoinositide-3-kinases (PI3K) are important oncology targets due to the deregulation of this signaling pathway in a wide variety of human cancers. A series of 2-morpholino, 4-substituted, 6-(3-hydroxyphenyl) pyrimidines have been reported as potent inhibitors of PI3Ks. Herein, we describe the structure-guided optimization of these pyrimidines with a focus on replacing the phenol moiety, while maintaining potent target inhibition and improving in vivo properties. A series of 2-morpholino, 4-substituted, 6-heterocyclic pyrimidines, which potently inhibit PI3K, were discovered. Within this series a compound, 17, was identified with suitable pharmacokinetic (PK) properties, which allowed for the establishment of a PI3K PK/pharmacodynamic−efficacy relationship as determined by in vivo inhibition of AKTSer473 phosphorylation and tumor growth inhibition in a mouse A2780 tumor xenograft model.
Keywords: phosphoinositide 3-kinase alpha, PI3K/AKT pathway
The phospoinositide-3-kinase (PI3K) family of lipid kinases is involved in a diverse set of cellular functions, including cell growth, proliferation, motility, differentiation, glucose transport, survival intracellular trafficking, and membrane ruffling.1 PI3Ks can be categorized in class I, II, or III, depending on their subunit structure, regulation, and substrate selectivity.2 Class IA PI3Ks are activated by receptor tyrosine kinases and consist of a regulatory subunit (p85) and a catalytic subunit (p110). There are three catalytic isoforms: p110 α, β, and δ. A single class IB PI3K, activated by G protein-coupled receptor, consists of only one member: a p110 γ catalytic subunit and a p101 regulatory subunit. The primary in vivo substrate of the class I PI3Ks is phosphatidylinositol (4,5) diphosphate, which, upon phosphorylation at the 3-position of the inositol ring to form phosphatidylinositol triphosphate (3,4,5)P3, serves as a second messenger by activating a series of downstream effectors that mediate the cellular functions mentioned above. The PI3K isoforms have different distributions and share similar cellular functions, which are context dependent. In particular, p110α pathway deregulation has been demonstrated in ovarian, breast, colon, and brain cancers.3,4 Inhibitors of PI3Kα represent an intriguing therapeutic modality for these indications, and as such, there is much interest in generating suitable molecules to test this hypothesis in the clinic.5−9
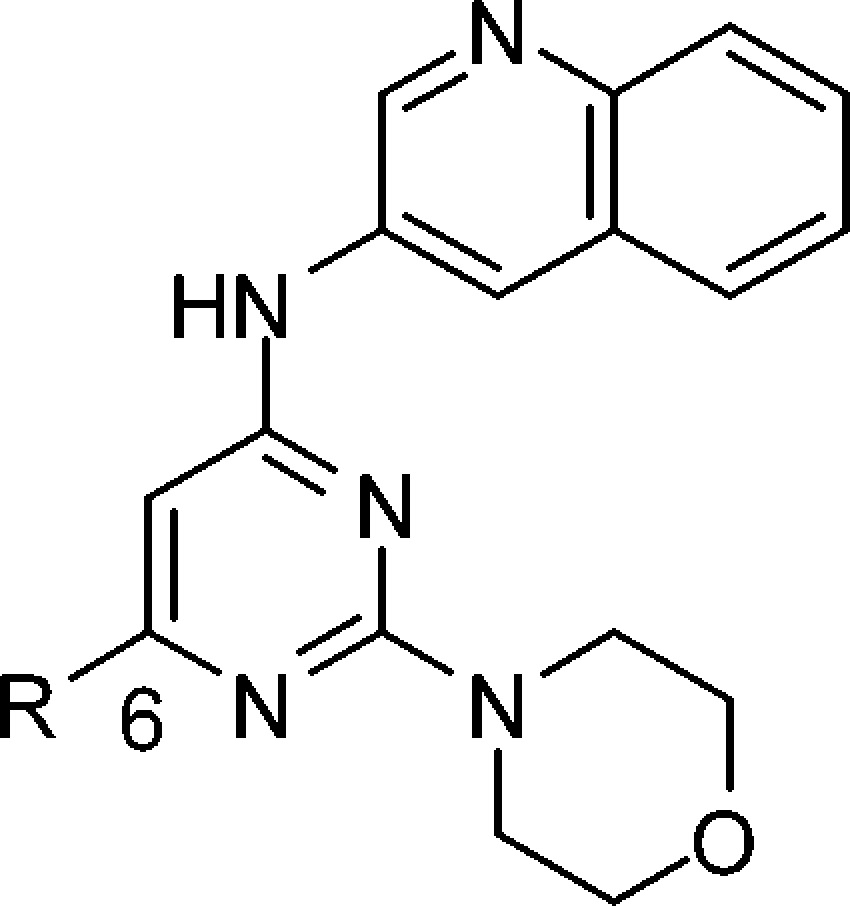
We have reported phenolic mopholino pyrimidines,10 such as compound 1 (Figure 1), as potent pan class I PI3K inhibitors that exhibit high selectivity toward other serine/threonine as well as tyrosine kinases. While exhibiting potent in vitro properties, the in vivo potential of such compounds may be limited due to the presence of the phenol moiety. Described herein are our efforts to identify potent morpholino pyrimidinyl inhibitors of PI3K that do not require a phenol group and exhibit PK properties suitable for achieving in vivo target modulation and efficacy.
Figure 1.

PI3Kα enzymatic potency and rat PK properties of 6-substituted, 4-(aminopyrid-3-yl), 2-morpholino pyrimidines.
The importance of the phenol moiety in 1 for PI3K binding as well as the phenols effect on in vivo properties can be seen in contrasting the properties of phenol 1 with the trifluoromethylphenyl analogue 2. The trifluoromethylphenyl analogue 2 is 60-fold less active against PI3Kα, while its rat PK is improved relative to phenol 1 when considering the % F, area under the curve (AUC), and iv t1/2. Thus, the challenge for compound optimization that we faced was to mimic the phenol binding interaction with a group that would not adversely affect the pharmacokinetic (PK) properties.
To approach this challenge, we turned to the cocrystal structure of compound 1 in PI3Kγ to gain an understanding of the phenol OH's binding interactions.11 Given the high homology between the α and the γ isoforms and approximately the same potency of 1 against the two isoforms, p110γ was used as a surrogate for p110α. The cocrystal structure of 1 in the ATP binding site of PI3Kγ, Figure 2, indicates the key binding contacts being made by the phenol as well as the morpholine group. The morpholine oxygen forms a hydrogen bond to the hinge Val882 NH. The phenol hydroxyl makes hydrogen bonds with Asp841 and Tyr867. The C4 aminopyridyl substituent extends out toward solvent and does not appear to make any specific hydrogen bonds.
Figure 2.
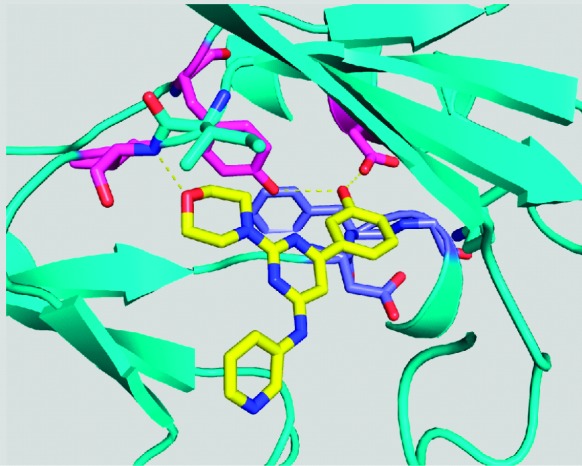
Structure of 1 in PI3Kγ.
With this structural insight, our strategy to identify phenol replacements was to survey a variety of heterocycles at the pyrimidine C6 position that would have the ability to make hydrogen-bonding interactions with the Asp841 and Tyr867 residues, identify such groups, and then profile their PK properties in rat. In this C6 survey, the morpholine at C2 and a 3-aminoquinoline at C4 were held constant. Upon identification of a suitable C6 phenol replacement, further optimization at the C4 position to modulate druglike properties and maintain potency was envisioned since the cocrystal structure indicated that this position extends out toward solvent and would tolerate a range of substituents.
C6-modified 2-morpholino pyrimidines III were synthesized by several routes as depicted in Scheme 1.12 Sequential nucleophilic substitution of 2,4,6-tribromopyrimidine, I, at the 4-position with a range of amines, then at the 2-position by morpholine yielded bromopyrimidine II. Subseqeunt Suzuki reaction yielded target compounds III. Alternatively, condensation of morpholine formamidine hydrobromide IV and diethylmalonate followed by refluxing in POCl3 yielded 2-morpholino 4,6-dichloropyrimidine V, which could undergo amination and Suzuki reaction to access target compounds. For preparing a series of C4 analogues with a fixed C6 heterocycle, the Suzuki step was performed first to yield chloropyrimidine VI prior to the Buchwald step yielding target compounds III. When necessary, the heterocyclic boronate esters used to install the C6 heteroaryl groups were prepared from the corresponding heteroarylbromides.
Scheme 1. Synthesis of 4,6-Substituted 2-Morpholino Pyrimidines.

Reagents: (a) H2NR1R2, DIEA, MeCN, 45 °C. (b) Morpholine, 45 °C. (c) HetB(OR)2, Pd(dppf)Cl2, DME, 2 M Na2CO3, 110 °C. (d) NaOEt, diethyl malonate, reflux. (e) POCl3, reflux. (f) Amine, Pd(OAc)2, BINAP, Cs2CO3, THF, 110 °C.
Prepared compounds were initially screened in biochemical PI3K assays, and compounds with PI3Kα IC50 values < 100 nM were tested in the A2780 ovarian carcinoma cell line (where the PI3K pathway is deregulated due to PTEN deletion) for inhibition of cell proliferation and phosphorylation of AKTSer473 as a target modulation readout. The results of the C6 substituent survey, Table 1, indicate that a 3-pyridyl group 4 is potent, being only 2-fold less active against PI3Kα than the phenol starting point 3. Substituted pyridines that place hydrogen bond donors ortho to the ring nitrogen were evaluated, and the aminopyridine 5 exhibits increased potency relative to the 3-pyridyl compound 4, being equipotent to the phenol. Introduction of the 5-pyrimidyl increases potency relative to the 3-pyridyl, with compound 10 being more potent than the phenol 3. In contrast, pyrazine 8 exhibits comparable potency to the 3-pyridyl 4. When the 2-amino group was introduced into the C6 pyrimidine, further potency enhancements were observed with the aminopyrimidine 12 being ≥30-fold more active than the starting phenol 3 against PI3Kα. Interestingly, the pyridone 7, which mimics the meta orientation of phenol 3, exhibited reduced activity relative to 3, highlighting the subtleties of trying to mimic a phenolic interaction.
Table 1. Biochemical Inhibition of PI3Kα, Inhibition of AKTSer473 Phosphorylation, and Antiproliferative Effect in A2780 Cells by 6-Substituted 2-Morpholinopyrimidin-4-yl)quinolin-3-amines.
| no. | R | PI3Kα IC50 (μM) | pAKTSer473 A2780 EC50 (μM) | A2780 EC50 (μM) |
|---|---|---|---|---|
| 3 | 3-phenol | 0.061 | 0.37 | 0.33 |
| 4 | pyridin-3-yl | 0.135 | 0.65 | 0.33 |
| 5 | 6-aminopyridin-3-yl | 0.055 | 0.31 | 0.45 |
| 6 | 1H-pyrazolo[3,4-b]pyridin-5-yl | 0.066 | 3.73 | |
| 7 | 2-hydroxypyridin-4-yl | 0.253 | 0.31 | |
| 8 | pyrazin-2-yl | 0.231 | 0.39 | 0.59 |
| 9 | 5-aminopyrazin-2-yl | 0.044 | 3.30 | 0.36 |
| 10 | pyrimidin-5-yl | 0.008 | 0.45 | 4.84 |
| 11 | 2-hydroxypyrimidin-5-yl | 0.774 | ||
| 12 | 2-aminopyrimidin-5-yl | <0.002 | 0.04 | 0.15 |
| 13 | 2-methylaminopyrimidin-5-yl | 0.007 | 0.11 | 0.14 |
| 14 | 2-dimethylaminopyrimidin-5-yl | 0.247 | 0.67 |
Upon identification of phenol replacements with maintained (aminopyridine) or improved (aminopyrimidine) in vitro potency, the aminopyridine- and aminopyrimidine-containing PI3K inhibitors 4, 5, and 12 were assessed in rat PK. As indicated in Table 2, the PK properties were improved relative to the phenol 3. The % F was increased from 1% to acceptable levels (23−56%), and the clearance value was reduced substantially. The improvement was greatest for aminopyridine 5 with 56% F, CL = 5 mL/min/kg, and iv t1/2 = 103 min. The comparison of PK properties between the phenolic compounds 1 (Figure 1) and 3 (Table 2), where the % F went from 9 to 1%, indicated that PK properties with a fixed C6 group could be modulated by the C4 substituent and suggested that the PK properties of the C6 aminopyridine or aminopyrimidines could be further improved by optimization of the C4 substituent.
Table 2. Rat PK Propertiesa of Phenol Replacements.
| no. | iv t1/2 (min) | CL (mL/min/kg) | oral AUC (μM h) | Vss (L/kg) | po % F |
|---|---|---|---|---|---|
| 3 | 30 | 179 | 0.03 | 6.9 | 1 |
| 4 | 55 | 26 | 16 | 2.4 | 46 |
| 5 | 103 | 5 | 93 | 0.8 | 56 |
| 12 | 43 | 37 | 6 | 1.9 | 23 |
Amounts: 5 mpk iv and 20 mpk po.
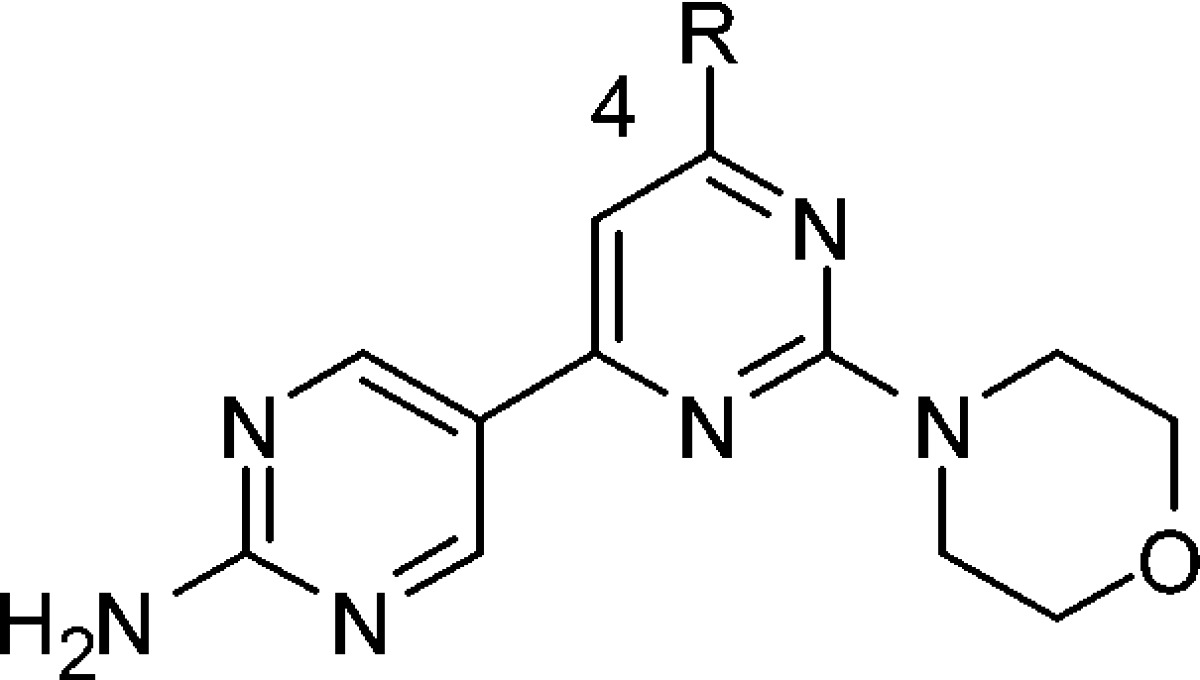
With a phenol replacement that improved both in vitro and in vivo properties identified, a C4 survey where the 2-morpholinyl and 6-(5-substituted-2-aminopyrimididyl) groups of the central pyrimidine were held fixed was conducted. As may be expected from the cocrystal structure of compound 1 with PI3Kγ (Figure 2), a variety of groups are tolerated at the C4 position (Table 3). The aminoquinoline 12 as well as the 6-substituted 3-aminopyridyl-substituted compounds 15−17 were extremely potent, being active at the limit of detection of the enzymatic assay. Additionally, of note is the potency and ligand efficiency13 of the 4-H-substituted 20 (ΔG = −0.56 kcal mol−1 per non-H atom!), which inhibited PI3Kα with an IC50 = 14 nm, A2780 pAKT473 EC50 = 132 nM, and A2780 EC50 = 5 μM. Additionally, the C2 symmetric bis aminopyrimidine-substituted morpholino pyrimidine 18 was potent, IC50 = 6 nm and A2780 EC50 = 0.23 μM.
Table 3. Biochemical Inhibition of PI3Kα, Inhibition of AKTSer473 Phosphorylation, and Antiproliferative Effect in A2780 Cells by 4-Substituted, 2-Morpholino, 6-(2-Aminopyrimid-5-yl) Pyrimidines.
| no. | R | PI3Kα IC50 (μM) | pAKTSer473 A2780 EC50 (μM) | A2780 EC50 (μM) |
|---|---|---|---|---|
| 12 | quinolin-3-yl-amino | <0.002 | 0.04 | 0.15 |
| 15 | 6-phenoxypyridin-3-yl-amino | <0.002 | 0.11 | 1.13 |
| 16 | 6-(1-methylpiperidin-4-yloxy)pyridin-3-yl-amino | <0.002 | 0.01 | 0.14 |
| 17 | 6-methoxypyridin-3-yl-amino | <0.002 | 0.09 | 0.73 |
| 18 | 2-amino-pyrimid-5-yl | 0.006 | 0.23 | |
| 19 | tetrahydro-2H-pyran-4-yl-amino | 0.013 | 0.22 | 1.96 |
| 20 | H | 0.014 | 0.13 | 4.98 |
Compounds with a positive combination of enzyme inhibition, cell target modulation, antiproliferative activity, and solubility were profiled further in rat PK studies. One such compound, 17,14 exhibited reasonable rat PK (5 mpk iv, 20 mpk oral, oral t1/2 = 77 min, % F = 89, CL = 79 mL/min/kg, Vss = 2.6 L/kg, and oral AUC = 9 μM h) and was studied further in mouse PK/PD and efficacy studies.15 Modulation of AKTThr308 and AKTSer473 phosphorylation was examined in A2780 xenograft tumors at time points ranging from 30 min to 24 h after a single 100 mg/kg dose of compound 17. As can be seen in Figure 3, at 8 h, >50% of target inhibition was achieved. The target modulation decreased as the compound exposure decreased, with the modulation approaching the vehicle level at 24 h.
Figure 3.
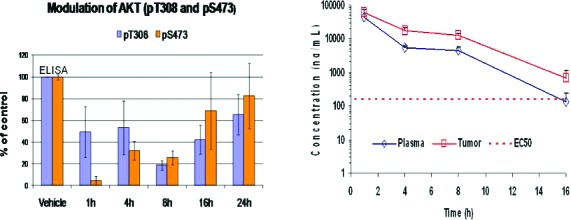
PKPD relationship of compound 17 in the A2780 xenograft model.
Efficacy experiments were then conducted in the A2780 tumor xenograft model, where tumor-bearing mice were administered compound 17 twice daily at 10 and 60 mg/kg. Tumor growth inhibition (50%) was observed at the 60 mg/kg dose level, while at 10 mg/kg, no inhibitory activity was observed (Figure 4).
Figure 4.
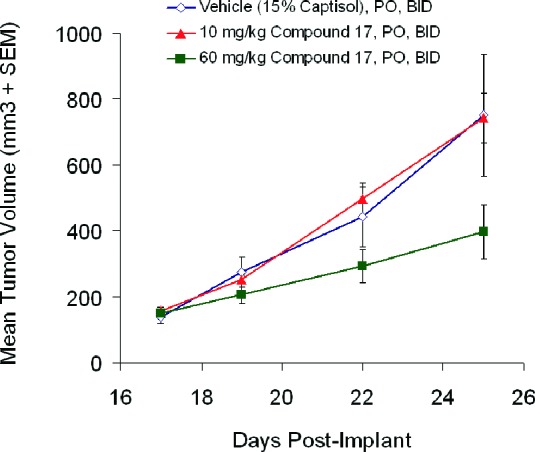
Efficacy of compound 17 in the A2780 xenograft model.
While the in vivo antiproliferative effect of compound 17 did not result in complete stasis or regression, the data support the notion that inhibition of PI3K and phosporylation of AKTSer473 in vivo with a compound from this series has an effect on tumor growth.
In summary, the structure-guided evolution of a series of in vitro potent 6-phenolic, 4-substituted, 2-morpholinopyrimidinyl PI3K inhibitors lacking suitable properties for in vivo activity into a series of 6-heterocyclic, 4-substituted, 2-morpholino pyrimidines with properties sufficient for in vivo PI3K activity, as evidenced by the modulation of phoshporylation of AKTSer473 and tumor growth inhibition in A2780 tumor-bearing mice, has been described. Compounds from this series that inhibit PI3K in vitro, have a more pronounced effect on the phosphorylation of AKTSer473 in vivo and show enhanced efficacy in PI3K-driven tumor models will be reported in due course.
Supporting Information Available
Experimental details for the synthesis and characterization of all compounds, biological assay, and pharmacology model procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Engelman J. A.; Luo J.; Cantley L. C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [DOI] [PubMed] [Google Scholar]
- Katso R.; Okkenhaug K.; Ahmandi K; White S.; Timms J.; et al. Cellular function of phosphoinositide 3-kinases: Implications for development, homeostasis and cancer. Annu. Rev. Cell Dev. Biol. 2000, 17, 615–675. [DOI] [PubMed] [Google Scholar]
- Samuels Y.; Wang Z.; Bardelli A.; Silliman N.; Ptak J.; Szabo S.; Yan H.; Gasdar A.; Powell S. M.; Riggins G. J.; Willson J. K.; Markowitz S.; Kinzler K. W.; Vogelstein B.; Velculescu V. E. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [DOI] [PubMed] [Google Scholar]
- Leslie N. R.; Downes C. P. PTEN function: how normal cells control it and tumour cells lose it. Biochem. J. 2004, 382, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P.; Cheng H.; Roberts T. M.; Zhao J. J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discovery 2009, 8, 627–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuss J. M.; Tsuhako A. L.; Anand N. K. In Annual Reports in Medicinal Chemistry; Macor J. E., Ed.; Academic Press: Oxford, 2009; Vol. 44, pp 339−351. [Google Scholar]
- Maira S.-M.; Stauffer F.; Brueggen J.; Furet P.; Schnell C.; Fritsch C.; Brachmann S.; Chene P.; De Pover A.; Schoemaker K.; Fabbro D.; Gabriel D.; Simonen M.; Murphy L.; Finan P.; Sellers W.; Garcia-Echeverria C. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 2008, 7, 1851–1863. [DOI] [PubMed] [Google Scholar]
- Folkes A. J.; Ahmadi K.; Alderton W. K.; Alix S.; Baker S. J.; Box G.; Chuckowree I. S.; Clarke P. A.; Depledge P.; Eccles S. A.; Friedman L. S.; Hayes A.; Hancox T. C.; Kugendradas A.; Lensun L.; Moore P.; Olivero A. G.; Pang J.; Patel S.; Pergl-Wilson G. H.; Raynaud F. I.; Robson A.; Saghir N.; Salphati L.; Sohal S.; Ultsch M. H.; Valenti M.; Wallweber H. J. A.; Wan N. C.; Wiesmann C.; Workman P.; Zhyvoloup A.; Zvelebil M. J.; Shuttleworth S. J. The Identification of 2-(1H-Indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a Potent, Selective, Orally Bioavailable Inhibitor of Class I PI3 Kinase for the Treatment of Cancer. J. Med. Chem. 2008, 51, 5522–5532. [DOI] [PubMed] [Google Scholar]
- Knight S. D.; Adams N. D.; Burgess J. L.; Chaudhari A. M.; Darcy M. G.; Donatelli C. A.; Luengo J. E.; Newlander K. A.; Parrish C. A.; Ridgers L. H.; Sarpong M. A.; Schmidt S. J.; Van Aller G. S.; Carson J. D.; Diamond M. A.; Elkins P. A.; Gardiner C. M.; Garver E.; Gilbert S. A.; Gontarek R. R.; Jackson J. R.; Kershner K. L.; Luo L.; Raha K.; Sherk C. S.; Sung C. M.; Sutton D.; Tummino P. J.; Wegrzyn R. J.; Auger K. R.; Dhanak D. Discovery of GSK2126458, a Highly Potent Inhibitor of PI3K and the Mammalian Target of Rapamycin. ACS Med. Chem. Lett. 2010, 1, 39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecchi S.; Renhowe P. A.; Taylor C.; Kaufman S.; Merrit H.; Wiesmann M.; Shoemaker K.; Knapp M. S.; Hendrickson T. F.; Fantl W.; Voliva C. F.. Identification and structure-activity relationship of 2-morpholino 6-(3-hydroxyphenyl)pyrimidines, a class of potent and selective PI3-kinase inhibitors. Bioorg. Med. Chem. Lett. 2010, accepted for publication. [DOI] [PubMed] [Google Scholar]
- The structure of 1 in PI3Kγ has been deposited in the RCSB Protein Data Bank under the accession code 3P2B.
- Burger M.; Ni Z.-J.; Pecchi S. Atallah G.; Bartulis S.; Frazier K.; Smith A.; Verhagen J.; Zhang Y.; Wagman A.; Ng S.; Pfister K.; Poon D.; Louie A.; Pick Y.; Barsanti P.; Iwanowicz E.; Fantl W.; Hendrickson T.; Knapp M.; Merritt H.; Voliva C.; Wiesmann M.; Xin X.. Pyrimidine derivatives used as PI3K inhibitors. WO2007/084786 A1.
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency; a useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- Compound 17 is a pan class 1 PI3K inhibitor (PI3K α, β, γ, and δ IC50 values = 0.001, 0.092, 0.009, and 0.020 μM; PI4Kβ, mTOR, and VPS34 IC50 values = 5, 4, and >9 μM; pAKTThr308 A2780 EC50 = 0.6 μM.
- Mouse PK parameters at 5 mpk iv and 10 mpk po; oral t1/2 = 282 min, % F = 87, and CL = 99 mL/min/kg. Vss = 1.1 L/kg, and oral AUC = 2 μM h.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.