

Abstract
Following from the evaluation of different types of electrophiles, combined modeling and crystallographic analyses are used to generate potent boronic acid based inhibitors of a penicillin binding protein. The results suggest that a structurally informed approach to penicillin binding protein inhibition will be useful for the development of both improved reversibly binding inhibitors, including boronic acids, and acylating inhibitors, such as β-lactams.
Keywords: Penicillin binding proteins, boronic acids, antibiotics, antibiotic-resistance, β-lactams, transpeptidase-inhibition
Penicillin-binding proteins (PBPs) catalyze steps in the biosynthesis of peptidoglycan, which is a major component of the bacterial cell wall (for reviews, see refs (1) and (2)), and their inhibition causes irregularities in cell wall structure, lysis, and eventual cell death.3 PBPs are inhibited by the β-lactam antibiotics (including penicillins, cephalosporins, monobactams, and carbapenems, Figure 1a). From the 1940s onward, very substantial synthetic and screening efforts were made to optimize the side chains of β-lactam antibiotics with a view to improving their potency, spectrum of activity, and pharmacokinetics (for review, see ref (4)). The majority of this work was carried out in the absence of detailed structural knowledge on PBPs, which has begun to emerge over the past decade or so.1,2,5
Figure 1.

Irreversible and reversible inhibition of PBPs. (a) Penicillin and cephalosporin antibiotics; (b) reaction of β-lactam antibiotics with a PBP or β-lactamase; (c) the intermediate analogue approach; (d) structures of intermediate analogs in the initial screen.
The continuing and increasing problem of resistance to β-lactam antibiotics due to β-lactamases has motivated work toward the identification of non-hydrolyzable PBP inhibitors.6 One approach involves the use of appropriately functionalized electrophiles (sometimes referred to as transition state analogues) that react reversibly with the nucleophilic active site serine of the PBPs. Peptide-based inhibitors of PBPs from Streptomyces (R61),7Escherichia coli (PBP 5),8Neisseria gonorrheae (PBP 3/4),9 and Actinomadura (R39)10 have been reported. Following these pioneering efforts, it has recently been reported that aryl boronic acids inhibit R39.11 Here, we report the identification and structural analysis of potent nonacylating, reversible transition state inhibitiors of R39. Following from work with different types of electrophiles, the activity of acetamido-boronic acids was optimized by computational and crystallographic analyses. The results demonstrate how a structurally informed approach can be applied to PBP inhibition. An analogous approach should be applicable to other types of PBP inhibitors, including β-lactams.
Catalysis by both PBPs and the serine subfamilies of β-lactmases proceeds via an acyl−enzyme complex; “tetrahedral” intermediates are involved in the formation and breakdown of this complex (Figure 1b).7−10 Several classes of electrophiles are proposed to act as structural analogues of these tetrahedral intermediates, including trifluomethylketones, phosphonates, and boronic acids (Figure 1c). Using a crystal structure of the acyl−enzyme complex formed by reaction of nitrocefin with R39 (PDB ID 1W8Y)12 for guidance (Figure 2a), three types of electrophile were identified and then synthesized as potential inhibitors (compounds 1−17, Figure 1d); for synthetic details, see the Supporting Information. The chosen side chains reflect those in β-lactam antibiotics, that is, the thiopheneacetyl group of cefalotin and the 2,6-dimethoxyphenyl group of methicillin (of interest as the bulk of this side chain confers some β-lactamase resistance). Screening of these potential inhibitors against R39 revealed little or no activity for the trifluoromethyl ketones and the phosphonates (Supporting Information Table S1) but identified alkyl boronic acids 10 and 11 as potential lead structures, with IC50 values of 33 and 75 μM, respectively (Supporting Information Table S1).
Figure 2.

Active site views from structures of R39 (green) bound to nitrocefin (magenta, PDB ID 1W8Y(12)) and 10 (blue, PDB ID 2XLN). (a) R39−nitrocefin acyl−enzyme complex. The side-chain amide of nitrocefin is positioned to form hydrogen bonds with the side chain of Asn300. The C-3 side chain of nitrocefin is not shown. (b) Formation of a tetrahedral adduct between the boron of 10 and Oγ atom of Ser49. The bond angle values around boron are 113.5° (C1−B−OH1), 112.0° (C1−B−OH2), 103.9° (OH1−B−OH2), 103.8° (OH1−B−Oγ), 116.1°(OH1−B−Oγ), and 107.4° (C1−B−Oγ). (c) Superimposition of the nitrocefin (magenta) and 10 (blue) complexes showing the active site surface.
We then determined a crystal structure of 10 in complex with R39 to investigate its binding mode (PDB ID 2XLN, Figure 2b and Supporting Information Figure S1a). The structure reveals reaction of the electrophilic boron atom with the Oγ atom of active site Ser49 to form a tetrahedral adduct. This complex is apparently stabilized by hydrogen-bonding interactions between one of the boronate hydroxyl group and the backbone nitrogen atoms of Ser49 and Thr413 (which forms an “oxyanion hole”), in a manner analogous to that observed between the nitrocefin acyl−enzyme carbonyl oxygen and the oxyanion hole (Figure 2a). The C-α group of 10 is positioned next to that of Leu349, possibly explaining why analogues 12−17 with substituents bulkier than a methyl group are inactive. The side chain amides of nitrocefin and 10 are positioned to make similar hydrogen-bonding interactions to the Asn300 side chain and Thr413 backbone carbonyl. Although the overall structures for the complexes with nitrocefin and 10 are very similar, in the case of 10 the side chain of Tyr147 is rotated to enable an apparent π-stacking interaction with the phenyl ring of 10.
The structural data suggest that 10 reacts with R39 to form a reasonable mimic of an intermediate complex; hence, it was selected as a starting point for further optimization, using the inhibitor design software SPROUT.13,14 In this strategy, the protein is first examined for potential ligand interaction sites. Fragments are then “added” to the lead structure. A scoring system based on predicted binding affinity, structural complexity, and synthetic accessibility guides the “identification” of potentially improved inhibitors (Supporting Information Figure S2).
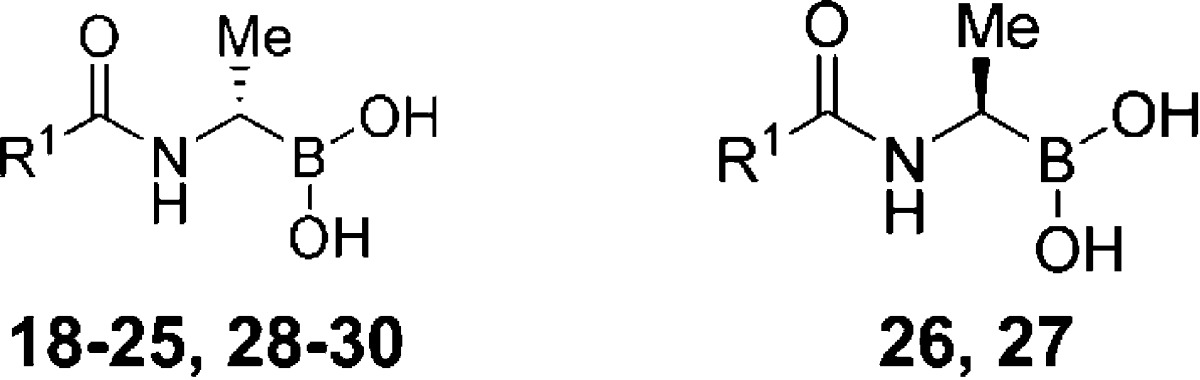
Initial computational analysis of 10 and nitrocefin R39 complexes reveals two regions of relatively high hydrophobicity which we considered could be utilized for structural modification. One region (A) is adjacent to the 4- and 5-positions of the phenyl ring of 10, and the other (B) is a relatively large area of space formed by Trp139, Asp142, Tyr147, Arg351, and Met414, which is occupied by the nitrocefin C-7 side chain (Figure 2). A series of second-generation boronic acid inhibitors 18−27 (Table 1) was identified by computationally modifying 10 using a virtual fragment library of mainly hydrophobic elements. Boronic acids 18−27 were then prepared to explore structure−activity relationships in regions A and B, respectively.
Table 1. Activity of Computationally Designed Second- (18−27) and Third-Generation (28−30) Boronic Acids against R39a.
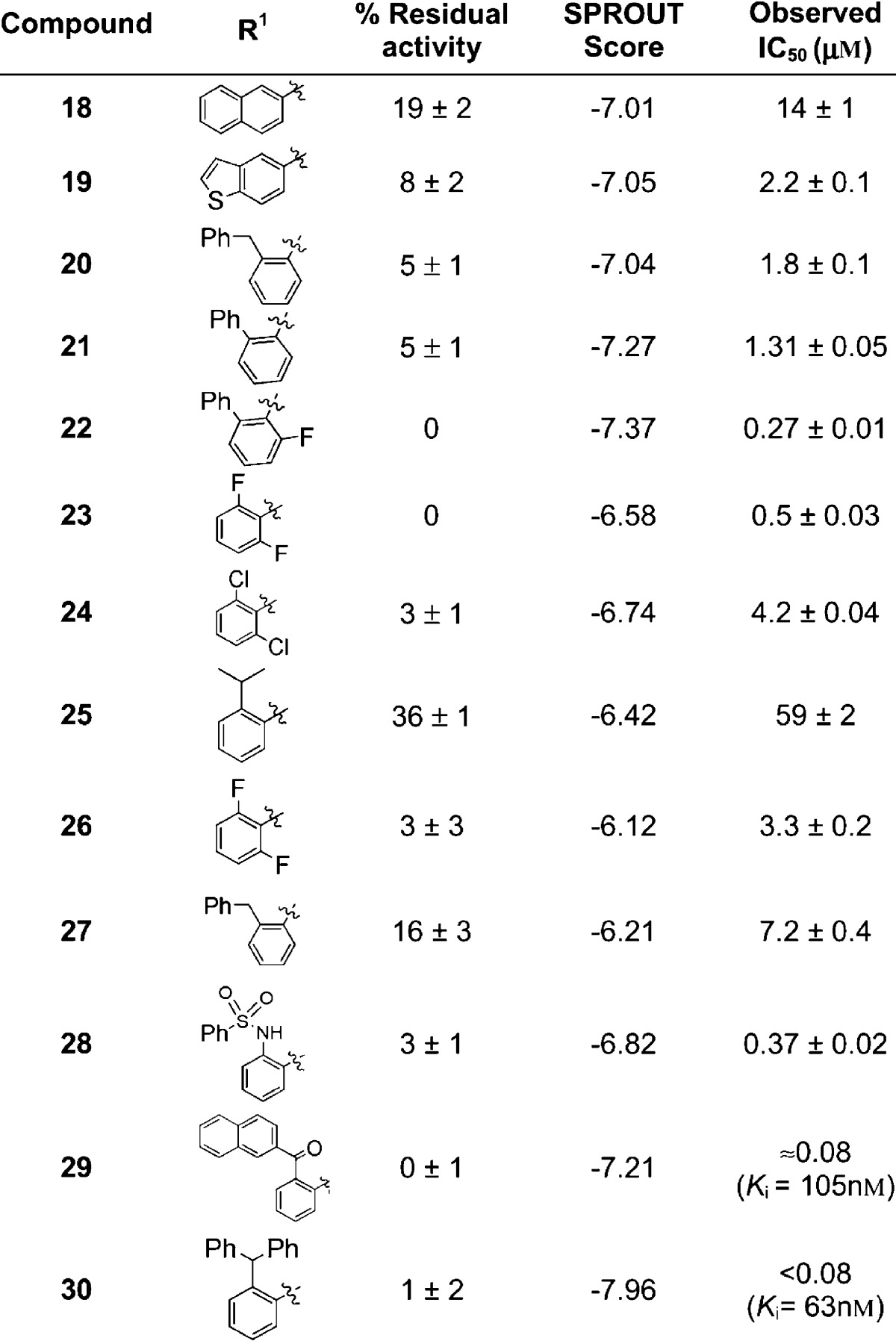 |
Percentage residual activities were determined in triplicate at 100 μM.
The synthesis of the boronic acids 18−27 was adapted from the strategy of Matteson et al.,15 starting from boronate esters formed from (+)- or (-)-pinanediol (Scheme 1). Key steps are the stereoselective chain homologation using dichloromethyllithium and nucleophilic displacement of chloride with bistrimethylsilylamide. Deprotection of the N-α-acetyl boronate ester was achieved via the trifluoroborate intermediate, formed by reaction of the boronate esters with KHF2, followed by acid hydrolysis.16,17
Scheme 1. Synthesis of Boronic Acid Inhibitors.

Conditions: (a) (−)-pinanediol, THF, RT, 95%; (b) lithium diisopropylamide, CH2Cl2, ZnCl2, THF, −78 °C to rt, 50−80%; (c) LiN(TMS)2, THF, −78 °C to rt, 60−80%; (d) HCl/Et2O, 95%; (e) R2COCl, 4-dimethylaminopyridine, Et3N, THF, DMF, 0 °C to rt, 50−90%; (f) R2CO2H, (2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate), 1-hydroxybenzotriazole, N-methylmorpholine, THF, DMF, 0 °C to rt, 50−90%; (g) KHF2, MeOH, 70−90%; (h) chlorotrimethylsilane, MeCN, H2O, 60−90%.
The computationally designed compounds 18−24, 26, and 27 (IC50 values 0.27−14 μM) are more active against R39 than 10 (IC50 = 33 μM) (Table 1). The benzothiazole ring in 19 (IC50 = 2.2 μM) is predicted to fit snugly within the smaller region A. Consistent with this proposal, an analogue 18, with a larger naphthyl ring, was less active (IC50 = 14 μM). The presence of bulky groups at the ortho-position generally improves potency as demonstrated by the results for 20−22 (SPROUT score = −7.04, −7.27, −7.37, respectively; observed IC50 = 1.80, 1.31, and 0.27 μM, respectively). An exception is the ortho-substituted isopropyl derivative 25. The most potent compounds in this series are compounds 22 and 23, with IC50 values of 0.27 and 0.50 μM, respectively.
A crystal structure was then obtained with 20 complexed to R39 (PDB ID 2XK1, Figure 3, Supporting Information Figure S1b). The mode of binding of the boronic acid with Ser49 is very similar to that of 10 (Figure 2b), with all apparent hydrogen bonds being conserved. The structure implies that the increased potencies of the S-enantiomers (10, 20, and 23) compared to the R-enantiomers (11, 27, and 26) are due to better fit of the C-α group within a small pocket adjacent to region A. Interestingly, and as predicted from the modeling studies, the presence of the bulkier side chain of 20 (compared to 10) causes it to adopt a different conformation, wherein the benzyl group occupies a space defined by residues Ser49, Glu178, Phe297, and Ser298 (region C). In order for the benzyl group of 20 to “navigate” the sharp bend into region C, the phenyl ring occupies region B and the methylene bridge of 20 “twists” to avoid steric interaction with the active site surface.
Figure 3.
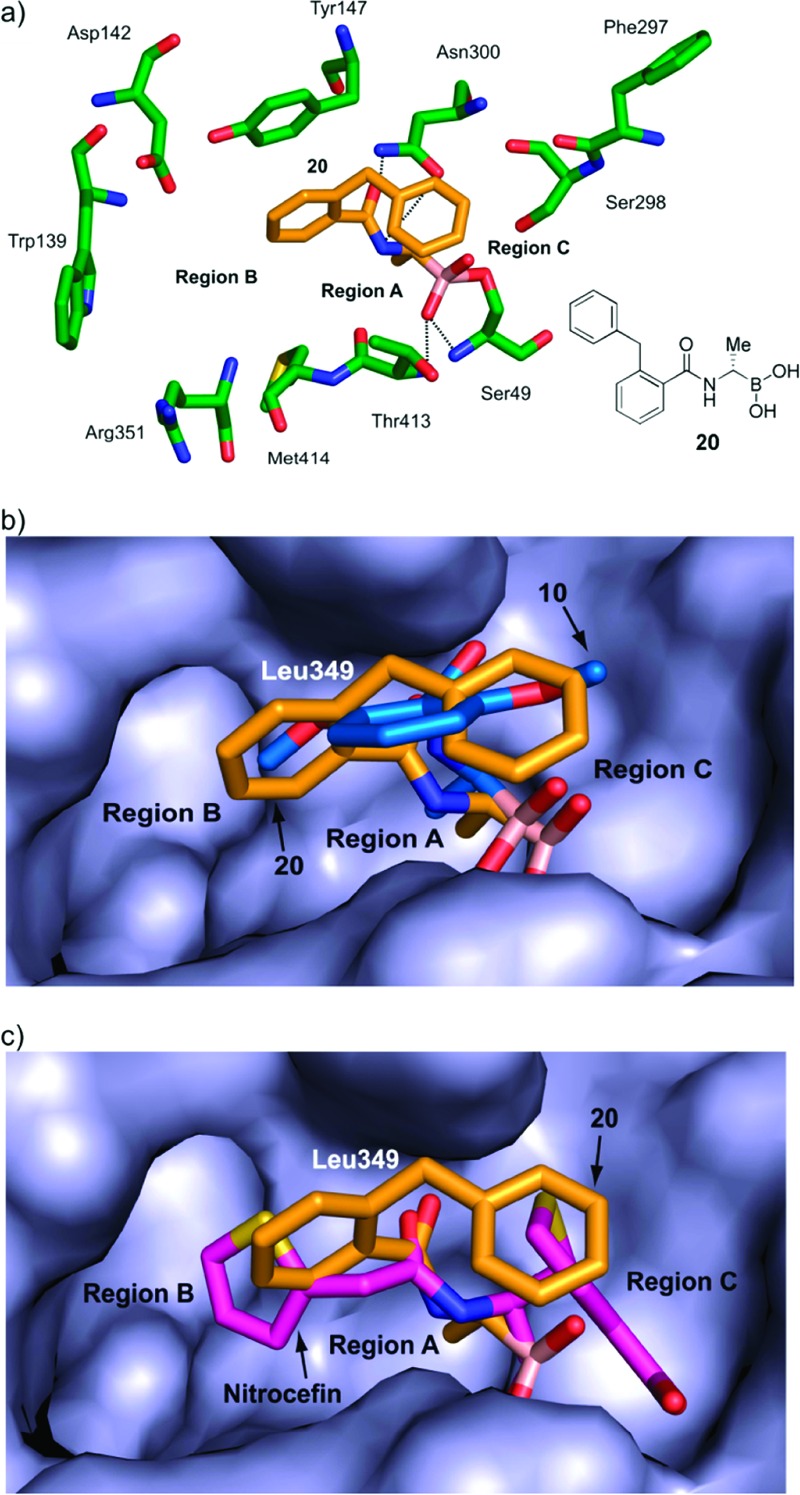
Views from a crystal structure of 20 (gold) bound to R39 (green) (PDB ID 2XK1). (a) The apparent hydrogen bonds for 10 (Figure 2) are conserved. The benzyl group of 20 occupies region C (in the nitrocefin structure, this is occupied by the dihydrothiazolidine ring). (b) Superimposition of the structures of 10 (blue) and 20 (gold). The phenyl group of 20 and the methoxy group of 10 occupy region B. (c) Superimposition of the nitrocefin (magenta) and 20 (gold) structures.
Compound 20 was considered an attractive lead with respect to directing substituents into both regions B and C. Modeling resulted in the proposal of a third set of inhibitors 28−30 (Table 1). These compounds were designed to have restricted flexibility because of their ortho-substituents and were predicted to have better binding affinities compared to 20 (Supporting Information Figure S3). Indeed, following their synthesis, assays showed significant improvements in their potencies (IC50 = 0.37 μM, Ki = 105 and 63 nM, respectively) (Table 1), hence validating the structure-based modeling approach for side chain optimization.
Overall, we have demonstrated how a combination of crystallographic analyses coupled to modeling can significantly enhance the potency of the lead PBP inhibitor to generate some of the most potent, non-β-lactam PBP inhibitors yet reported. Our sytematic, structurally informed approach also reveals how a previously unexplored region in the PBP active site can be used to achieve potent inhibition by reversibly binding low molecular weight boronic acids. This approach may be useful for the development of both improved β-lactam and reversibly binding, non-β-lactam inhibitors. Detailed structural information on PBP and β-lactamases is now available, and a challenge will be to use this to achieve the breadth of selectivity and potency required for useful antibiotics.
Acknowledgments
We thank the staff of ESRF BM30a for assistance in data collection, R. Herman for expert work in protein crystallization, and A. Dessen for discussion.
Supporting Information Available
Chemical synthesis and compound characterizations, inhibition assay methods, protein purification, crystallization and structure solution methods, and SPROUT modeling methods. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
Coordinates for R39 in complexed with 10 and 20 have been deposited with the RCSB Protein Data Bank under the codes 2XLN and 2XK1, respectively.
This work was supported by the European Union (European Community Sixth Framework Programme) via EUR-INTAFAR, by the Fonds de la Recherche Scientifique (IISN 4.4505.00, IISN4.4509.09), and the University of Liège (Fonds spéciaux, Crédit classique, 2009).
Supplementary Material
References
- Macheboeuf P.; Contreras-Martel C.; Job V.; Dideberg O.; Dessen A. Penicillin binding proteins: key players in bacterial cell cycle and drug resistance processes. FEMS Microbiol. Rev. 2006, 30, 673–691. [DOI] [PubMed] [Google Scholar]
- Sauvage E.; Kerff F.; Terrak M.; Ayala J. A.; Charlier P. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008, 32, 234–258. [DOI] [PubMed] [Google Scholar]
- Höltje J. V. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol. Mol. Biol. Rev. 1998, 62, 181–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin J. E.; Schofield C. J.. The biosynthesis of β-lactams. In The Chemistry of β-Lactams; Page M. I., Ed.; Blackie Academic and Professional: London, 1992; pp 1−78. [Google Scholar]
- Massova I.; Mobashery S. Kinship and diversification of bacterial penicillin-binding proteins and ss-lactamases. Antimicrob. Agents Chemother. 1998, 42, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher J. F.; Meroueh S. O.; Mobashery S. Bacterial resistance to beta-lactam antibiotics: compelling opportunism, compelling opportunity. Chem. Rev. 2005, 105, 395–424. [DOI] [PubMed] [Google Scholar]
- Silvaggi N. R.; Anderson J. W.; Brinsmade S. R.; Pratt R. F.; Kelly J. A. The crystal structure of phosphonate-inhibited d-Ala-d-Ala peptidase reveals an analogue of a tetrahedral transition state. Biochemistry 2003, 42, 1199–1208. [DOI] [PubMed] [Google Scholar]
- Nicola G.; Peddi S.; Stefanova M.; Nicholas R. A.; Gutheil W. G.; Davies C. Crystal structure of Escherichia coli penicillin-binding protein 5 bound to a tripeptide boronic acid inhibitor: a role for Ser-110 in deacylation. Biochemistry 2005, 44, 8207–8217. [DOI] [PubMed] [Google Scholar]
- Pechenov A.; Stefanova M. E.; Nicholas R. A.; Peddi S.; Gutheil W. G. Potential transition state analogue inhibitors for the penicillin-binding proteins. Biochemistry 2003, 42, 579–588. [DOI] [PubMed] [Google Scholar]
- Dzhekieva L.; Rocaboy M.; Kerff F.; Charlier P.; Sauvage E.; Pratt R. F. Crystal structure of a complex between the Actinomadura R39 DD-peptidase and a peptidoglycan-mimetic boronate inhibitor: interpretation of a transition state analogue in terms of catalytic mechanism. Biochemistry 2010, 49, 6411–6419. [DOI] [PubMed] [Google Scholar]
- Inglis S. R.; Zervosen A.; Woon E. C. Y.; Gerards T.; Teller N.; Fischer D. S.; Luxen A.; Schofield C. J. Synthesis and evaluation of 3-(dihydroxyboryl)benzoic acids as D,D-carboxypeptidase R39 inhibitors. J. Med. Chem. 2009, 52, 6097–6106. [DOI] [PubMed] [Google Scholar]
- Sauvage E.; Herman R.; Petrella S.; Duez C.; Bouillenne F.; Frere J.-M.; Charlier P. Crystal structure of the Actinomadura R39 DD-peptidase reveals new domains in penicillin-binding proteins. J. Biol. Chem. 2005, 280, 31249–31256. [DOI] [PubMed] [Google Scholar]
- Gillet V.; Johnson A. P.; Mata P.; Sike S.; Williams P. SPROUT-a programme for structure generation. J. Comput.-Aided Mol. Des. 1993, 7, 127–153. [DOI] [PubMed] [Google Scholar]
- Law J. M. S.; Fung D. Y. K.; Zsoldos Z.; Simon A.; Szabo Z.; Csizmadia I. G.; Johnson A. P. Validation of the SPROUT de novo design program. THEOCHEM 2003, 666, 651–657. [Google Scholar]
- Matteson D. S.; Ray R.; Rocks R. R.; Tsai D. J. S. Directed chiral synthesis by way of a-chloro boronic esters. Organometallics 1983, 2, 1536–1543. [Google Scholar]
- Inglis S. R.; Woon E. C. Y.; Thompson A. L.; Schofield C. J. Observations on the deprotection of pinanediol and pinacol boronate esters via fluorinated intermediates. J. Org. Chem. 2010, 75, 468–471. [DOI] [PubMed] [Google Scholar]
- Yuen A. K. L.; Hutton C. A. Deprotection of pinacolyl boronate esters via hydrolysis of intermediate potassium trifluoroborates. Tetrahedron Lett. 2005, 46, 7899–7903. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.