

Abstract
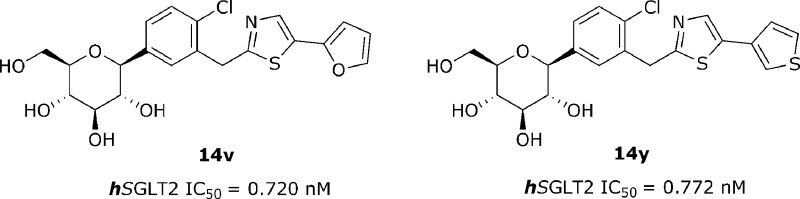
Novel C-aryl glucoside SGLT2 inhibitors containing the thiazole motif were designed and synthesized for biological evaluation. Among the compounds assayed, thiazole containing furanyl moiety 14v and thiophenyl moiety 14y demonstrated the best in vitro inhibitory activity against SGLT2 in this series to date (IC50 = 0.720 nM for 14v and IC50 = 0.772 nM for 14y). Both of these compounds have been further evaluated on a urinary glucose excretion test and the urine volumes excreted.
Keywords: Thiazole, SAR, SGLT2, glucoside, diabetes
Diabetes has become an increasing concern worldwide. In 2010, approximately 285 million people around the world will have diabetes, corresponding to 6.4% of the world’s adult population, with a prediction that by 2030 the number of people with diabetes will have grown to 438 million.1 Type 2 diabetes is the most common disorder of glucose homeostasis, accounting for nearly 90−95% of all cases of diabetes.2
Sodium-dependent glucose cotransporters (SGLTs) couple the transport of glucose against a concentration gradient with the simultaneous transport of Na+ down a concentration gradient.3 It is estimated that 90% of renal glucose reabsorption is facilitated by SGLT2.4 SGLT2 inhibitors have attracted considerable attention because there is need for novel antidiabetic agents with a unique mode of action to compensate for existing therapies and avert potential side effects.5
Bristol-Myers Squibb and Johnson & Johnson have identified, respectively, dapagliflozin 1 and canagliflozin 2, potent, selective SGLT2 inhibitors for the treatment of type 2 diabetes.6−9 At present, dapagliflozin is the most advanced SGLT2 inhibitor in clinical trials (Phase 3), and canagliflozin is the second most advanced clinical candidate. In addition, Boehringer Ingelheim, Lexicon, Astellas, and Pfizer are reported to be in various phases of clinical trials.10−13
We have explored metabolically more stable C-glucosides bearing a heteroaromatic ring in order to develop novel SGLT2 targeting antidiabetic agents since C-glucosides are more stable than O-glucoside derivatives. We envisioned that replacement of the distal ring or proximal ring of dapagliflozin 1 with a heterocyclic ring might improve the physicochemical properties while maintaining a similar level of antidiabetic activity, thereby providing a high possibility for clinical development.14−17 For instance, C log P values of this series of compounds would routinely show 2.7−3.0, while that of dapagliflozin (1) is shown to be 3.4. Along this line, the structure of dapagliflozin (1) was modified into 5, bearing a thiazole ring at the distal ring position as shown in Figure 1. Herein, we report the synthesis and structure−activity relationship (SAR) evaluation of thiazolylmethylphenyl glucoside congeners.
Figure 1.
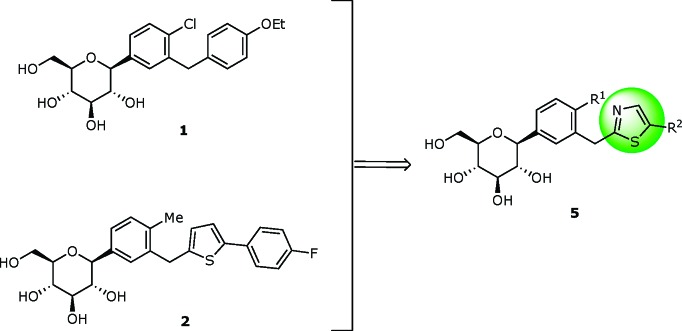
Exploration of C-glucoside bearing thiazole at the distal ring.
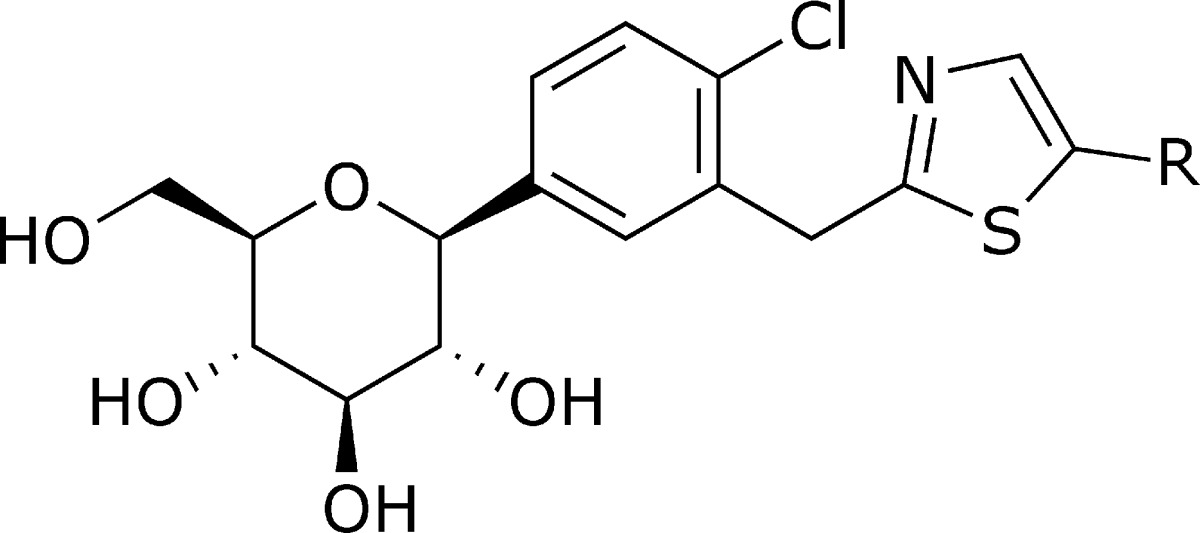
Preparation of the thiazole compound is described in Scheme 1. Thus, previously reported carboxylic acid 6(14,15) was coupled with 2-amino-1-(furan-2-yl)ethanone hydrochloride in the presence of EDCI, HOBt, and NMM to provide the corresponding amide 7 in 82% yield. Amide 7 smoothly underwent thionation and subsequent cyclization by the action of Lawesson reagent in refluxing THF to lead to thiazole 8. Finally, total removal of benzyl protection was affected with TMSI at mild heating for 15 h to produce the target compound 14v in 73% yield.
Scheme 1.

The cell-based SGLT2 AMG (methyl-α-d-glucopyranoside) inhibition assay was performed to evaluate the inhibitory effects of all prepared compounds on hSGLT2 activities.18,19 Exploration of the SAR began by replacing the phenyl moiety at the distal ring position of dapagliflozin 1 with the 2-thiazolyl moiety. Table 1 shows the structure−activity relationship upon alteration of the substituent at the distal thiazole ring employing only the β-anomer. Initially, ethyl on the C-5 thiazole ring showed decent activity (14a, IC50 = 16.7 nM). However, as the number of carbons increases, the inhibitory activity against hSGLT2 fluctuates. For example, a 7-fold drop in activity is observed by varying the length of the alkyl chain from ethyl (14a, IC50 = 16.7 nM) to n-butyl (14b, IC50 = 119 nM), whereas a 9-fold increase in activity is observed from n-butyl (14b, IC50 = 119 nM) to n-pentyl (14c, IC50 = 13.3 nM). Additional 3-fold increase in activity is observed from n-pentyl (14c) to n-hexyl (14d, IC50 = 5.0 nM). A branched alkyl group at the position appears to decrease activity (14f, IC50 = 51.4 nM), whereas the conjugated alkenyl group exemplified in 14g restores inhibitory activity against hSGLT2 (IC50 = 4.69 nM). Carbocycle at the position also appears to be tolerant (14i, IC50 = 9.71 nM). Alkoxy groups on the thiazole were also explored. These alkoxides (14k−14n) appear to be tolerated across the board (IC50 = 8.2 nM−19.6 nM). Meanwhile, isosteric replacement of alkoxy groups with alkylthio groups for this region appears to diminish inhibitory activity in approximately 3-fold, primarily implying that the bulky sulfur atom is less favored at this location. Replacement of this moiety with phenyl group 14s improved the inhibitory activity against hSGLT2 (IC50 = 13.3 nM). Interestingly, the somewhat big biphenyl group 14u showed good inhibitory activity (IC50 = 9.31 nM). Meanwhile, substitution of phenyl with 2-furyl (14v) or 3-thiophenyl (14y) dramatically improved the inhibitory activity against hSGLT2 (14v, IC50 = 0.720 nM vs 14y, IC50 = 0.772 nM). Substituted thiophene 14z decreases activity 10-fold. However, thiazole 14aa and thiadiazole 14ab demonstrated lower in vitro activity (14aa, IC50 = 3.13 nM; 14ab, IC50 = 14.0 nM) than furan 14v or thiophene 14y, likely suggesting that increased polarity is not favorable in the region. This phenomenon is unequivocally observed in carboxylic acid 14ad, which is completely devoid of any activity against hSGLT2 (IC50 > 10,000 nM).
Table 1. In Vitro Screening Data for hSGLT2 Inhibitory Activity.
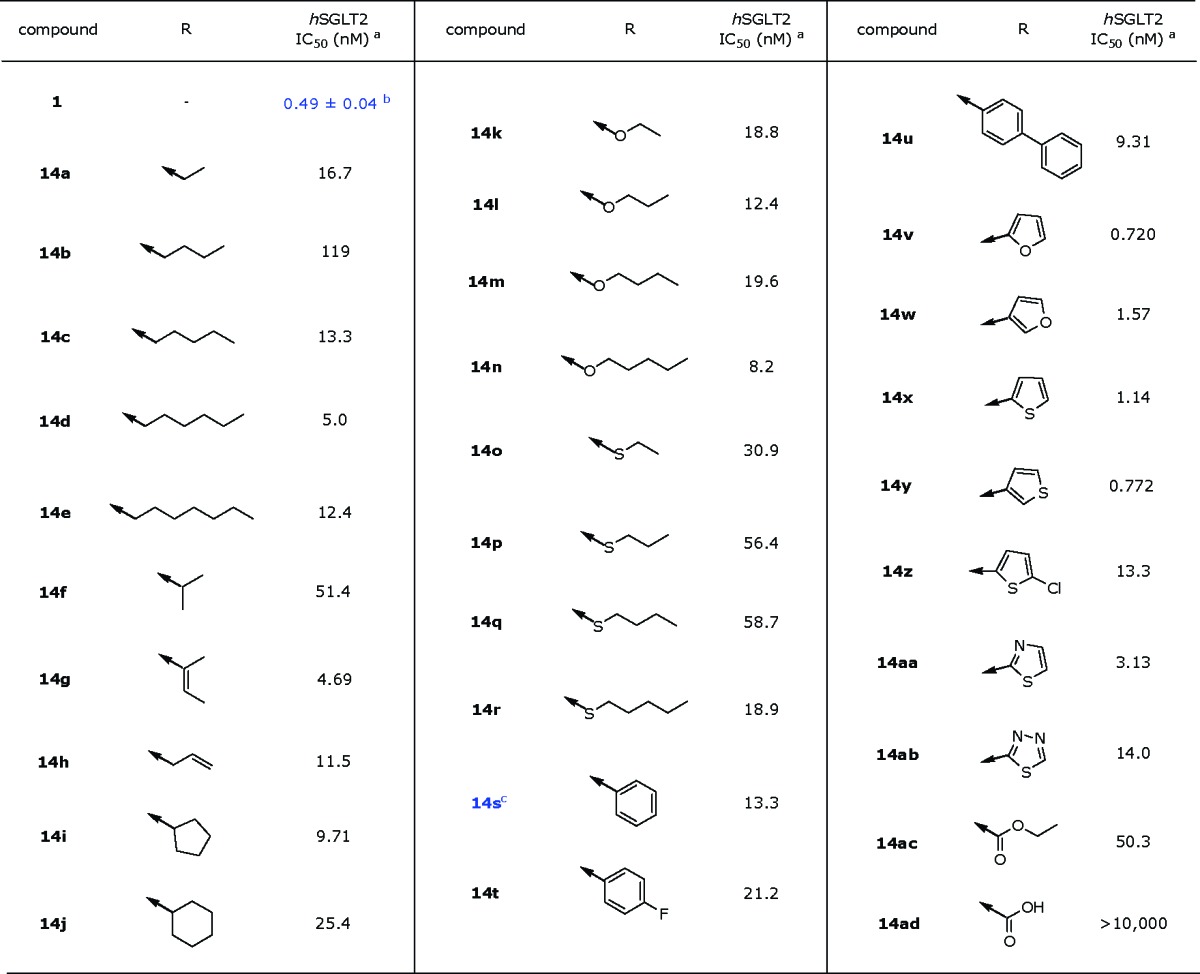 |
These data were obtained by single determinations.
The IC50 value was obtained by in-house multiple determinations.
Compound 14s had been disclosed in WO2005012326.22
It was encouraging to note that considerable inhibitory activity against hSGLT2 was already evident in compounds 14v and 14y. At this juncture, we proceeded to explore the C-4 region of the proximal phenyl ring by using 5-(furan-2-yl)thiazolyl and 5-(thiophen-3-yl)thiazolyl as lead scaffolds. The inhibitory activity data is shown in Table 2. The small methyl group (15a and 15b) or other halogen atoms (15c−15f) appear to maintain a similar level of activity, albeit in slightly decreased potency. On the contrary, hydrogen 15i, cyano 15j, 15k, or cyclopropyl 15l demonstrated a loss of activity in an order of magnitude, suggesting that merely small lipophilic groups are favored at the C-4 position of the proximal phenyl ring.
Table 2. In Vitro Inhibitory Activity against hSGLT2.
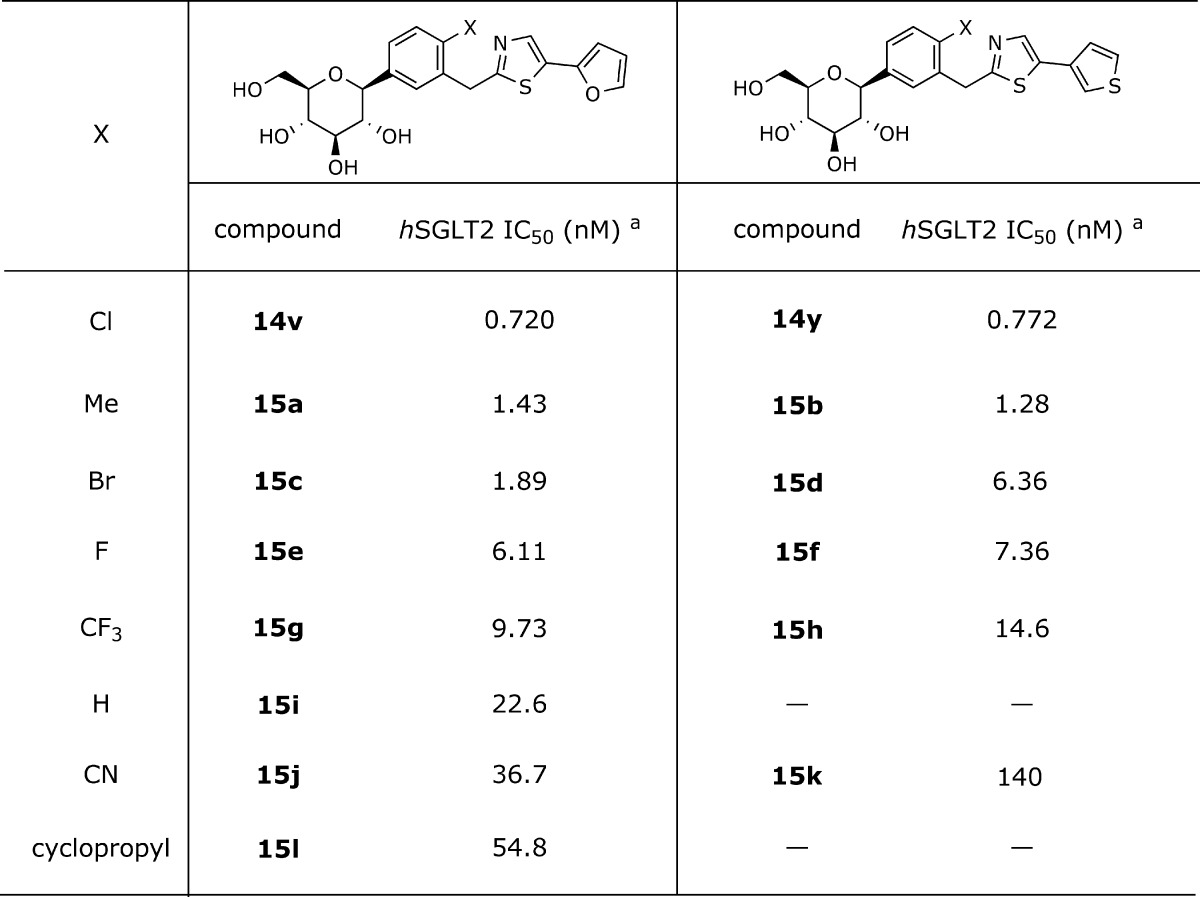 |
These data were obtained by single determinations.
Next, an isomeric thiazole series of compounds was explored by using 2-furanyl and 3-thiophenyl as lead scaffolds (16a−16d, Table 3). This series exhibited a slight decrease in inhibitory activities against hSGLT2 compared with that of the previous thiazole series. For instance, 2-(furan-2-yl)thiazol-5-yl 16a (IC50 = 1.42 nM) showed a 2-fold decrease in activity than the counterpart 14v (IC50 = 0.720 nM). As a more clear demonstration, two pairs of examples are presented, including furans 14w (IC50 = 1.57 nM), 16b (IC50 = 4.47 nM), and thiophenes 14x (IC50 = 1.14 nM) and 16c (IC50 = 2.79 nM), demonstrating close to a 3-fold decrease of activity, respectively.
Table 3. In Vitro Inhibitory Activity against hSGLT2.
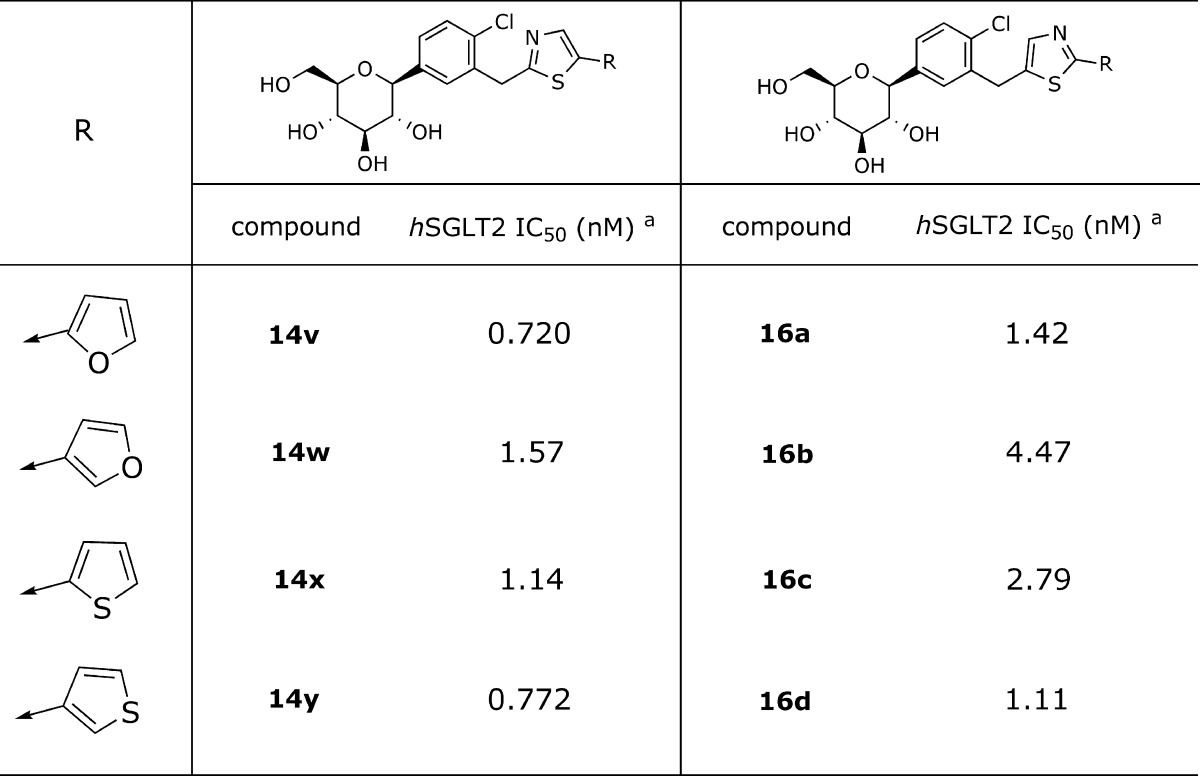 |
These data were obtained by single determinations.
Selected promising compounds (14v and 14y) were tested on animal models for in vivo efficacy. Urinary glucose excretion was evaluated in normal SD rats. As shown in Figure 2, a single oral dose of thiazole 14y (1 mg/kg or 10 mg/kg) increased urinary glucose excretion in normal SD rats, resulting in 160-fold (1 mg/kg) or 730-fold (10 mg/kg) elevation in glucose disposal relative to that of vehicle controls. However, a single oral dose of thiazole 14v (1 mg/kg or 10 mg/kg) increased urinary glucose excretion in normal SD rats, resulting in 230-fold (1 mg/kg) or 700-fold (10 mg/kg) elevation in glucose disposal relative to that of vehicle controls. Urine volumes excreted in normal SD rats are also shown in Figure 2. Dapagliflozin 1 (1 mg/kg) caused increased urine volume over that of the vehicle by 5.7-fold, while thiazole 14y (1 mg/kg or 10 mg/kg) increased urine volume by 2.5-fold (1 mg/kg) or by 4.2-fold, respectively. Thiazole 14v increased urine volume to a lesser degree at a dose of 10 mg/kg (3.7-fold increase).
Figure 2.

Effects of dapagliflozin, compound 14y and 14v on urine glucose excretion and urine volume in normal Spraue−Dawley rats. All results are expressed as means ± SEM. The statistical analysis was performed by one-way ANOVA followed by Dunnett’s posthoc test. *P < 0.05 vs vehicle. **P < 0.01 vs vehicle.
In order to further evaluate this series, the PK properties of 14y and 14v were measured in male SD rats. After oral administration of 5 mg/kg of 14y to rats, a Cmax of 1.54 μg/mL was obtained at 0.38 h. The elimination half-life of 14y following oral administration was 2.51 h. Compound 14y showed moderate oral bioavailability (F = 15%). Furanyl compound 14v also demonstrated PK profiles similar to those of compound 14y, showing slightly better oral bioavailability (F = 20.6%). For comparison, in-house oral administration of 5 mg/kg of dapagliflozin 1 to rats showed a Cmax of 2.50 μg/mL at 0.67 h. The elimination half-life of 1 following oral administration was 4.01 h. Dapagliflozin 1 showed superior oral bioavailability (F = 88%). Therefore, it is concluded that the decreased in vivo efficacy of the current SGLT2 inhibitors 14v and 14y, compared with that of dapagliflozin 1, should be attributed to the 1.5-fold lower in vitro potencies as well as their unfavorable PK properties.
In summary, the potential of metabolically more stable C-glucosides bearing a thiazole ring as antidiabetic agents was exploited. Among the compounds tested, thiazole containing furanyl moiety 14v or thiophenyl moiety 14y showed the best in vitro inhibitory activity against hSGLT2 in this series to date (IC50 = 0.720 nM for 14v; IC50 = 0.772 nM for 14y). These compounds demonstrated reasonable glucose excretion and glucosuria in normal SD rats along with moderate PK profiles. Further work concerning optimization of SGLT2 inhibitors suitable for development is ongoing on the basis of this series.
Acknowledgments
We appreciate Dr. Eun Chul Huh for his leadership and support as CTO, Green Cross Corporation (GCC).
Supporting Information Available
Experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- International Diabetes Federation. Diabetes Atlas, 4th ed.; International Diabetes Federation: Montreal, Canada, 2009. [Google Scholar]
- Dwarakanathan A. Diabetes update. J. Insur. Med. 2006, 38, 20–30. [PubMed] [Google Scholar]
- Mackenzie B.; Loo D. D.; Panayotova-Heiermann M.; Wright E. M. Biophysical characteristics of the pig kidney Na+/glucose cotransporter SGLT2 reveal a common mechanism for SGLT1 and SGLT2. J. Biol. Chem. 1996, 271, 32678–32683. [DOI] [PubMed] [Google Scholar]
- Moe O. W.; Berry C. A.; Rector F. C.. The Kidney, 5th ed.; Brenner B. M., Rector F. C., Eds.; WB Saunders Co.: Philadelphia, PA, 2000; pp 375−415. [Google Scholar]
- Washburn W. Evolution of sodium glucose co-transporter 2 inhibitors as anti-diabetic agents. Expert Opin. Ther. Patents 2009, 25, 1485–1499. [DOI] [PubMed] [Google Scholar]
- Ellsworth B. A.; Meng W.; Patel M.; Girotra R. N.; Wu G; Sher P.; Hagan D.; Obermeier M.; Humphreys W. G.; Robertson J. G.; Wang A.; Han S.; Waldron T.; Morgan N. N.; Whaley J. M.; Washburn W. N. Aglycone exploration of C-arylglucoside inhibitors of renal sodium-dependent glucose transporter SGLT2. Bioorg. Med. Chem. Lett. 2008, 18, 4770–4773. [DOI] [PubMed] [Google Scholar]
- Meng W.; Ellsworth B. A.; Nirschl A. A.; McCann P. J.; Patel M.; Girotra R. N.; Wu G.; Sher P. M.; Morrison E. P.; Biller A. A.; Zahler R.; Deshpande P. P.; Pullockaran A.; Hagan D. L.; Morgan N.; Taylor J. R.; Obermeier M. T.; Humphreys W. G.; Khanna A.; Discenza L.; Robertson J. G.; Wang A.; Han S.; Wetterau J. R.; Janovitz E. B.; Flint O. P.; Whaley J. M.; Washburn W. N. Discovery of dapagliflozin: A potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. J. Med. Chem. 2008, 51, 1145–1149. [DOI] [PubMed] [Google Scholar]
- Washburn W. N. Development of the renal glucose reabsorption inhibitors: A new mechanism for the pharmacotherapy of diabetes mellitus type 2. J. Med. Chem. 2009, 52, 1785–1794. [DOI] [PubMed] [Google Scholar]
- Nomura S.; Sakamaki S.; Hongu M.; Kawanshi E.; Koga Y.; Sakamoto T.; Yamamoto Y.; Ueta K.; Kimata H.; Nakayama K.; Tsuda-Tsukimoto M. Discovery of canagliflozin, a novel C-glucoside with thiophene ring, as sodium-dependent glucose cotransporter 2 inhibitor for the treatment of type 2 diabetes mellitus. J. Med. Chem. 2010, 53, 6355–6360. [DOI] [PubMed] [Google Scholar]
- Boehringer Ingelheim Pharmaceuticals, www.clinicaltrials.gov, 2010; NCT01164501.
- Lexicon Pharmaceuticals, www.clinicaltrials.gov, 2009; NCT00962065. 12.
- Astellas Pharma Inc., www.clinicaltrials.gov, 2010; NCT01135433.
- Mascitti V. Presented at the 240th American Chemical Society National Meeting and Exposition, Boston, MA, Aug 22−26, 2010; Medi-151. [Google Scholar]
- Lee J.; Lee S.-H.; Seo H. J.; Son E.-J.; Lee S. H.; Jung M. E.; Lee M.; Han H.-K.; Kim J.; Kang J.; Lee J. Novel C-aryl glucoside SGLT2 inhibitors as potential antidiabetic agents: 1,3,4-thiadiazolylmethylphenyl glucoside congeners. Bioorg. Med. Chem. 2010, 18, 2178–2194. [DOI] [PubMed] [Google Scholar]
- Kim M. J.; Lee J.; Kang S. Y.; Lee S.-H.; Son E.-J.; Jung M. E.; Lee S. H.; Song K.-S.; Lee M.; Han H.-K.; Kim J.; Lee J. Novel C-aryl glucoside SGLT2 inhibitors as potential antidiabetic agents: pyridazinylmethylphenyl glucoside congeners. Bioorg. Med. Chem. Lett. 2010, 20, 3420–3425. [DOI] [PubMed] [Google Scholar]
- Kang S. Y.; Song K.-S.; Lee J.; Lee S.-H.; Lee J. Synthesis of pyridazine and thiazole analogs as SGLT2 inhibitors. Bioorg. Med. Chem. 2010, 18, 6069–6079. [DOI] [PubMed] [Google Scholar]
- Lee J.; Kim J. Y.; Choi J.; Lee S.-H.; Kim J.; Lee J. Pyrimidinylmethylphenyl glucoside as novel C-aryl glucoside SGLT2 inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 7046–7049. [DOI] [PubMed] [Google Scholar]
- For cloning and cell line construction for human SGLT2, The hSGLT2 sequence was cloned into pcDNA3.1(+) for mammalian expression and were stably transfected into chinese hamster ovary (CHO) cells.
- For the sodium-dependent glucose transport assay, IC50 was determined by nonlinear regression analysis using GraphPad PRISM.20,21
- Han S.; Hagan D. L.; Taylor J. R.; Xin L.; Meng W.; Biller S. A.; Wetterau J. R.; Washburn W. N.; Whaley J. M. Dapagliflozin, a selective SGLT2 inhibitor, improves glucose homeostasis in normal and diabetic rats. Diabetes 2008, 57, 1723–1729. [DOI] [PubMed] [Google Scholar]
- Katsuno K.; Fujimori Y.; Takemura Y.; Hiratochi M.; Itoh F.; Komatsu Y.; Fujikura H.; Isaji M. Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level. J. Pharmacol. Exp. Ther. 2007, 320, 323–330. [DOI] [PubMed] [Google Scholar]
- Nomura S.; Kawanishi E.; Ueta K. Novel Compounds Having Inhibitory Activity Against Sodium-Dependent Transporter. PCT Int. Appl. WO2005012326.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.