

Abstract
Attenuation of fructose metabolism by the inhibition of ketohexokinase (KHK; fructokinase) should reduce body weight, free fatty acids, and triglycerides, thereby offering a novel approach to treat diabetes and obesity in response to modern diets. We have identified potent, selective inhibitors of human hepatic KHK within a series of pyrimidinopyrimidines (1). For example, 8, 38, and 47 exhibited KHK IC50 values of 12, 7, and 8 nM, respectively, and also showed potent cellular KHK inhibition (IC50 < 500 nM), which relates to their intrinsic potency vs KHK and their ability to penetrate cells. X-ray cocrystal structures of KHK complexes of 3, 8, and 47 revealed the important interactions within the enzyme's adenosine 5'-triphosphate (ATP)-binding pocket.
Keywords: Kinase, diabetes, obesity, crystal structure, fructose metabolism
Noninsulin-dependent diabetes mellitus (NIDDM), which is characterized by hyperglycemia and insulin resistance, is frequently accompanied by obesity and cardiovascular disease.1−3 There is a marked positive correlation between the increased energy intake in the form of highly refined sugars and the prevalence of NIDDM and obesity.4 Diets high in fructose promote various metabolic disturbances in animal models, including weight gain, hyperlipidemia, hypertension, and insulin resistance.5−9 Also, in overweight humans, the long-term consumption of fructose is found to increase energy intake, body weight, fat mass, blood pressure, and plasma triglycerides.10,11
Excessive fructose ingestion stimulates de novo lipogenesis via upregulation of gene expression,12,13 favors re-esterification of fatty acids, and increases the production of very low-density lipoprotein (VLDL) particles.14 Chronic high-fructose diets elevate free fatty acids and triglycerides, which impair glucose utilization in muscle tissue and increase the rate of lipolysis in adipose tissue.14 Elevated triglycerides can impede insulin-signaling pathways,15−17 support chronic inflammation,18−20 and lead to glucolipid toxicity21 with possible failure of pancreatic β-cells.22
Fructose is readily absorbed from the diet and rapidly metabolized in the liver by fructokinase, also known as ketohexokinase (KHK; EC 2.7.1.3).1 The hepatic enzyme KHK phosphorylates fructose on position C1 with the aid of adenosine 5′-triphosphate (ATP) to yield fructose-1-phosphate (F1P), which enters normal metabolic pathways.23−23c However, in contrast to the tight regulation of glucose pathways, the fructose metabolism lacks robust control mechanisms.14 KHK has a high KM, is not inhibited by product, and is not allosterically regulated.24 As a consequence, high concentrations of fructose can rapidly flow into glycolytic pathways to provide the components of triglycerides.24 In this context, targeting fructose metabolism by inhibition of KHK should reduce body weight, free fatty acids, and triglycerides, thereby offering a novel approach for treating NIDDM and obesity amidst the modern diets of postindustrial societies.
There is human genetic validation of KHK as a therapeutic target on the basis of mutations that cause essential fructosuria, an autosomal recessive disorder.25 Individuals with this benign condition have inactive isoforms of hepatic KHK, such that ingestion of fructose, sucrose, or sorbitol results in a persistent rise in blood fructose levels and increased excretion of fructose into the urine. Thus, a KHK inhibitor would eliminate excess carbohydrate without a mechanism-based safety issue.
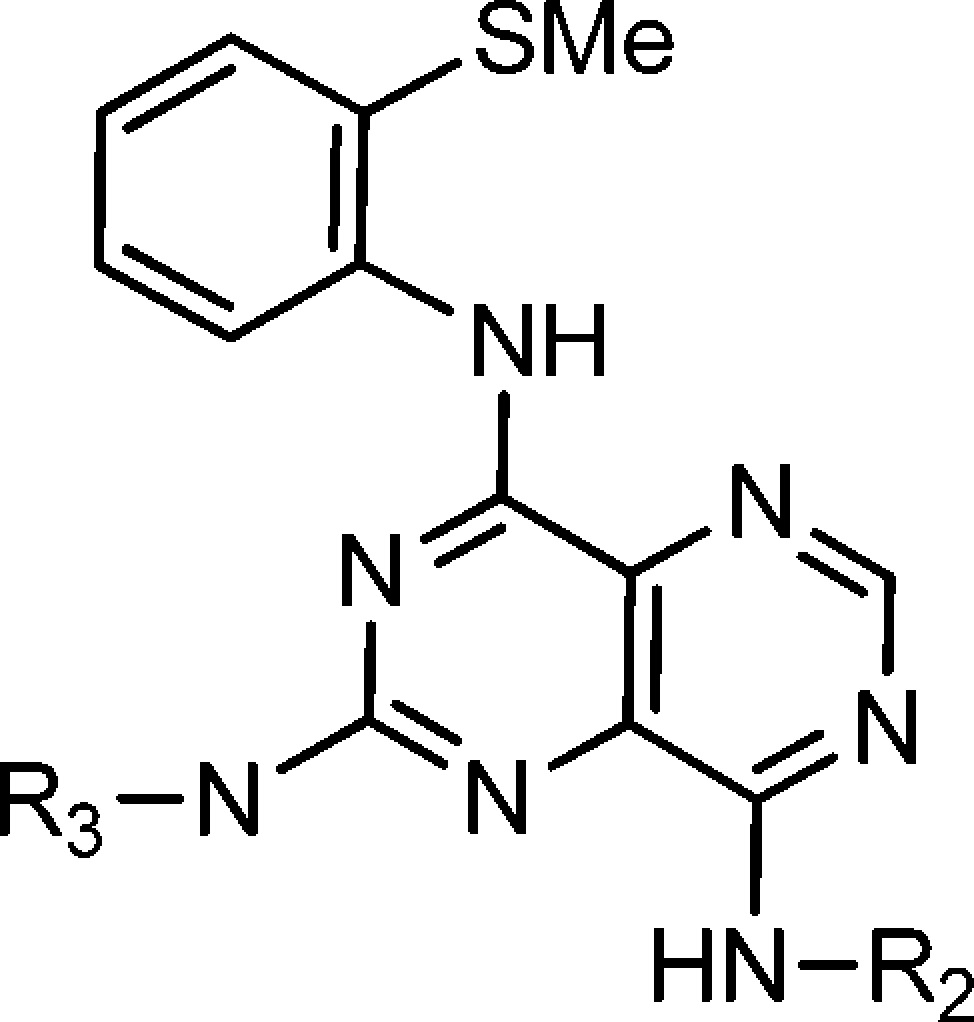
Herein, we report potent, selective inhibitors of human hepatic KHK (KHK-C isoform25−26d), which function by docking within the ATP-binding site. By using a combination of high-throughput screening (HTS) and structure-based drug design, we discovered and optimized a series of pyrimidinopyrimidines (1). We also obtained X-ray cocrystal structures of KHK, as a pseudohomodimer (a and b subunits) with each subunit occupied by an inhibitor ligand, thereby illustrating the key intermolecular interactions.
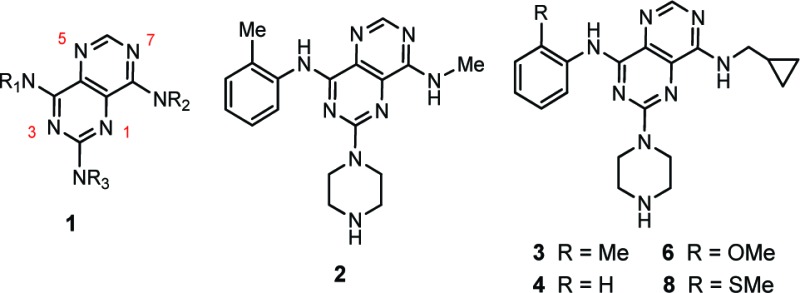
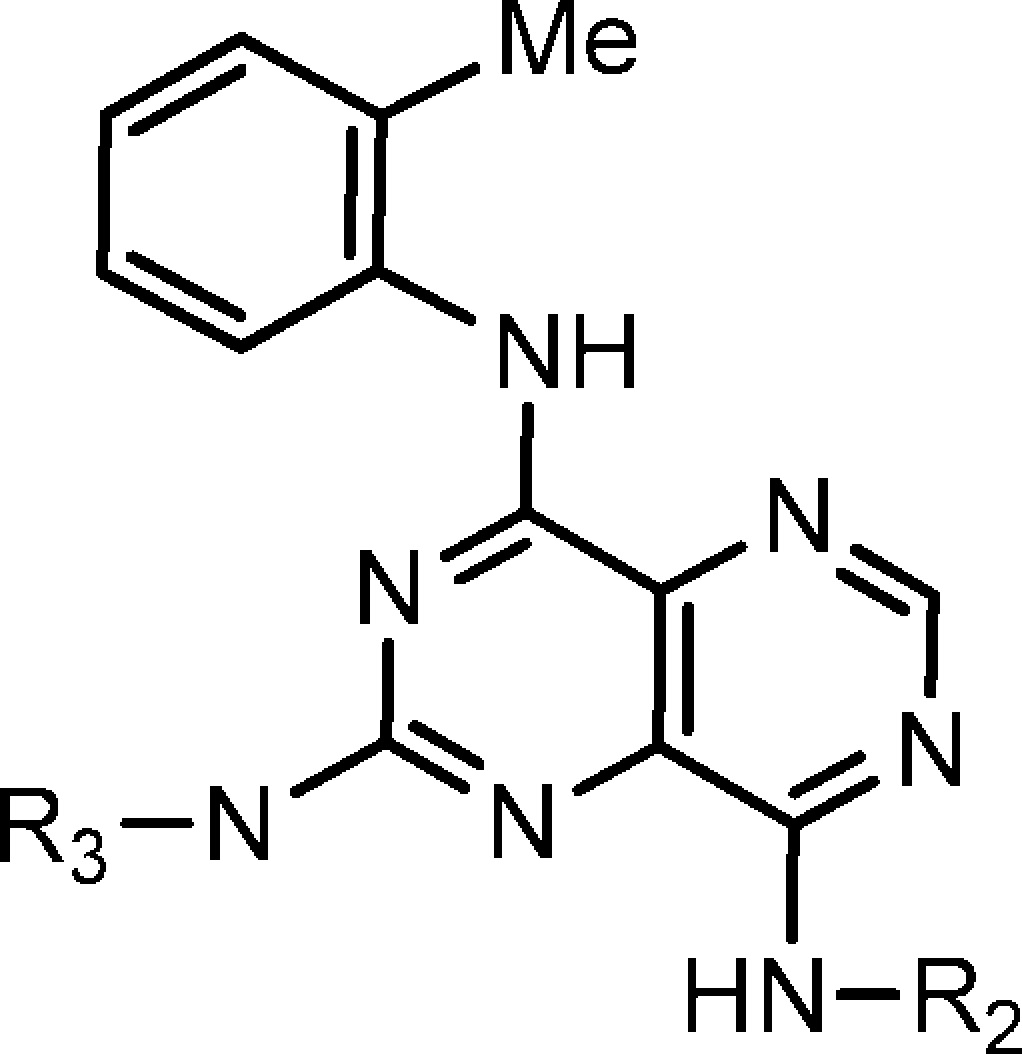
The activity of KHK can be assessed in vitro with a pyruvate oxidase-coupled enzyme assay,23a but this approach to screening for inhibitors is not readily adaptable to a HTS protocol. Although we developed a ThermoFluor assay27 to assess direct ligand binding to KHK, we elected to conduct a HTS campaign by using a fluorescence polarization (FP) assay to measure the course of the enzymatic reaction. We employed the transcreener ADP assay, which is a homogeneous, competitive method that specifically detects adenosine 5′-diphosphate (ADP), a primary KHK reaction product,28 in conjunction with KHK-C that was recombinantly expressed and purified to homogeneity.25,26c,26d,29a,29b Our HTS campaign involving ca. 800 000 compounds, with a wide range of structures, yielded several chemotypes as possible drug discovery starting points. A promising avenue for exploration was the pyrimidino[5,4-d]pyrimidine series, 1, since several representative compounds were on hand from an earlier chemical library enhancement project.30 Confirmed hit 2, with an IC50 value of 400 nM, served as a basis for further study. The initial group of confirmed hits revealed that NH-cyclopropylmethyl was one of the better groups for NR2, with analogue 3 having an IC50 value of 210 nM. The corresponding compound that lacks a 2-methyl group (4) had an IC50 of 3200 nM, and 3-methyl congener 5 had an IC50 of 2800 nM (Table 1).
Table 1. Structures and KHK Inhibition Results for Derivatives of 1 with Variation of R1.
| compda | R1 | R2 | IC50 (nM)b |
|---|---|---|---|
| 2 | 2-MeC6H4 | Me | 400 |
| 3 | 2-MeC6H4 | CH2-c-Pr | 210 |
| 4 | Ph | CH2-c-Pr | 3200 |
| 5 | 3-MeC6H4 | CH2-c-Pr | 2800 |
| 6 | 2-MeOC6H4 | CH2-c-Pr | 100 |
| 7 | 2-EtOC6H4 | CH2-c-Pr | 200 |
| 8 | 2-MeSC6H4 | CH2-c-Pr | 12 |
| 9 | 3-MeSC6H4 | CH2-c-Pr | 2000 |
| 10 | 4-MeSC6H4 | CH2-c-Pr | >9000 |
| 11 | 2-MeSO2C6H4 | CH2-c-Pr | >9000 |
| 12 | 2-EtSC6H4 | CH2-c-Pr | >9000 |
| 13 | 2-CF3SC6H4 | CH2-c-Pr | 3000 |
| 14 | 2-EtC6H4 | CH2-c-Pr | 130 |
| 15 | 2-(i-Pr)C6H4 | CH2-c-Pr | 5000 |
| 16 | 2-(c-Pr)C6H4 | CH2-c-Pr | 380 |
| 17 | 2-FC6H4 | CH2-c-Pr | 1500 |
| 18 | 2-ClC6H4 | CH2-c-Pr | 540 |
| 19 | 2-BrC6H4 | CH2-c-Pr | 170 |
| 20 | c-Pr | CH2-c-Pr | >9000 |
| 21 | c-hexyl | CH2-c-Pr | >9000 |
New compounds were purified by HPLC and characterized by ESI-MS and 1H NMR (see the Supporting Information).
Inhibition of recombinant human hepatic KHK (KHK-C) in terms of IC50 values; >9000 nM relates to <50% inhibition at 9 μM.
To prepare analogues for biological study, including gram quantities, we used a general synthetic route, which is exemplified for 3 (Scheme 1).30 Key trichloro intermediate III, from chlorination of II, was reacted with amine substrates to introduce groups into the R1, R2, and R3 sites sequentially (III → IV → V → VI). The Boc protecting group in VI was removed with CF3CO2H to give final product 3.
Scheme 1. Synthesis of 3.

Thus, we prepared a set of close analogues of 3 (Table 1; 6–21) and obtained particularly impressive potency with the 2-methylthio group (8; IC50 = 12 nM). Derivatives with R1 varied (Table 1) served to define a preliminary structure–activity relationship (SAR). It is evident from this set of compounds that an appropriate ortho substituent is important for potent KHK inhibition (cf. 3–8, 8–10, and 17–19) and that the size of this group is crucial, with steric bulk being a serious limitation (cf. 8, 12; 3, 14, 16; and 17–19). While 2-SMe (8) was an ideal substitutent, moderately sized groups, such as Me (3), Br (19), MeO (6), EtO (7), and Et (14), gave reasonable IC50 values of 100–200 nM. It also appears that groups with a strong electrostatic field are less favorable (17 and 18 vs 19).
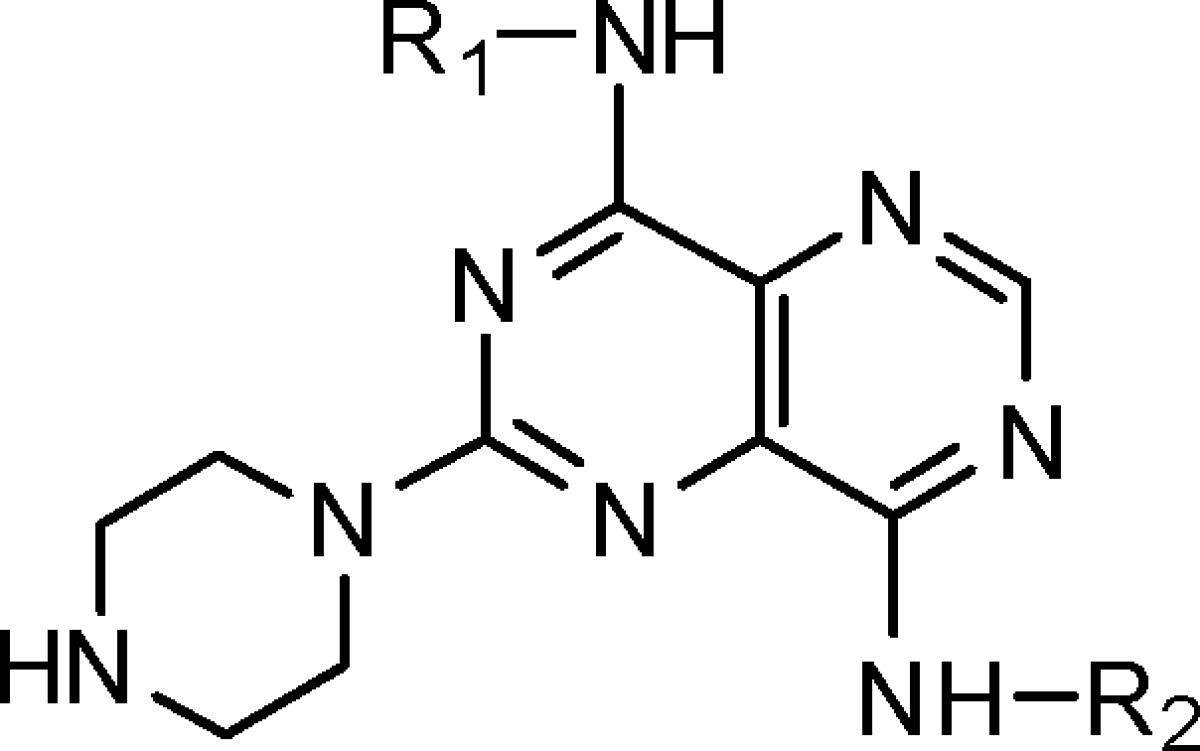
With the 2-methyl group held constant in R1, we probed the effects of altering R2 and R3 (Table 2). These results indicate that the R2 group can vary widely in size and type (2, 3, and 22–29); however, large alkyl (23 and 24) and disubstitution (25) are disfavored. For R3, groups bearing NH2+ or NH3+ are favored (cf. 3 and 30–35). Analogues of 8 were then examined to fine-tune KHK inhibitory potency for advancing to further studies (Table 3). Several compounds had IC50 values of ≤50 nM (36–47), and besides 8, compounds 36, 38, 40–43, 45, and 47 had IC50 values of ≤20 nM. These results support the view, as noted above, that R2 can vary widely in size and type and that R3 can benefit by having an ammonium group with more than one proton (NH2+ or NH3+) (cf. 8 and 50; 45 and 49). Also, R3 can tolerate conformational constraint (cf. 8 and 47).
Table 2. Structures and KHK Inhibition Results for Analogues of 3 with Variation of R2 and R3.
| compda | N-R2 | N-R3 | IC50 (nM)b |
|---|---|---|---|
| 2 | NH-Me | piperazino | 400 |
| 22 | NH-Pr | piperazino | 400 |
| 23 | NH-hexyl | piperazino | 1600 |
| 24 | NH-(c-hexyl) | piperazino | 2300 |
| 25 | NEt2 | piperazino | 1700 |
| 26 | NH–CH2C≡CH | piperazino | 300 |
| 27 | NH–CH2Ph | piperazino | 400 |
| 28 | NH–CH2(2-thienyl) | piperazino | 300 |
| 29 | NH–CH2(2-thiazolyl) | piperazino | 60 |
| 30 | NH(CH2-c-Pr) | homopiperazino | 300 |
| 31 | NH(CH2-c-Pr) | N-Me-piperazino | 1500 |
| 32 | NH(CH2-c-Pr) | morpholino | >7000 |
| 33 | NH(CH2-c-Pr) | 4-(NH2CH2)-piperidino | 70 |
| 34 | NH(CH2-c-Pr) | 4-(NH2)-piperidino | 200 |
| 35 | NH(CH2-c-Pr) | 4-piperidinyl-NH | 710 |
See Table 1.
Inhibition of recombinant human KHK-C; >7000 nM relates to <50% inhibition at 7 μM.
Table 3. Structures and KHK Inhibition Results for Analogues of 7 with Variation of R2 and R3.
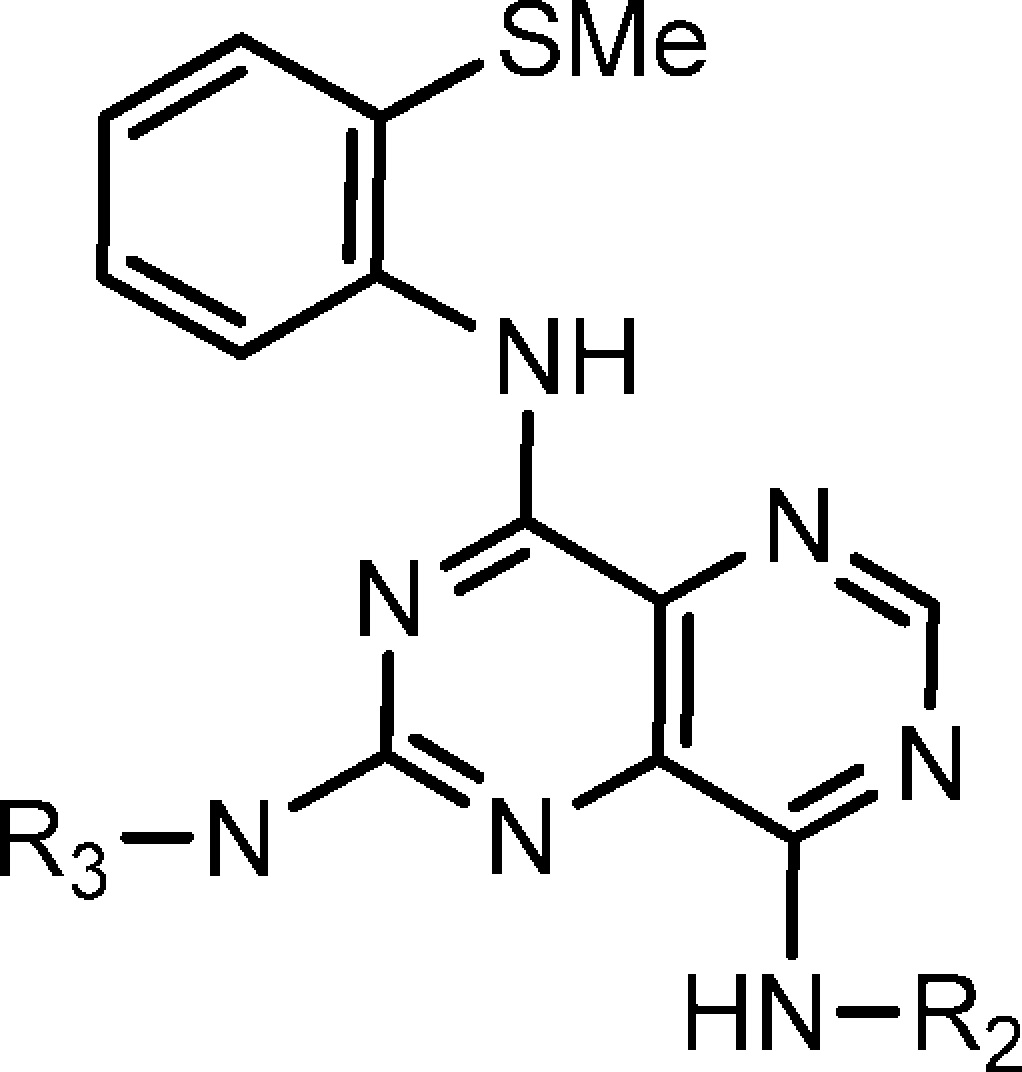
| compda | R2 | N-R3 | IC50 (nM)b |
|---|---|---|---|
| 36 | CH2-c-Bu | piperazino | 18 |
| 37 | CH2CH2-c-Pr | piperazino | 50 |
| 38 | CH2CH2OMe | piperazino | 7.0 |
| 39 | CH2(2-thienyl) | piperazino | 30 |
| 40 | CH2(2-thiazolyl) | piperazino | 16 |
| 41 | CH2(2-pyridyl) | piperazino | 9.8 |
| 42 | H | piperazino | 7.1 |
| 43 | CH2-c-Pr | (R)-3-(NH2)-piperidino | 18 |
| 44 | CH2-c-Pr | (S)-3-(NH2)-piperidino | 23 |
| 45 | CH2-c-Pr | 4-(NH2CH2)-piperidino | 10 |
| 46 | CH2-c-Pr | 3-(NH2CH2)-azetidino | 30 |
| 47 | CH2-c-Pr | 2,6-diazaspiro[3.3]hept-2-yl | 8.0 |
| 48 | CH2-c-Pr | MeNHCH2CH2NMe- | 130 |
| 49 | CH2-c-Pr | 4-(Me2NCH2)-piperidino | 140 |
| 50 | CH2-c-Pr | N-Me-piperazino | 110 |
See Table 1.
Inhibition of recombinant human KHK-C.
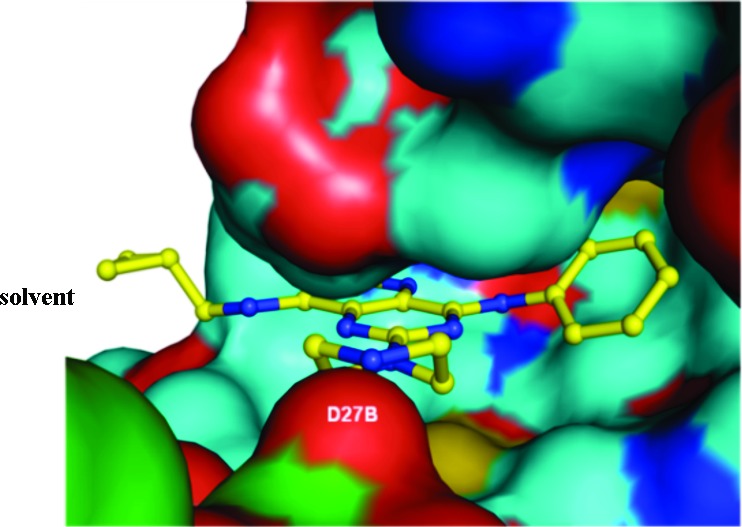
We investigated KHK–ligand complexes via X-ray crystallography to gain an understanding of the key intermolecular interactions.31−31c The X-ray cocrystal structures for 3·KHK (2.8 Å resolution) and 8·KHK (2.8 Å) showed that the ligand (3 or 8) was present in each ATP-binding site of the KHK pseudohomodimer (subunits a and b).32 Considering the a subunit, there is a notable protein–ligand interaction mediated by a conserved water molecule, which hydrogen bonds to ring nitrogen N3 (viz. 1) of the ligand and Phe-245 Nα of KHK. The o-tolyl group occupies a hydrophobic region that is largely defined by Phe-260, and the piperazine NH2+ interacts with a carboxylate group from Asp-27 in KHK-b (“Asp-27B”) (Figures 1 and 2). For both structures, the distances for the two key H-bonds, N3/water oxygen and piperazine nitrogen/Asp-27 Oδ, were 3.1 and 2.8 Å, respectively (Figure 1). We suggest that the enhanced potency for 8 may relate to the MeS group being poised to interact in an optimal manner with a hydrophobic cleft or patch on the enzyme's surface, proximal to Phe-260. We also obtained a cocrystal structure for 47·KHK (2.7 Å). The a and b subunits with 47 in the ATP-binding site are displayed in Figure 3. In subunit a, the terminal nitrogen of the spiro-bis-azetidine is hydrogen bonded with Asp-27B (3.1 Å) and Asn-107 (2.9 Å) (Figure S1 in the Supporting Information),33 whereas that interaction is absent in subunit b because of its “open” conformation (Figure 3).26c
Figure 1.
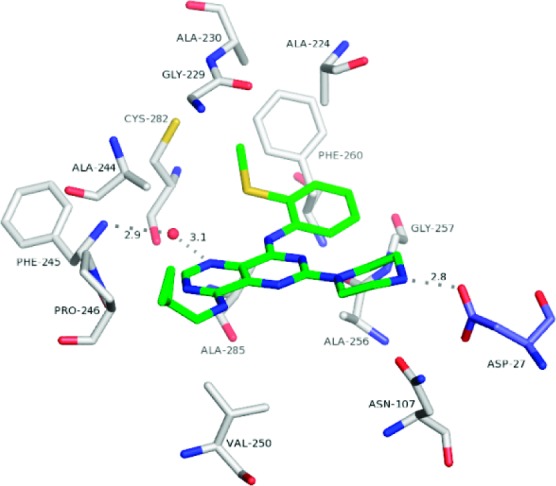
Crystal structure of the 8·KHK complex. View of 8 (stick model: C, green; N, blue; and S, yellow) and neighboring KHK residues (labeled) in subunit a (stick models: C, white; N, blue; O, red; and S, yellow) and Asp-27 in subunit b (“Asp-27B”) (stick model: C, light blue; N, blue; and O, red). The conserved water molecule is shown as a red sphere. The H-bonds between N3 and the water oxygen (3.1 Å), Phe-245 Nα and the water oxygen (2.9 Å), and the piperazine nitrogen and Asp-27B Oδ (2.8 Å) are denoted by dashed lines.
Figure 2.
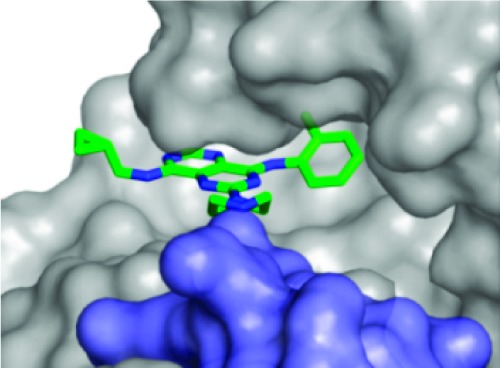
Crystal structure of the 3·KHK complex. View of 3 (stick model: C, green; and N, blue) within the KHK ATP-binding pocket of subunit a (Connelly surface model: gray for subunit a; and light blue for subunit b). The cyclopropyl group extends beyond the pocket toward the solvent. The Asp-27 carboxylate of the b subunit (“Asp-27B”) interacts with the piperazine NH2+.
Figure 3.
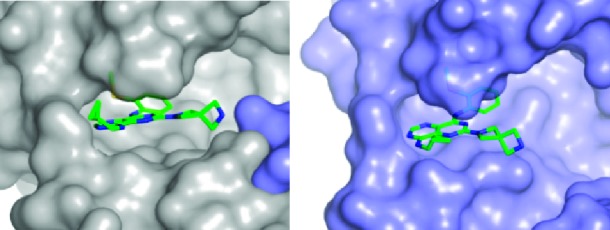
Crystal structure of the 47·KHK complex. View of 47 (stick model: C, green; N, blue; and S, yellow) in the ATP-binding pocket of subunit a (left panel; Connelly surface model, gray; Asp-27B, light blue) and subunit b (right panel; Connelly surface model, light blue).
Certain compounds with potent KHK inhibition were studied in a cellular assay that measured the level of KHK product F1P in cell lysates by using LC-MS to quantify F1P (Table 4).34 Compounds 8, 38, 40–42, and 47 exhibited reasonably potent cellular KHK inhibition (IC50 < 500 nM), which relates to their intrinsic potency vs KHK and their ability to enter cells. Compound 42 is noteworthy given its IC50 value below 100 nM. The much weaker cell inhibition result for 3 vs 8 is consistent with the relative KHK potencies (IC50 values of 210 and 12 nM, respectively).
Table 4. Cellular Functional Results.
| compd | R2 | N-R3 | IC50 (nM)a |
|---|---|---|---|
| 8 | CH2-c-Pr | piperazino | 400 |
| 38 | CH2CH2OMe | piperazino | 140 |
| 40 | CH2(2-thiazolyl) | piperazino | 270 |
| 41 | CH2(2-pyridyl) | piperazino | 270 |
| 42 | H | piperazino | 78 |
| 46 | CH2-c-Pr | 3-(NH2CH2)-azetidino | 590 |
| 47 | CH2-c-Pr | 2,6-diazaspiro[3.3]hept-2-yl | 360 |
| 3b | CH2-c-Pr | piperazino | 2400 |
Inhibition of F1P production in HepG2 cell lysates. Number of experiments (n) and standard deviations are given in the Supporting Information.
The SMe in 8 is replaced by Me.
Lead compound 8 was examined in a diverse panel of 31 protein kinases, representing different families, for off-target kinase inhibition at a concentration of 10 μM (Invitrogen; with 100 μM ATP).35 None of the 31 kinases was inhibited >40% at this high concentration of 8 (IC50 ≫ 10 μM for 31 kinases). For three off-target metabolic kinases, ribokinase, hexokinase, and adenosine kinase, the selectivity of 8 was found to be at least 50-fold (relative to KHK inhibition).36,37
Compound 8 has useful solubility (e.g., 1.5 mg/mL in 10% aqueous Solutol), an acceptable clog D5.0 of 2.9, and favorable CaCO-2 cell permeability data (×10–6 cm/s: A → B, 0.3; B → A, 1.0). It was stable in human and rat liver microsome preparations (88 and 72% remaining at 10 min) and did not significantly inhibit cytochrome P450s from human liver microsomes (1A2, 2C19, 2D6, 2C9, and 3A4). Compound 8 exhibited reasonable oral bioavailability in rats (F = 34%; oral t1/2 = 4 h), but it had a high volume of distribution (Vdss = 32 L/kg) and a high rate of clearance (CL = 160 mL/min/kg).38 Thus, at an oral dose of 10 mg/kg, its plasma Cmax was just 0.16 μM.39
In conclusion, we have identified potent, selective inhibitors of human ketohexokinase (KHK-C isoform) by virtue of a novel pyrimidinopyrimidine compound series (viz. 1). An in vitro assay based on the direct detection of ADP was very useful for developing the SAR. By appropriate substitution of the heterocyclic nucleus, we obtained low-nanomolar inhibitors that operate by docking within the ATP-binding pocket of KHK. Compound 8 (IC50 = 12 nM) was effective in a cellular functional assay (IC50 = 400 nM) and was orally bioavailable in rats (F = 34%). Our X-ray cocrystal structures of 3·KHK, 8·KHK, and 47·KHK display the important intermolecular interactions between the ligand pharmacophore and the enzyme. The availability of potent, selective KHK inhibitors should facilitate future studies concerning the role of fructose on metabolic function in normal animals and in disease models.
Acknowledgments
We are grateful to the spectroscopy group for analytical characterization data for new compounds; to Keli Dzordzorme, Shariff Bayoumy, and Robyn Williams for cloning and expressing KHK; to Alexander Barnakov and Ludmila Barnakova for purifying KHK; to Cynthia Milligan for KHK crystallization; to Edward Lawson and Gregory Leo for assistance in preparing the Supporting Information; and to Prof. Erick Carriera (ETH, Zürich, CH) for a generous gift of N-Boc-2,6-diazaspiro[3.3]heptane, used in the synthesis of 47. We thank the staff at IMCA-CAT for their support during the collection of X-ray diffraction data.
Supporting Information Available
Details for the synthetic procedures; characterization of final products; production and purification of KHK; biological assay procedures and results; CEREP profile, kinase profile, X-ray crystallography, and Figure S1. This material is available free of charge via the Internet at http://pubs.acs.org.
Use of the IMCA-CAT beamline BM-17 at the Advanced Photon Source was supported by companies of the Industrial Macromolecular Crystallography Association by a contract with Hauptman-Woodward Medical Research Institute. The Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences (Contract No. W-31-109-Eng-38).
Author Present Address
† Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, United States. Pennsylvania Drug Discovery Institute, 3805 Old Easton Road, Doylestown, Pennsylvania 18902, United States.
Supplementary Material
References
- Basciano H.; Federico L.; Adeli K. Fructose, insulin resistance, and metabolic dyslipidemia. Nutr. Metab. 2005, 2, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmet P.; Alberi K. G.; Shaw J. Global and societal implications of the diabetes epidemic. Nature 2001, 414, 782–787. [DOI] [PubMed] [Google Scholar]
- Songer T. J. The economic cost of NIDDM. Diabetes Metab. Rev. 1992, 8, 389–404. [DOI] [PubMed] [Google Scholar]
- Gross L. S.; Li L.; Ford E. S.; Liu S. Increased consumption of refined carbohydrates and the epidemic of type 2 diabetes in the United States: An ecological assessment. Am. J. Clin. Nutr. 2004, 79, 774–779. [DOI] [PubMed] [Google Scholar]
- Kasim-Karakas S. E.; Vriend H.; Almario R.; Chow L. C.; Goodman M. N. Effects of dietary carbohydrates on glucose and lipid metabolism in golden Syrian hamsters. J. Lab. Clin. Med. 1996, 128, 208–213. [DOI] [PubMed] [Google Scholar]
- Hwang I. S.; Ho H.; Hoffman B. B.; Reaven G. M. Fructose-induced insulin resistance and hypertension in rats. Hypertension 1987, 10, 512–516. [DOI] [PubMed] [Google Scholar]
- Reiser S.; Hallfrisch J. Insulin sensitivity and adipose tissue weight of rats fed starch or sucrose diets ad libitum or in meals. J. Nutr. 1977, 107, 147–155. [DOI] [PubMed] [Google Scholar]
- Zavaroni I.; Sander S.; Scott S.; Reaven G. M. Effect of fructose-feeding on insulin secretion and insulin action in the rat. Metabolism 1980, 29, 970–973. [DOI] [PubMed] [Google Scholar]
- Martinez F. J.; Rizza R. A.; Romero J. C. High-fructose feeding elicits insulin resistance, hyperinsulinemia, and hypertension in normal mongrel dogs. Hypertension 1994, 23, 456–463. [DOI] [PubMed] [Google Scholar]
- Raben A.; Vasilaras T. H.; Moller A. C.; Astrup A. Sucrose compared with artificial sweeteners, different effects on ad libitum food intake and body weight after 10 wk of supplementation in overweight subjects. Am. J. Clin. Nutr. 2002, 76, 721–729. [DOI] [PubMed] [Google Scholar]
- Teff K. L.; Elliott S. S.; Tschop M.; Kieffer T. J.; Rader D.; Heiman M.; Townsens R. R.; Keim N. L.; D'Alessio; Havel P. J. Dietary fructose reduces circulating insulin and leptin, attenuates postprandial suppression of ghrelin, and increases triglycerides in women. J. Clin. Endocrinol. Metab. 2004, 89, 2963–2972. [DOI] [PubMed] [Google Scholar]
- Kok N.; Roberfroid M.; Delzenne M. Dietary oligofructose modifies the impact of fructose on hepatic triacylglycerol metabolism. Metabolism 1996, 45, 1547–1550. [DOI] [PubMed] [Google Scholar]
- Miyazaki M.; Dobrzyn A.; Man W. C.; Chu K.; Sampath H.; Kim H. J.; Ntambi J. M. Stearoyl-CoA desaturase I gene expression is necessary for fructose-mediated induction of lipogenic gene expression by sterol regulatory element-binding protein-1c-dependent and -independent mechanisms. J. Biol. Chem. 2004, 279, 25164–25171. [DOI] [PubMed] [Google Scholar]
- Mayes P. A. Intermediary metabolism of fructose. Am. J. Clin. Nutr. 1993, 58, 754S–765S. [DOI] [PubMed] [Google Scholar]
- Dresner A.; Laurent D.; Marcucci M.; Griffin M. E.; Dufour S.; Cline G. W.; Slezak L. A.; Andersen D. K.; Hundal R. S.; Rothman D. L.; Petersen K. F.; Shulman G. I. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J. Clin. Invest. 1999, 103, 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno M.; Bezerra R. M.; Silva M. S.; Tavares D. Q.; Carvalho C. R.; Saad M. J. A high-fructose diet induces changes in pp185 phosphorylation in muscle and liver of rats. Braz. J. Med. Biol. Res. 2000, 33, 1421–1427. [DOI] [PubMed] [Google Scholar]
- Koteish A.; Diehl A. M. Animal models of steatosis. Semin. Liver Dis. 2001, 21, 89–104. [DOI] [PubMed] [Google Scholar]
- Wu T.; Giovannucci E.; Pishon T.; Hankinson S. E.; Ma J.; Rifai N.; Rimm E. B. Fructose, glycemic load, and quantity and quality of carbohydrate in relation to plasma C-peptide concentrations in US women. Am. J. Clin. Nutr. 2004, 80, 1043–1049. [DOI] [PubMed] [Google Scholar]
- Oron-Herman M.; Rosenthal T.; Sela B. A. Hyperhomocysteinemia as a component of syndrome X. Metabolism 2003, 52, 1491–1495. [DOI] [PubMed] [Google Scholar]
- Holven K. B.; Aukrust P.; Retterstol K.; Hagve T. A.; Morkrid L.; Ose L.; Nenseter M. S. Increased levels of C-reactive protein and interleukin-6 in hyperhomocysteinemic subjects. Scand. J. Clin. Lab. Invest. 2006, 66, 45–54. [DOI] [PubMed] [Google Scholar]
- Swanson J. E.; Laine D. C.; Thomas W.; Bantle J. P. Metabolic effects of dietary fructose in healthy subjects. Am. J. Clin. Nutr. 1992, 58, 851–856. [DOI] [PubMed] [Google Scholar]
- Lewis G. F.; Carpentier A.; Adeli K.; Giacca A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr. Rev. 2002, 23, 201–229. [DOI] [PubMed] [Google Scholar]
- Raushel F. M.; Cleland W. W. Bovine liver fructokinase: Purification and kinetic properties. Biochemistry 1977, 16, 2169–2175. [DOI] [PubMed] [Google Scholar]
- Raushel F. M.; Cleland W. W. Determination of the rate-limiting steps and chemical mechanism of fructokinase by isotope exchange, isotope partitioning, and pH studies. Biochemistry 1977, 16, 2176–2181. [DOI] [PubMed] [Google Scholar]
- Raushel F. M.; Cleland W. W. Substrate and anomeric specificity of fructokinase. J. Biol. Chem. 1973, 248, 8174–8177. [PubMed] [Google Scholar]
- Elliott S. S.; Keim N. L.; Stern J. S.; Teff K.; Havel P. J. Fructose, weight gain, and the insulin resistance syndrome. Am. J. Clin. Nutr. 2002, 76, 911–922. [DOI] [PubMed] [Google Scholar]
- Asipu A.; Hayward B. E.; O'Reilly J.; Bonthron D. T. Properties of normal and mutant recombinant human ketohexokinases and implications for the pathogenensis of essential fructosuria. Diabetes 2003, 52, 2426–2432. [DOI] [PubMed] [Google Scholar]
- Bonthron D. T.; Brady N.; Donaldson I. A.; Steinmann B. Molecular basis of essential fructosuria: Molecular cloning and mutational analysis of human ketohexokinase (fructokinase). Hum. Mol. Genet. 1994, 3, 1627–1631. [DOI] [PubMed] [Google Scholar]
- Trinh C. H.; Asipu A.; Bonthron D. T.; Phillips S. E. V. Structures of alternatively spliced isoforms of human ketohexokinase. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2009, D65, 201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs A. C.; Abad M. C.; Zhang X.; Tounge B. A.; Lewandowski F. A.; Struble G. T.; Sun W.; Sui Z.; Kuo L. C. Electron density guided fragment-based lead discovery of ketohexokinase inhibitors. J. Med. Chem. 2010, 53, 7979–7991. [DOI] [PubMed] [Google Scholar]
- Details for KHK-C cloning and expression are given in the Supporting Information (see the paragraph at the end of this paper).
- Matulis D.; Kranz J. K.; Salemme F. R.; Todd M. J. Thermodynamic stability of carbonic anhydrase: measurements of binding affinity and stoichiometry using ThermoFluor. Biochemistry 2005, 44, 5258–5266. [DOI] [PubMed] [Google Scholar]
- Purchased from BellBrook Laboratories, Madison, NJ.
- Kozak M.; Hayward B.; Borek D.; Bonthron D. T.; Jaskolski M. Expression, purification and preliminary crystallographic studies of human ketohexokinase. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2001, D57, 586–588. [DOI] [PubMed] [Google Scholar]
- Details on the screening assay method are given in the Supporting Information (see the paragraph at the end of this paper).
- El-Araby M.; Pottorf R. S.; Player M. R. Synthesis of a 2,4,8-trisubstituted pyrimidino[5,4-d]pyrimidine library via sequential SNAr reactions on solid-phase. Combin. Chem. High Throughput Screening 2004, 7, 413–421. [DOI] [PubMed] [Google Scholar]
- Structural information on KHK from X-ray crystallography has been reported (ref (26b) and (26c)).
- Details for our X-ray crystallography are given in the Supporting Information (see the paragraph at the end of this paper).
- The atomic coordinates and structure factors for KHK complexes with 3, 8, and 47 were deposited in the Protein Data Bank, with the accession codes 3QA2, 3Q92, and 3QAI, respectively [Protein Data Bank, Research Collaboratory for Structural Bioinformatics (http://www.rcsb.org)].
- Native KHK is a functional dimer with two subunits that are basically the same (homodimer) (refs (23a), (26a), and (26c)) except for their spatial disposition due to conformational differences (hence, our use of the term “pseudohomodimer”) (refs (26b), (26c), and this work). The subunits are designated a and b herein.
- See the paragraph about Supporting Information at the end of this paper.
- Details on the cellular assay procedure, including a negative control, are given in the Supporting Information.33
- The compound kinase specificity was assessed against a panel of 31 kinases (see the Supporting Information) with a FRET assay platform by Invitrogen (http://www.invitrogen.com).33 Representative kinases across the various kinase families were tested with 10 μM compound and 100 μM ATP. Inhibition activities were ranked based on percent inhibition at 10 μM. Compounds with inhibition of less than 25% at 10 μM were classified as selective for KHK.
- Compounds of interest were tested for inhibition of metabolic kinase activity via ribokinase (EC 2.7.1.15), hexokinase (EC 2.7.1.1), and adenosine kinase (EC 2.7.1.20). These kinases were selected because of their structural or sequence similarity to KHK.
- A CEREP panel for receptors and ion channels was performed (http://www.cerep.fr/cerep/users/index.asp).33
- Experimental details are given in Supporting Information.33
- After iv dosing to rats at 2 mg/kg, 8 had a C0 of 0.6 μM and a t1/2 of 2 h.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.