

Abstract

Structural optimization of salicylamide-based hemagglutinin (HA) inhibitor 1 resulted in the identification of cis-3-(5-hydroxy-1,3,3-trimethylcyclohexylmethylamino)benzenesulfonamide 28 and its derivatives as potent anti-influenza agents. The lead compound 28 and its 2-chloro analogue 40 can effectively prevent cytopathic effects (CPE) caused by infection of influenza A/Weiss/43 strain (H1N1) with EC50 values of 210 and 86 nM, respectively. Mechanism of action studies indicate that 40 and its analogues inhibit the virus fusion with host endosome membrane by binding to HA and stabilizing the prefusion HA structure. With significantly improved metabolic stability, the reported series represents the first generation of orally bioavailable HA inhibitors that have a good selectivity window and potential for further development as novel anti-influenza agents.
Keywords: Anti-influenza, hemagglutinin, benzenesulfonamide, inhibitor
Influenza-caused acute respiratory diseases in humans, birds, and other mammals represent one of the major threats to public health.1 As a genus of the Orthomyxoviridae family, the influenza A viruses have eight segmented RNA genomes and are the most virulent human pathogens among the three types of influenza viruses. Through frequent genetic drift and reassortment of different viral strains, influenza A viruses rapidly and continuously accumulate mutations to evade prevailing immunities in the general population. Influenza A viruses not only cause seasonal epidemics in the winter but also are able to unleash a major pandemic with a greater mortality and morbidity. In the most recent influenza pandemic, the 2009 influenza A/H1N1 strain likely emerged from a genetic reassortment of human, swine, and avian strains.2 Beyond those seasonal flu epidemics and the recent H1N1 pandemic, there also have been concerns about the far more lethal H5N1 avian flu strains and the possibility of breakthrough of interspecies and interhuman transmission barriers through viral genetic mutations, which could have a devastating effect on public health.3
Currently, the prevention and treatment of influenza A infection rely on vaccination and a limited number of antiflu drugs that inhibit two viral targets, M2 ion channel and neuraminidase. Amantadine and rimantadine block the M2 ion channel but have limited clinical use due to rapid emergence of drug resistance.4 Among the marketed neuraminidase inhibitors, oseltamivir is the sole oral drug that has been widely used for the treatment of flu. Nevertheless, the recently emerged oseltamivir-resistant H1N1 strain in the 2008–2009 flu season has been a cause for concern.5 In light of the persistent threat posed by influenza A viruses to the public, there is an urgent need to develop new anti-influenza agents with different mechanisms of action for therapeutic or prophylactic purpose.
As with many other enveloped viruses, membrane fusion of the influenza virus with a host cell is a key biological process required for the successful release of viral genome into the cytoplasm.6 Hemagglutinin (HA), one of the influenza envelope proteins, is a mushroomlike glycoprotein on the surface of viruses that plays a pivotal role in host cell recognition and membrane fusion. It has long been regarded as a potential target for anti-influenza therapies. The stalk region of HA is genetically stable among all 16 HA subtypes, and it constitutes the core fusion machinery that undergoes irreversible structural rearrangements and facilitates the fusion of viral envelope with host cell endosome membrane. It has been recently demonstrated that human monoclonal antibodies binding to the stalk region of HA are able to inhibit the fusion process and demonstrate broad spectrum activities against HA phenotype group 1 influenza A viruses, including human H1N1 and avian H5N1 strains.7,8 Besides new vaccine strategies based on the consensus sequence of HA, there has been particular interest in identifying and developing novel small molecule HA inhibitors as well as antibodies against a broad variety of flu viruses.9
The development of orally bioavailable small molecule HA inhibitors, especially those targeting the conserved stalk region of HA, is expected to have enormous value for the treatment of drug-resistant influenza infections. Many small molecule HA inhibitors such as cis-2-hydroxy-N-(5-hydroxy-1,3,3-trimethyl-cyclohexylmethyl)-benzamide (1),10 podocarpate ester (2),11 triperiden analogue (3),12 and 1-phenyl-cycloalkane carbamide (4)13 have been implicated to have potential anti-influenza effects by stabilizing the HA structure and preventing its conformational change during the membrane fusion process (Figure 1). On the basis of affinity-labeling studies and docking predictions, 1 and its analogues were claimed to bind to a hydrophobic pocket in the stalk region of HA and demonstrated good anti-influenza potency.14 In this study, we aim to identify highly potent HA inhibitors with 1 as a starting point and address potential DMPK liabilities of the salicylamide functional group. Our preliminary structure–activity relationship (SAR) studies of 1 suggested that the salicylamide group is more important for conformational control of the molecule versus a role in potential hydrogen bonding with HA protein. Thus, we designed ring-cyclized analogues, such as 1H-indazol-3-ylamine 7, which could mimic the salicylamide structure while retaining antiviral activity and exhibiting better metabolic stability than 1.
Figure 1.

Structures of HA inhibitors 1–4.
We explored bicyclic aryl derivatives first while keeping the cis-aliphatic amino group constant. The synthesis of 7 was carried out from cis-amine 5(10) as shown in Scheme 1. Briefly, the coupling reaction of 5 and 2-fluoro-benzoic acid with HATU afforded the amide intermediate, which was then treated with Lawesson's reagent to give thioamide 6. A mixture of 6 and hydrazine hydrate in DMSO was heated at 150 °C to yield designed indazole analogue 7. It was evaluated for anti-influenza activity in the CPE assay using Madin–Darby canine kidney (MDCK) cells. Indazole 7 was found to be able to inhibit the replication of the influenza A/Weiss/43 strain and prevent the viral cytopathic effect with an EC50 of 6.9 μM (Table 1). Because 7 also demonstrated improved mouse liver microsomal stability (MLM = 45.2 mL/min/kg) as compared to 1 (MLM = 76.7 mL/min/kg),15 it was used as a template for additional structural modifications.
Scheme 1. General Synthesis for HA Inhibitors 7–28.

Reagents and conditions: (a) 2-Fluorobenzoic acid, HATU, NEt3, DCM, room temperature. (b) Lawesson's reagent, toluene, refluxing. (c) NH2NH2·H2O, DMSO, 150 °C. (d) Pyridine, DMSO, microwave, 150 °C. (e) CuI, K3PO4, l-proline, microwave, DMSO, 80–120 °C.
Table 1. Anti-Influenza Activities of Compounds 7–28.
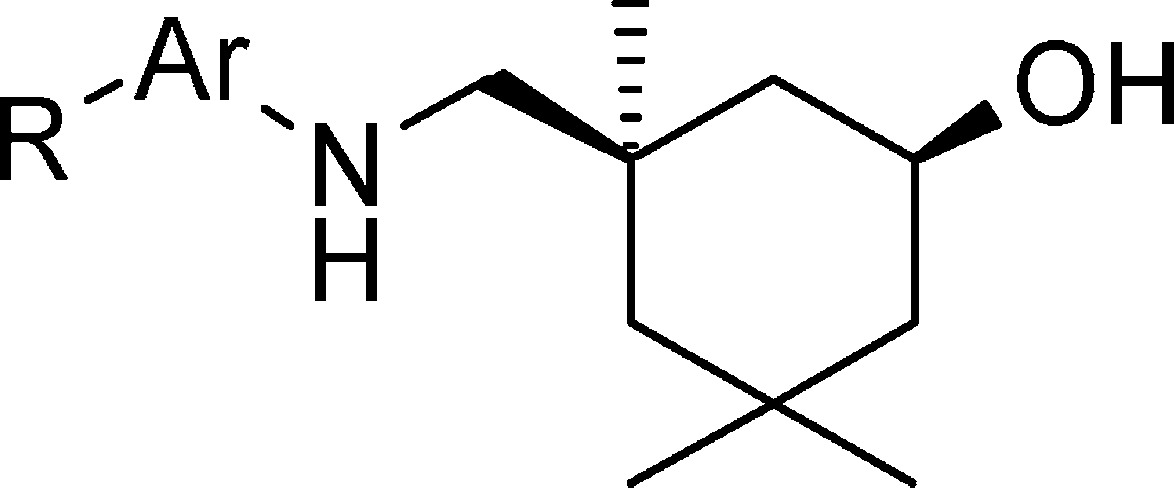
| compd ID | Ar | R | EC50 (CPE, μM) a | CC50 (μM) b | MLM CLh (mL/min/kg) c |
|---|---|---|---|---|---|
| 1 | salicylamide | H | 0.030 | >50 | 76.7 |
| 7 | 1H-indazol-3-yl | H | 6.90 | >100 | 45.2 |
| 8 | quinazolin-2-yl | H | 8.5 | 26.7 | 82.9 |
| 9 | benzothiazol-2-yl | H | 2.12 | 36.8 | 85.4 |
| 10 | benzothiazol-2-yl | 6-F | 2.5 | 16.2 | 78.2 |
| 11 | benzothiazol-2-yl | 6-Cl | >50 | 4.2 | |
| 12 | pyridin-2-yl | H | 4.43 | >100 | 80.6 |
| 13 | pyridin-2-yl | 5-CF3 | 3.39 | 30.5 | |
| 14 | pyridin-2-yl | 6-CF3 | 0.22 | 13.5 | 82.3 |
| 15 | Ph | H | 0.43 | 37 | 85.6 |
| 16 | Ph | 2-Cl | 0.52 | 6.9 | |
| 17 | Ph | 3-Cl | 0.057 | 9.7 | 86.1 |
| 18 | Ph | 4-Cl | 0.44 | 8.1 | |
| 19 | Ph | 2-CF3 | 0.15 | 6.6 | |
| 20 | Ph | 3-CF3 | 0.042 | 7.8 | 83.8 |
| 21 | Ph | 3-CF3, 5-Cl | 0.044 | 6.7 | 49.8 |
| 22 | Ph | 3-Cl, 5-F | 0.015 | 4.4 | 85.1 |
| 23 | Ph | 3-CF3, 4-Cl | 0.018 | 2.8 | 62.4 |
| 24 | Ph | 3-CN | 0.25 | 46.9 | 84 |
| 25 | Ph | 3-PhO | >100 | 6.0 | |
| 26 | Ph | 3-CONH2 | 37.9 | >100 | 43.0 |
| 27 | Ph | 3-SO2Me | 0.93 | >50 | 65.3 |
| 28 | Ph | 3-SO2NH2 | 0.21 | >100 | 37.2 |
Compound concentration to inhibit 50% of the cytopathic effect based on OD measurement. Values are the average of ≥two determinations run in duplicate, SD ± 15%.
Compound concentration to inhibit 50% of MDCK cell growth relative to control.
In vitro mouse liver microsome clearance calculated using the well-stirred model. See ref (15) for details.
To test the influence of geometry and the substitution pattern of the aromatic rings on the antiviral activity, analogues 8–11 were made by nucleophilic replacement of various bicyclic aryl chlorides with amine 5 in pyridine. Those bicyclic aromatic analogues were tested for their antiviral potency and cytotoxicity in MDCK cells using the CPE assay. It quickly became apparent that analogues with quinazoline or benzothiazole replacements have moderate activity, and the substitution on the aromatic groups is generally not tolerated, thus leaving little space for further structural optimizations. For example, analogue 11 with 6-Cl substitution was inactive in the CPE assay, but 9 and 10 (R = H and F) showed an EC50 around 2 μM. In addition, many analogues had a marginal selectivity window and a high intrinsic clearance probably due to increased hydrophobicity. Therefore, new scaffolds with a less lipophilic ring such as pyridine or phenyl were synthesized to evaluate the possibility to increase potency and metabolic stability.
Among the aminopyridine analogues, 14 with 6-CF3 is over 15 times more potent than analogues 12 and 13 but demonstrated poor microsomal stability. The synthesis of aniline analogues 15–28 was carried out by the copper-catalyzed cross-coupling of substituted phenyl bromides with 5.16 Aniline analogues 17 and 20 with 3-Cl and 3-CF3 substitution groups demonstrated much better antiviral potencies than others with an EC50 of 57 and 42 nM, respectively (see 12–20). When a second small hydrophobic group was introduced, it gave highly potent analogues 22 and 23 with EC50 below 20 nM. Both bulky hydrophobic groups and polar substituents such as amides abolished anti-influenza activity. Further chemistry explorations led to sulfonamide 28, which showed submicromolar antiviral activities and a good selectivity window in MDCK cells. As compared to other derivatives, 28 also showed good water solubility (141 μg/mL in phosphate buffer, pH 6.5) and a significantly improved stability in human and mouse liver microsomes with HLM of 6.6 mL/min/kg and MLM of 37.2 mL/min/kg, respectively.
On the basis of the SAR and metabolite analysis of the aniline series, disubstituted benzenesulfonamide analogues were designed and synthesized (Scheme 2). Substituted benzenesulfonamide 30 was prepared from aniline 29 by a Sandmeyer reaction and subsequent treatment of the resulting benzenesulfonyl chloride with ammonia. The cross-coupling of 30 with 5 was carried out with CuI, l-proline, and K3PO4 in DMSO at 120 °C to give compounds 31–35. However, because of favorable ortho-substitution of 30 (R = 2-Cl), an alternative method was developed for the synthesis of 40. Briefly, sulfonamide 37 was prepared from 5-fluoro-2-nitro-phenylamine 36, and it was treated with amine 5 under basic conditions to afford aniline 38. Then, the nitro group of 38 was reduced by tin(II) chloride under refluxing conditions to benzene-1,4-diamine 39. A selective Sandmeyer reaction of 39 with copper(II) chloride and tert-butyl nitrite gave 2-chloride analogue 40 in moderate yield.
Scheme 2. Synthesis of Substituted Benzenesulfonamides 31–35 and 40.

Reagents and conditions: (a) (1) NaNO2, HCl; (2) CuCl, AcOH, SO2. (b) NH3, DCM. (c) CuI, K3PO4, l-proline, microwave, DMSO, 120 °C. (d) Compound 5, K2CO3, DMSO, 50 °C, 43%. (e) SnCl2·H2O, EtOH, reflux, 99%. (f) CuCl2·2H2O, t-BuONO, DMF, 45 °C, 22%.
As expected, the derivatives 31, 32, and 40 with additional F- or Cl-substitution showed a 3–5-fold increase in inhibitory potency as compared to the parent compound 28 (Table 2). On the other hand, extra substitution groups such as methyl, methoxyl, and CF3 generally led to reduced antiviral activities. It is noteworthy that both 31 and 40 showed over 600-fold selectivity (CC50/EC50) against MDCK cells in the CPE assay.
Table 2. Anti-Influenza Activities of Sulfonamide Analogues and Their DMPK Profiles.
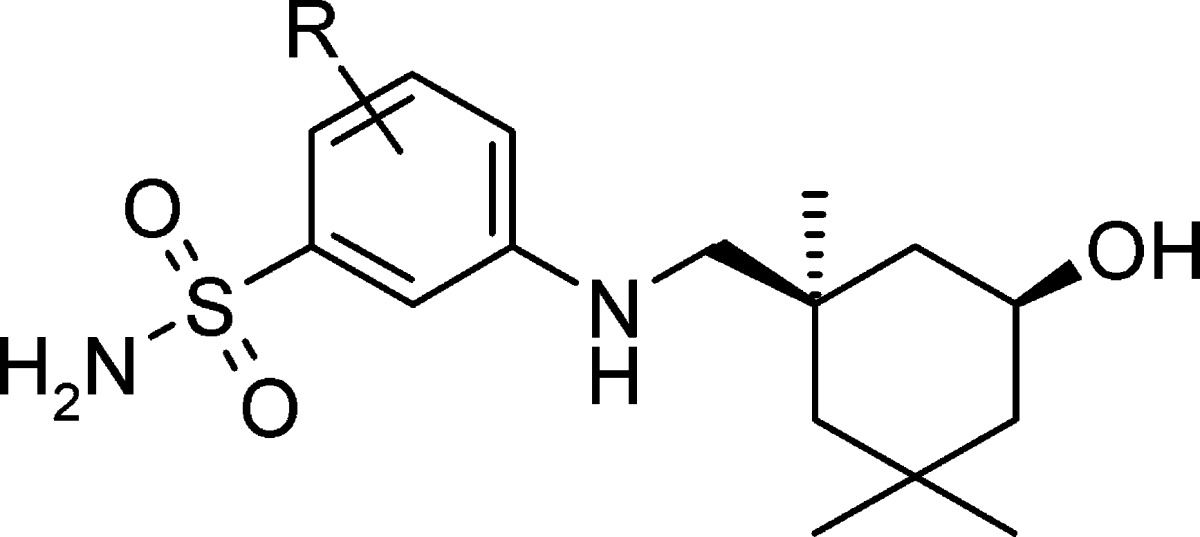
| compd | R | EC50 (CPE, μM) a | CC50 (μM) b | IC50 (μM) c | HLM CLh (mL/min/kg) d | MLM CLh (mL/min/kg) b |
|---|---|---|---|---|---|---|
| 31 | 3-F | 0.089 | 55.0 | 0.58 | 7.2 | 42.3 |
| 32 | 3-Cl | 0.044 | 8.4 | 0.23 | 12.1 | 51.8 |
| 33 | 3-CF3 | 1.36 | 19.5 | 2.91 | 5.1 | 39.6 |
| 34 | 2-Me | 0.56 | 81.5 | 1.13 | 8.5 | 43.7 |
| 35 | 2-OMe | 2.62 | >100 | 4.16 | 10.0 | 66.4 |
| 40 | 2-Cl | 0.086 | 71.2 | 0.25 | 8.4 | 42.5 |
Compound concentration to inhibit the cytopathic effect based on OD measurement. Values are means of three determinations run in duplicate, SD ± 15%.
See the notes in Table 1.
Half maximal inhibition concentration to prevent hemolysis of chicken RBCs; values are the average of two duplicate experiments, SD ± 20%.
In vitro human liver microsome clearance calculated using the well-stirred model.
To confirm the novel series indeed targets influenza HA protein and blocks virus fusion as expected, the inhibitory effect of representative analogues on the hemolysis of chicken red blood cells (RBCs) was tested. As one of the most potent compounds in the CPE assay, 23 effectively blocked the hemolysis of RBCs with an IC50 of 0.06 μM. In accord with the CPE results, analogues 11, 25, and 26 did not have an obvious impact on the hemolysis with IC50 values above 31.6 μM. Sulfonamide 40 showed potent hemolysis inhibition with an IC50 of 0.25 μM but did not have an effect on hemagglutination, in which only viral HA-cellular receptor binding is involved. This result suggests that 40 prevents hemolysis very likely by binding to the stalk region of HA and thus interfering with crucial conformational changes of HA normally triggered by a low pH environment.
It has been well-documented that the low pH-triggered conformational changes of HA expose its internal trypsin cleavage sites and render HA sensitive to trypsin. Thus, the mechanism of action of 40 was further verified in an HA trypsin sensitivity assay with 1 as a positive control (Figure 2). The purified HA, in the presence or absence of compounds, was subjected to a brief low pH treatment followed by trypsin digestion. It was clearly demonstrated that 40 protects HA from low pH-induced trypsin degradation (Figure 2, lane 6). Taken together, the mechanism of action studies and the CPE data indicate that 40 inhibits the virus fusion with host endosome membrane by binding to HA and preventing the conformational changes of the prefusion HA structure.
Figure 2.
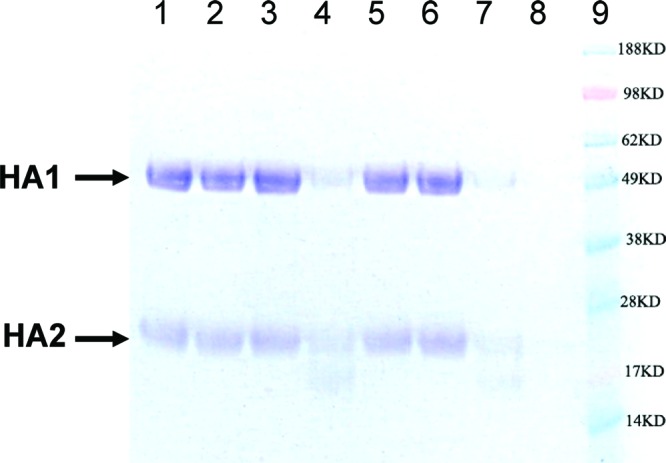
SDS-PAGE analysis of trypsin sensitivity assay showing 1 and 40 protected purified HA from trypsin digestion. Lane 1, purified HA; lane 2, HA treated with trypsin without a prior acidification step; lane 3, acidified HA without trypin treatment; lane 4, DMSO control; lane 5, compound 1 (10 μM); lane 6, compound 40 (10 μM); lane 7, ribavirin (10 μM); lane 8, trypsin only; and lane 9, molecular markers. Lanes 4–7 included all steps and components in a typical trypsin sensitivity assay. Positions of HA1 and HA2 are marked.
The single dose pharmacokinetic properties of a few sulfonamide analogues were analyzed in CD-1 mice. They all demonstrated good oral availabilities with F ≥ 55% and significantly improved in vivo stability and longer terminal half-life as compared to 1. The chloro substitution on the phenyl ring resulted in reduced clearance of 32 (CL = 42 mL/min/kg) and 40 (CL = 53 mL/min/kg) when compared to the parent compound 28, which had a clearance of 82 mL/min/kg in mice. Furthermore, 32 and 40 did not significantly inhibit five major CYP isoforms, with IC50 values above 10 μM.
Viral specificity studies showed that 40 had similar antiviral potency against another group 1 influenza strain A/PR/8/34 (H1N1) but was inactive against influenza A/Hongkong/8/68 virus, a H3N2 strain that belongs to the HA phenotype group 2 influenza viruses. This selectivity between group 1 and 2 viruses is probably due to the structural difference at the binding site in the HA stalk region, as revealed by recent cocrystal studies of HA complexed with group 1 broadly neutralizing antibodies7,8 and group 2 specific HA inhibitor tert-butyl hydroquinone.17 Additional biostructure analysis of the stalk region of HA proteins indicates that this region is highly conserved and stable within each group and thus provides an attractive target for antiviral drug discovery. Highly potent HA inhibitors with sufficient metabolic stability and selectivity window like 40 could be very valuable in the ongoing battle against the vicious influenza A pathogens including seasonal H1N1 and avian H5N1 strains.
In summary, a novel series of potent and orally available anti-influenza inhibitors were identified through structural modifications of salicylamide 1. The representative analogue 40 demonstrated potent antiviral activities with an adequate selectivity window and had significantly improved metabolic stability in mouse PK studies. The identification of benzenesulfonamide-based HA inhibitors and their mechanism of action studies warrant further structural optimizations and development for the treatment of flu.
Acknowledgments
We acknowledge Jason Wong and Shuhai Zhao for many scientific discussions. We also thank Yongguo Li, Wenzhi Chen, Peilan Ding, Qingshan Gao, Rong Zhao, and Sheng Zhong for their analytical assistance.
Supporting Information Available
Detailed experimental procedures for the synthesis of compounds 7, 28, 31–35, and 40 and the biological assays (CPE, RBC hemolysis, and trypsin sensitivity). This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- World Health Organization, http://www.who.int.
- Smith G. J. D.; Vijaykrishna D.; Bahl J.; Lycett S. J.; Worobey M.; Pybus O. G.; Ma S. K.; Cheung C. L.; Raghwani J.; Bhatt S.; Peiris J. S. M.; Guan Y.; Rambaut A. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 2009, 459, 1122. [DOI] [PubMed] [Google Scholar]
- Fauci A. S. Pandemic influenza threat and preparedness. Emerging Infect. Dis. 2006, 12, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright R. A.; Shay D. K.; Shu B.; Cox N. J.; Klimov A. I. Adamantane resistance among influenza A viruses isolated early during the 2005–2006 influenza season in the United States. J. Am. Med. Assoc. 2006, 295, 891. [DOI] [PubMed] [Google Scholar]
- Weinstock D. M.; Zuccotti G. The evolution of influenza resistance and treatment. J. Am. Med. Assoc. 2009, 301, 1066. [DOI] [PubMed] [Google Scholar]
- Dimitrov D. S. Virus entry: Molecular mechanisms and biomedical applications. Nat. Rev. Microbiol. 2004, 2, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui J.; Hwang W.; Perez S.; Wei G.; Aird D.; Chen L.; Santelli E.; Stec B.; Cadwell G.; Ali M.; Wan H.; Murakami A.; Yammanuru A.; Han T.; Cox N. J.; Bankston L. A.; Donis R. O.; Liddington R. C.; Marasco W. A. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 2009, 16, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekiert D. C.; Bhabha G.; Elsliger M.; Friesen R. H. E.; Jongeneelen M.; Throsby M.; Goudsmit J.; Wilson I. A. Antibody recognition of a highly conserved influenza virus epitope. Science 2009, 324, 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boriskin Y. S.; Leneva I. A.; Pecheur E.-I.; Polyak S. J. Arbidol: A broad-spectrum antiviral compound that blocks viral fusion. Curr. Med. Chem. 2008, 15, 997. [DOI] [PubMed] [Google Scholar]
- Deshpande M. S.; Wei J.; Luo G.; Cianci C.; Danetz S.; Torri A.; Tiley L.; Krystal M.; Yu K.; Huang S.; Gao Q.; Meanwell N. A. An approach to the identification of potent inhibitors of influenza virus fusion using parallel synthesis methodology. Bioorg. Med. Chem. Lett. 2001, 11, 2393. [DOI] [PubMed] [Google Scholar]
- Staschke K. A.; Hatch S. D.; Tang J. C.; Hornback W. J.; Munroe J. E.; Colacino J. M.; Muesing M. A. Inhibition of influenza virus hemagglutinin-mediated membrane fusion by a compound related to podocarpic acid. Virology 1998, 248, 264. [DOI] [PubMed] [Google Scholar]
- Oka M.; Ishiwata Y.; Iwata N.; Honda N.; Kakigami T. Synthesis and anti-influenza virus activity of tricyclic compounds with a unique amine moiety. Chem. Pharm. Bull. 2001, 49, 379. [DOI] [PubMed] [Google Scholar]
- Tang G.; Qiu Z.; Lin X.; Li W.; Zhu L.; Li S.; Li H.; Wang L.; Chen L.; Wu Z. J.; Yang W. Discovery of novel 1-phenyl-cycloalkane carbamides as potent and selective influenza fusion inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 3507. [DOI] [PubMed] [Google Scholar]
- Cianci C.; Yu K.; Dischino D.; Harte W.; Deshpande M.; Luo G.; Colonno R.; Meanwell N.; Krystal M. pH-dependent changes in photoaffinity labeling patterns of the H1 influenza virus hemagglutinin by using an inhibitor of viral fusion. J. Virol. 1999, 73, 1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obach R. B.; Baxter J. G.; Liston T. E.; Silber B. M.; Jones B. C.; MacIntyre F.; Rance D. J.; Wastall P. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther. 1997, 283, 46. [PubMed] [Google Scholar]
- Zhang H.; Cai Q.; Ma D. Amino acid promoted CuI-catalyzed C-N bond formation between aryl halides and amines or N-containing heterocycles. J. Org. Chem. 2005, 70, 5164. [DOI] [PubMed] [Google Scholar]
- Russell R. J.; Kerry P. S.; Stevens D. J.; Steinhauer D. A.; Martin S. R.; Gamblin S. J.; Skehel J. J. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 17736. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.