

Abstract
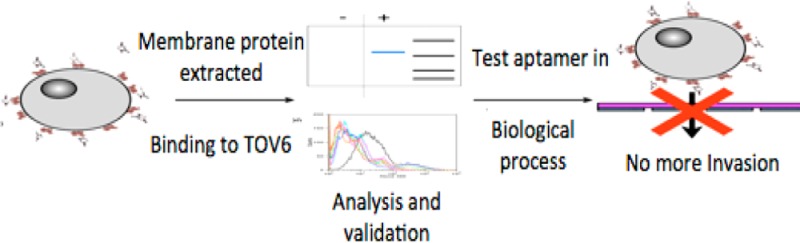
In this paper, we describe the elucidation of the target of an aptamer against ovarian cancer previously obtained by cell-SELEX (SELEX = systematic evolution of ligands by exponential enrichment). The target’s identity, stress-induced phosphoprotein 1 (STIP1), was determined by mass spectrometry and validated by flow cytometry, using siRNA silencing and protein blotting. Initial oncologic studies show that the aptamer inhibits cell invasion, indicating that STIP1, which is currently under investigation as a potential biomarker for ovarian cancer, plays a critical role in this process. These results serve as an excellent example of how protein target identification of aptamers obtained by cell-SELEX can serve as a means to identify promising biomarker candidates and can promote the development of aptamers as a new drug class to block important oncological processes.
Ovarian cancer is a lethal malignancy, which is typically diagnosed only at advanced stages, when metastasis has set in. Diagnosis at later stages significantly reduces treatment options. The identification of biomarkers that are specifically indicative of ovarian cancer would improve the chances for earlier detection, a principle that is true for any cancer. Indeed, disease biomarkers are currently in high demand to identify new drug leads, improve diagnostic techniques, and monitor treatment.1 As an alternative to antibody arrays, we introduce aptamer-assisted biomarker discovery. Aptamer TOV6 was used as a model throughout this study. This aptamer was previously selected by cell-SELEX (SELEX = systematic evolution of ligands by exponential enrichment) against ovarian cancer cell line TOV-21G.2,3
Membrane proteins represent a potentially important source of putative cancer biomarkers but are currently understudied due to their low solubility.1,4 They can be overexpressed in specific types of cancer and may play a critical role in oncogenesis.5 Studies show that membrane proteins are shed or secreted from the plasma membrane into conditioned media in cell lines and can also be found in the systemic blood circulation of cancer patients.6,7 Thus, their detection can serve as an important disease indicator. Significantly, membrane proteins form the targets of over half of the currently approved drugs.8 Therefore, the identification of molecules that bind to cancer cell membrane proteins could become a critical step toward developing new diagnostic or therapeutic approaches (e.g., by acting as protein antagonists).9
Ovarian cancer is the most lethal gynecological malignancy.10 About 90% of all ovarian cancers are adenocarcinoma,11 and the two large histological subgroups are serous (40%) and clear cell (5–25%) adenocarcinoma. Although pathological/histological examination is the only means now available to distinguish between the two subgroups, the different subclass responses to chemotherapy justify the need for a tool that can efficiently distinguish these two subgroups at the molecular level.12 Recently, selected DNA aptamers have shown the potential to be such a tool due to their ability to differentiate between the ovarian clear cell line TOV-21G and the ovarian serous adenocarcinoma CAOV3 at the molecular level. Since their target molecules are also cancer cell membrane proteins, it seems plausible that these aptamers may serve as significant reagents in the development of novel therapeutics.
In this paper we describe the methodology we developed to identify and further validate the protein target of aptamer TOV6, essentially by using the aptamer as a pull-down reagent for its cognate target after fixation with formaldehyde. In the past, such targets have proven difficult to isolate because of the low solubility and low abundance of membrane proteins. The instability of the aptamer–protein complex in a detergent system used for membrane protein solubilization and cell lysis adds an additional challenge to the identification of an aptamer’s target.13−16 Particularly, to bind with their cognate target with high affinity and specificity, aptamers must fold into tertiary structures with their target, a process that is dictated by a number of forces, including hydrogen bonding, electrostatic interactions, van der Waals forces, and stacking interactions.15 However, the extraction of membrane proteins requires the use of surfactants, which can seriously interfere with such interactions, which in turn makes the extraction and the identification of aptamer targets a frustrating task. To solve the problem of aptamer–protein complex instability during extraction, we previously proposed a chemical fixation between an aptamer and its target by incorporating nucleotides that cross-link with their target via UV irradiation.17 However, this method is labor-intensive and would make large-scale aptamer–target elucidation impractical.
Therefore, in the present work, formaldehyde was explored as an alternative cross-linker to circumvent these problems. Formaldehyde is a well-known reagent, used extensively in the study of the intracellular interactions between DNA and protein, as described in methods such as chromatin immunoprecipitation (ChIP).18 Similar to ChIP, this method utilized formaldehyde-induced cross-linking19 between the DNA aptamer and the target protein to enable subsequent protein identification by mass spectrometry.
After binding of TOV6 to its cognate target on the cell surface membrane, the TOV6/target interaction was fixed by formaldehyde. The protein–aptamer hybrid was then extracted from the cell lysate and recovered, and the protein was identified as stress-induced phosphoprotein 1 (STIP1) by mass spectrometry. The identity of the target was confirmed through siRNA silencing and antibody binding. By using Boyden chambers, it was also shown that STIP1 plays a role in cell invasion and that TOV6 is a potent inhibitor in this oncological process. Thus, we report here that the identity of the target molecule of aptamer TOV6 is STIP1, and by identifying the target of TOV6, a blocking effect in the metastatic process of TOV-21G could be identified. In short, STIP1 is part of a cell peripheral complex with heat shock protein 90 (HSP90) that activates matrix metalloproteases.7 This effect shows that TOV6 could be a therapeutic agent and that cell-SELEX can be important in the development of novel drugs against cell surface proteins.
The following paragraphs describe the steps leading to the identification and validation of aptamer TOV6 against its cognate target, STIP1. In addition, it will be demonstrated that STIP1 plays a pivotal role in the regulation of cell invasion and that aptamer TOV6 plays an equally important role in inhibiting this process.7,20
Aptamer TOV6 binds to the ovarian clear cell adenocarcinoma TOV-21G, but not to the cervical cancer cell line HeLa or the ovarian serous adenocarcinoma CAOV3. It also binds to the glioblastoma cell line A172, among others.2 As shown in Figure 1, formaldehyde cross-linking was able to maintain the interaction between the aptamer and the protein after extraction from the cells, as a clear band is visible in lane 8 around 78 kDa. When compared to lane 4, where no formaldehyde was applied to aptamer-bound cells, no such band is visible. Furthermore, the cross-linking time was found to be a critical factor in optimizing the functionality of aptamer TOV6 as a pull-down reagent for its cognate target (Supplemental Figure 2, Supporting Information). Following optimization, two samples from separate experiments were sent for protein sequencing by mass spectrometry (MS). In these two experiments, STIP1 was found within the top hits by our MS service facility (Supplemental Table 1, Supporting Information). The presence of STIP1 as a membrane protein has previously been reported in various ovarian cell lines, including TOV-21G,21 but also in the glioblastoma cell line A17222 and the pancreatic carcinoma cell line Panc-1.6
Figure 1.

Electropherogram of extracted protein obtained from the aptamer-mediated protein captured from cross-linked aptamer TOV6 or randomer (unless specified otherwise). After PAGE, the gel was stained with MS-compatible silver staining reagents. The ladder is represented in kilodaltons. Key: cell lysate, unbound fraction after incubation to beads; randomer, fraction of protein from randomer released from the beads after biotin incubation; eluate 1, fraction of protein from aptamer TOV6 released from the beads after a 1 h of biotin elution; eluate 2, fraction of protein from aptamer TOV6 released from the beads after the first elution buffer (12.5 mM biotin) incubation, obtained by further elution at 65 °C with elution buffer; eluate 3, fraction of protein from aptamer TOV6 released from the beads after elution buffer incubation, without cross-linking. The mass spectral analysis results from eluate 1 are presented in the Supporting Information (Supplemental Tables 1–5).
The binding of TOV6 to STIP1 was experimentally validated by STIP1 siRNA silencing on TOV-21G cells. Silencing with four different STIP1 siRNAs showed reduced binding of TOV6 compared with that with scrambled siRNA-treated cells. Since siRNA silencing can sometimes lead to unwanted compensation mechanisms by the cells,22,23 an additional control was included by monitoring the change of the fluorescence signal from tyrosine-protein kinase-like 7 (PTK7) protein aptamer Sgc8. As shown in Figure 2, compensation mechanisms were not observed, as the signal reported from binding to PTK7 was unaltered. Identical observations were made on A172 cells, where STIP1 was silenced, and the expression level of PTK7 was unaltered
Figure 2.
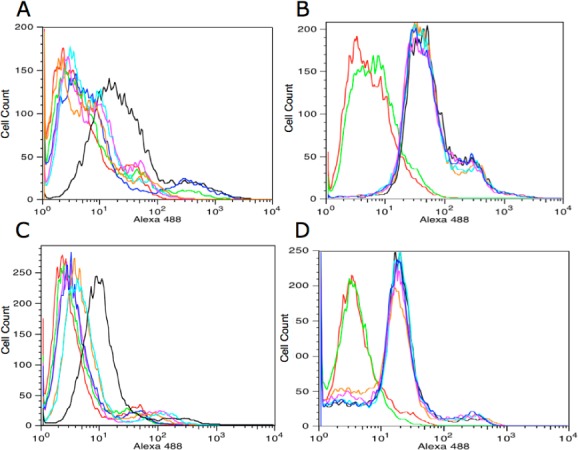
(A) Silencing of STIP1 in TOV-21G cells. The cells were tested for TOV6 binding after 72 h of siRNA treatment. (B) Absence of PTK7 silencing with STIP1 siRNA treatment in TOV-21G cells. The cells were tested for Sgc8 binding after 72 h of siRNA treatment. (C) Silencing of STIP1 in A172 cells. The cells were tested for TOV6 binding after 72 h of siRNA treatment. (D) Absence of PTK7 silencing with STIP1 siRNA treatment in A172 cells. The cells were tested for Sgc8 binding after 72 h of siRNA treatment. Key: red, A172 cells incubated with streptavidin–Alexa 488 only; green, library-incubated cells; black, scrambled siRNA-treated cells; dark blue, STIP1 siRNA 5; orange, STIP1 siRNA 6; light blue, STIP1 siRNA 10; magenta, STIP1 siRNA 11.
The STIP1 antibody could not bind to cells treated with STIP1 siRNA (Figure 3). Additional validation of the binding between TOV6 and STIP1 was obtained by performing an aptamer blot on rhSTIP1 (Supplemental Figure 3, Supporting Information). A clear signal from the recombinant protein was observed, while the aptamer was unable to blot bovine serum albumin (BSA). A panel of ovarian cancer cell lines was tested for binding with aptamer TOV6: SKOV3 (serous), C13 and TOV-112D (both endometroid), and OVCAR3 (serous) showed binding to the aptamer, while the aptamer failed to bind other ovarian cell lines, such as OVCAR8 (serous) and A2780 cP (undifferentiated) (Supplemental Table 2, Supporting Information). The expression of STIP1 on the cell surface was experimentally confirmed by Wang et al. in SKOV3, TOV-21G, and TOV-112D, which coincides with our binding experiment.21 These data indicate that there is heterogeneity within ovarian cancer related to the expression of STIP1 on the cell surface, as SKOV3 and OVCAR3 are both serous ovarian adenocarcinomas.24
Figure 3.
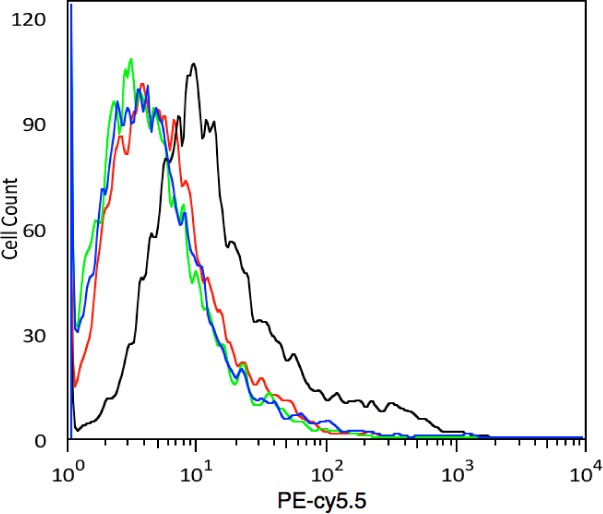
Effect of STIP1 silencing on STIP1 antibody binding. Key: red, TOV-21G cells incubated with streptavidin PE–Cy5.5 only; green, biotinylated IgG streptavidin PE–Cy5.5; black, biotinylated M33 antibody on scrambled siRNA-treated cells; blue, biotinylated M33 antibody on STIP1 siRNA 5.
In an earlier study, Walsh et al. investigated the role of STIP1 in relation to cell invasion in pancreatic cancer. They showed that STIP1 is found in the conditioned medium of Panc-1 cells, where STIP1 forms a complex with HSP90, which is critical in the regulation and activity of matrix metalloproteinase 2 (MMP-2), an enzyme that plays a key role in metastasis.6 This suggested that these three proteins play a key role in the onset or regulation of metastasis, a hallmark oncological process. They furthermore showed that the inhibition of HSP90 with the HSP90-specific inhibitor 17-AAG reduces the ability of cells to invade a Matrigel-layered Boyden chamber, suggesting the inactivation of the STIP1–MMP-2–HSP90 complex.6 When STIP1 was silenced, they also observed a significant reduction in cell invasion.
To verify if a similar function of STIP1 can be hypothesized for TOV-21G, a similar study was performed on TOV-21G cells. HSP90 inhibition has been reported to also lead to apoptosis;25,27 thus, the level of 17-AAG in the medium of TOV-21G needed to be minimized. The maximum tolerated concentration of 17-AAG that had a minimal effect on cell proliferation of TOV-21G was found to be 0.05 μM (Supplemental Figure 4, Supporting Information). Consequently, the effect of STIP1 silencing on cell migration and invasion was investigated. As shown in Figure 4, it was observed that STIP1 slightly affected the migration ability of TOV-21G, while HSP90 inhibition with 17-AAG had no influence on the cell’s propensity to transverse a semipermeable membrane. At 0.10 μM 17-AAG, a reduction in migration was observed in the STIP1 knockdown, but this result can be explained by reduction in cell proliferation at these levels. Of note is that this adverse effect is not seen when STIP1 protein is not silenced, which may indicate that STIP1 is able to partially compensate for HSP90.
Figure 4.

Migration of TOV-21G across a microporous membrane. STIP1 siRNA-treated cells migrate more slowly than scrambled siRNA-treated cells, indicating STIP1 silencing. The reduction of migration with 0.1 μM 17-AAG can be explained by the reduced viability of TOV-21G cells at this concentration. Error bars represent the standard deviation (n = 3).
When STIP1 expression was silenced, a reduction in invasiveness was observed (Figure 5). Cells incubated with 17-AAG showed a reduced ability to transverse the Matrigel membrane, and the effect of 17-AAG was enhanced when STIP1 was silenced. This suggests that not all the HSP90 located at the cell periphery is inhibited (or STIP1 is activating the matrix metalloproteinases (MMPs) in other forms, either by itself or by interaction with unknown proteins), and the removal of STIP1 by silencing reduces the presence of the active complex needed for metastasis even further. Therefore, our data suggest that STIP1 is involved in similar mechanisms needed for the invasiveness of TOV-21G, as described by Walsh and colleagues, as the invasiveness of the TOV-21G is reduced both by the inhibition of HSP90 and by the silencing of STIP1.
Figure 5.

Invasion assay of TOV-21G to determine the effect of HSP90 inhibition or STIP1 silencing on the ability of TOV-21G to cross a Matrigel layer. STIP1 siRNA-treated cells digest the Matrigel layer to a lesser extent, an effect that is amplified by 17-AAG inhibition, indicating the exhaustion of active HSP90–STIP1 complex at the cell periphery. Error bars represent the standard deviation (n = 3).
The fact that STIP1 was, to some extent, able to compensate for the inhibition of HSP90 led us to propose that STIP1 enables cell invasiveness, similarly to HSP90 at the cell periphery. HSP90 is known to be a critical factor in the activity of MMPs to enable the invasiveness of HT-1080, as antibodies were identified that can block HT-1080’s invasiveness.28,29 Because aptamer TOV6 binds to STIP1 (a protein that interacts with HSP90), the aptamer may be a plausible inhibitor to prevent this process. Thus, TOV-21G cells were incubated with an excess of TOV6 in the medium in the upper compartment of the Boyden chamber. The aptamer had no effect on inhibiting the migration of TOV-21G across a microporous membrane. However, when the invasion of the cells incubated with aptamer TOV6 was investigated, the invasion fell back to levels comparable to those of STIP1-silenced cells treated with 17-AAG (Figure 6), suggesting that TOV6 binding inhibits the physiological process involving STIP1.
Figure 6.

Effect of TOV6 on migration and invasion of TOV-21G cells. A 2 μM concentration of TOV6 has no effect on the migration ability of TOV-21G. A 2 μM concentration of aptamer TOV6 can prevent cell invasion to a degree equal to that of ∼0.05 μM 17-AAG with STIP1 silencing. Error bars represent the standard deviation (n = 3).
The work presented in this paper shows that aptamer selection can become a truly important tool toward biomarker discovery. The membrane proteome is challenging because of its low abundance and low solubility in 2D-Gel techniques.21,28 However, cross-linking aptamers with membrane proteins has been shown to efficiently circumvent some of these problems. By cross-linking the aptamer to its target, the available protein for analysis is maximized by increasing the solubility, thereby enabling mass spectral techniques to identify the protein binding to the aptamer. Also, cell-SELEX yields ligands that identify unique targets of significance, as the pools generated during the selection are more likely to bind to overexpressed cell surface proteins on cancer cells, while ligands to more general cell surface proteins are subtracted by negative selection.2 The aptamers that are generated can play specific roles in cancer diagnosis and therapy.
We have demonstrated a new method for easy identification of the target of aptamer TOV6 as STIP1, which plays a role in the invasiveness of TOV-21G. This technique, cell-SELEX-based biomarker discovery, is simple, rapid, and efficient, since it does not require any structural modification of the aptamer (e.g., incorporation of UV-cross-linkable nucleotides) and all conjugations are performed at termini of the aptamer sequence, which usually do not affect aptamer binding.17 STIP1 is currently being investigated as a potential biomarker for ovarian cancer in combination with CA125.21 Having demonstrated STIP1 as a potential biomarker for ovarian cancer using our approach, we then investigated the potential of aptamer TOV6 as a possible therapeutic tool, since the aptamer showed the ability to block cellular invasion. Aptamers that can block cellular invasion have been selected recently on the basis of phenotypic screens, but their protein targets have not yet been identified.26
Walsh et al. have shown that STIP1 is important for invasiveness in pancreatic cancer, and they also showed that STIP1’s interacting protein, the chaperone protein HSP90, is important in the regulation of MMPs.6,25 STIP1 is a cochaperone protein, and the overexpression of cochaperone proteins on the cancer cell membrane has previously been described.27 Such cochaperone proteins have also been shown to regulate important extracellular proteins needed for tumor metastasis (e.g., MMP2 for cell invasion).28 In a previous study, Eustace et al. screened antibody libraries for antibodies that reduce invasiveness, and they found one that can reduce invasiveness by binding to the HSP90 α-isoform, but not the β-isoform.25 Our data indicate that STIP1 is also involved in this process, as we see similar evidence of an STIP1–HSP90 complex. Our data also suggest that STIP1 is needed for the activity of this cell peripheral complex. Elucidating the exact mechanism by which this aptamer blocks cell invasion will require more intensive study, but it can be assumed that STIP1 and HSP90 play similar roles in view of their roles in activating the secretion of MMPs. The mechanism of TOV6’s involvement in this important oncological process will thus most likely be elucidated in the study of the activation of this class of enzymes; however, we speculate the aptamer disrupts the HSP90–STIP1 complex by either preventing the binding of STIP1 to HSP90 and taking away the right conformation of this complex to exert its function or preventing the activation of MMPs by binding in a pocket that is important for the activation of MMPs. The slight effects in migration observed might be explained by the reduced amount of secreted STIP1 when treated with siRNA. Excreted STIP1 binds to ALK2 and activates the SMAD1/SMAD5-ID3 pathway, which regulates migration, among other cell functions.29
Cell-SELEX-based biomarker discovery does not require prior information on the molecular composition of the cell surface, and it can yield valuable information about the membrane proteins overexpressed in tumors. The trillions of random DNA sequences in the initial DNA library, combined with the negative selection strategy, ensure that any molecules, previously known or unknown, will be identified as possible disease markers, as long as they are expressed in substantially different levels by disease and normal cells. A very important advantage accrues, because aptamers are generated during this process, and they can serve as specific high-affinity probes for the identified biomarkers for future diagnostic and potentially therapeutic applications. Because the cost and complexity are significantly lower than those of antibody-based techniques, our approach has the potential of wider application and may have a very positive impact on the discovery of biomarkers and drugs. Cell-SELEX-based biomarker discovery will also serve as a useful tool for the study of membrane proteins (especially those found to be overexpressed in tumors), a currently understudied class of proteins, as their physical properties hamper their analysis.
Experimental Section
Reagents and Cell Lines
Aptamer TOV6 (5′-ATC CAG AGT GAC GCA GCA CGG CAC TCA CTC TTT GTT AAG TGG TCT GCT TCT TAA CCT TCA TCG ACA CGG TGG CTT A-3′), SGC8 (5′-ATC TAA CTG CTG CGC CGC CGG GAA AAT ACT GTA CGG TTA GA-3′), and randomer (N76) were synthesized by standard phosphoramidite chemistry using a 3400 DNA synthesizer (Applied Biosystems) on biotin– or desthiobiotin–CPG (for protein capture only) (Glen Research) and were purified by reversed-phase HPLC (Varian Prostar). rhSTIP1 and STIP1 antibody M33 (clone 2E1) was purchased from Abnova (Taipei City, Taiwan), and the antibody was biotinylated with the EZ-link Sulfo-NHS-LC-Biotin Kit (Pierce). The capture of aptamer–protein hybrids was performed using Dynabeads M-280 streptavidin (Invitrogen). Streptavidin–Alexa 488 and streptavidin PE–Cy5.5 were purchased from Molecular Probes. Dr. Patricia Kruk, University of South Florida, kindly donated SKOV3, OVCAR3, OVCAR8, TOV-112D, C13, A2780 cP, and A2780s cells. The TOV-21G cell line was purchased from the American Type Cell Culture (ATCC), and all cell lines were maintained in culture with MCBD 105:medium 199 (1:1) supplemented with 10% FBS and 100 IU/mL Pen-Strep. The cells were cultured at 37 °C in a 5% CO2 atmosphere.
Buffers
For aptamer binding assays, cells were washed and treated with nonenzymatic cell dissociation buffer (Sigma) to remove them from the culture dish. Cells were further washed with washing buffer (WB) containing 4.5 g/L glucose and 5 mM MgCl2 in Dulbecco’s phosphate-buffered saline (PBS) with CaCl2 and MgCl2 (Sigma). Binding buffer (BB) used for aptamer binding was prepared by adding baker’s yeast tRNA (0.1 mg/mL, Sigma) and BSA (1 mg/mL, Fisher) to the washing buffer to reduce nonspecific binding. All chemicals used in the buffers were purchased from Sigma-Aldrich, unless otherwise specified. For cross-linking, 1% formaldehyde (Fisher) in PBS solution was used. The cell lysis buffer contained 2% Triton-X100 (Fisher), 1.5% Nonidet (Fisher), and 0.5% cholate in doubly deionized water. To wash the magnetic beads, a 10 mM HEPES/NaOH buffer (pH 7.8) was used with 100 mM NaCl, 2 mM ethylenediaminetetraacetic acid (EDTA), 1 mM ethylene glycol tetraacetic acid (EGTA), 0.2% sodium dodecyl sulfate (SDS), and 0.1% sodium lauroyl sarcosinate (SLS) (PI washing buffer). The elution buffer was composed of 12.5 mM biotin (diluted from B20656, Invitrogen) in 7.5 mM HEPES/NaOH buffer with 75 mM NaCl, 1.5% EDTA, 0.5% EGTA, 0.15% SDS, and 0.075% SLS. As a cross-link reversal solution, a mixture of 250 mM Tris buffer (pH 8.8) with 2% SDS and 0.2 M mercaptoethanol was used. All solutions, except WB and BB, contained 0.1 mM phenylmethanesulfonyl fluoride (PMSF). For SDS–PAGE, the SilverQuest staining kit was used to visualize the bands (Invitrogen).
Target Membrane Protein Extraction
TOV-21G cells (108) were incubated with 200 pmol of desthiobiotin–oligonucleotide (aptamer or randomer) according to Scheme 1. The cells were washed to remove excess aptamer, and the aptamer was then cross-linked to the cells by incubating them for 2 min (time was optimized; Supplemental Figure 2, Supporting Information) in a 1% formaldehyde/PBS solution. The cells were then immediately washed (three times) at 4 °C in PBS to remove the formaldehyde (as quenching the reaction with lysine, commonly used to stop a formaldehyde reaction, can make MS analysis a nearly impossible task). Subsequently, the cells were lysed in a Dounce homogenizer (Fisher) for 2 min (75 strokes per minute) in lysis buffer. The lysate was incubated overnight at room temperature in the presence of 200 μg of magnetic beads. The beads were then washed with PI washing buffer on a magnetic stand until any remaining membrane was washed from the beads, noticeable as the bead slurry became less viscous. Once the beads were clean, the protein–aptamer hybrid was eluted by incubation for 1 h with elution buffer (the biotin competes with the desthiobiotin from the aptamer conjugate and releases the aptamer–protein hybrid from the streptavidin beads) at room temperature. Trichloroacetic acid precipitation (final concentration 20%) and acetone washing at −20 °C was then used for further purification of the protein fraction from the biotin eluate. The acetone wash was repeated twice, and a pellet became visible after the second wash. This pellet was dissolved in cross-link reversal buffer (20 μL) and incubated at 98 °C for 1 h to reverse the cross-link between the aptamer and the protein, after which the sample was analyzed on an SDS–polyacrylamide electrophoresis gel and stained with an MS-compatible silver stain. Bands of interest were sequenced at the Taplin Mass Spectrometry Facility (Ross Tomaino) at the Harvard Medical School. They were determined by their intensity and by the ease in which they eluted by biotin-mediated elution. The protein hybrid would be eluted more quickly from streptavidin than other proteins. Proteins that bound to the streptavidin beads by nonspecific adsorption would desorb more slowly from the beads, as shown in Figure 1 (lane 7 versus lane 8, e.g., around 50 kDa).
Scheme 1. General Procedure for Protein Identification Using Aptamers.
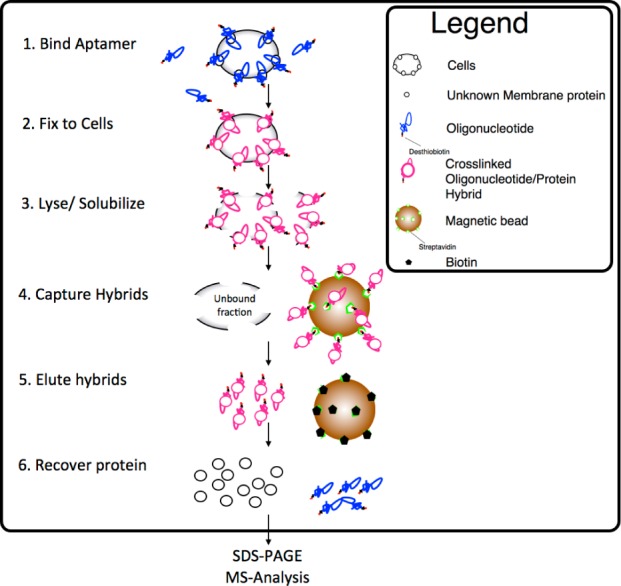
Key: (1) The aptamer is bound to the cells, and excess aptamer is removed with WB. (2) A 1% concentration of formaldehyde is added to the cells and allowed to cross-link for 2 min. Washing in WB dilutes the formaldehyde. (3) The cross-linked cells are homogenized in lysis buffer. (4) Magnetic beads are added, and the aptamer hybrid is captured on the beads. The beads are further washed, as removal of excess membrane significantly reduces the presence of background proteins. (5) The hybrid is eluted from its capture beads by incubation with a 12.5 mM biotin solution. (6) The cross-link is reversed, and the protein fraction is dissolved and separated by SDS–PAGE, after which the differential band is analyzed by mass spectrometry.
Aptamer and Antibody Binding with Flow Cytometry
To determine the binding of the aptamers on cells, the target cells (5 × 105) were incubated with aptamer (250 nM in 100 μL of BB) on ice for 30 min. To determine the binding of the antibodies on cells, the target cells (5 × 105) were incubated with antibody (5 μg in 100 μL of BB) on ice for 30 min. The cells were then washed twice with 500 μL of WB and suspended in 100 μL of WB containing streptavidin–Alexa 488 or streptavidin PE–Cy5.5. Cells were then washed twice with 500 μL of WB and suspended in 200 μL of WB for flow cytometric analysis using a FACScan cytometer (BD Immunocytometry Systems).
siRNA Transfection
Hs_STIP1, Hs_STIP5, Hs_STIP6, Hs_STIP10, and Hs_STIP11 (QAIGEN) siRNAs were used on 0.8 × 105 TOV-21G cells at a concentration of 1 nM with the HiPerfect transfection agent in antibiotic-free medium. Aptamer binding was verified 72 h postsilencing. The transfection efficiency was tested by a cell-death-positive control and a scrambled siRNA negative control to ensure proper cell viability and delivery efficiency, which could be easily verified under the microscope. The RNAi Human/Mouse Starter Kit (QAIGEN) provided these reagents, which were used according to the manufacturer’s recommendations.
STIP1 antibody (50 mg) was incubated with a 20 M excess of sulfo-NHS-biotin reagent, as described in the supplier’s guidelines (Pierce).
Aptamer Blotting
A 20 μg sample of rhSTIP1 or BSA was blotted as described elsewhere.30 In brief, the respective protein was adsorbed on a nitrocellulose membrane (Pierce) and blocked in 4% nonfat milk in PBS containing 0.05% (v/v) Tween 20 and 1 mM EDTA. Biotinylated TOV6 solution (250 nM in PBS) was then incubated on the membrane and washed four times with PBS, after which streptavidin–horseradish peroxidase (Invitrogen, 1:150000 dilution in PBS) was added. The chemiluminescent complex was then visualized with the ECL Plus Western blotting system (GE Healthcare).
Boyden Chamber Invasion Assays
The invasion assays were performed as described elsewhere.31 In brief, the direct invasiveness of the cells was evaluated with the BD Falcon FluoroBlok 24-multiwell insert system (BD Biosciences) precoated with Matrigel. A migration control was obtained on the same system without Matrigel coating (BD Biosciences). The top compartments of both systems was loaded with 60 000 cells per well in minimal medium (RPMI), and the lower compartment was filled with RPMI with 10% FBS (serving as a chemoattractant). Either aptamer TOV6 or 17-AAG was added to the minimal medium, which was prefiltered with 0.2 μm syringe filters (Fisher). The cells were allowed to migrate or invade by incubating the plates overnight in the cell incubator. The cells were labeled with calcein AM (Invitrogen) after the invasion or migration step by injecting the chemical into the lower trans well compartment. (The plates contain a filter that only allows detection of the cells in the lower compartment.) Migrated or invaded cells were read on a VERSAmax tunable microplate reader (Molecular Devices).
Acknowledgments
We thank Ross Tomaino from the Taplin Mass Spectrometry Facility at the Harvard Medical School for the analysis of the protein samples. This work is supported by grants awarded by the National Institutes of Health (GM079359 and CA133086), by the National Key Scientific Program of China (2011CB911000), NSFC grants (NSFC 21221003 and NSFC 21327009) and China National Instrumentation Program 2011YQ03012412.
Supporting Information Available
(Supplemental Scheme 1) chemistry of formaldehyde-mediated DNA–protein cross-linking, (Supplemental Figure 1) study of the effect of formaldehyde on streptavidin–fluorescent dye binding and recovery, (Supplemental Figure 2) optimization of cross-linking time with 1% cross-linking solution, (Supplemental Figure 3) chemiluminescent blot of rhSTIP1, (Supplemental Figure 4) normalized proliferation study of TOV-21G, (Supplemental Table 1) proteins found in aptamer TOV6 binding fractions (78 kDa), (Supplemental Table 2) proteins found in aptamer TOV6 binding fractions (110 kDa), (Supplemental Table 3) proteins found in aptamer TOV6 binding fractions (50 kDa), (Supplemental Table 4) proteins found in aptamer TOV6 binding fractions (46 kDa), (Supplemental Table 5) proteins found in aptamer TOV6 binding fractions (58 kDa), and (Supplemental Table 6) binding of aptamer TOV6 in ovarian cancer cell lines. This material is available free of charge via the Internet at http://pubs.acs.org/.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Barrera N. P.; Robinson C. V. Annu. Rev. Biochem. 2011, 80, 247–71. [DOI] [PubMed] [Google Scholar]
- Van Simaeys D.; López-Colón D.; Sefah K.; Sutphen R.; Jimenez E.; Tan W. PLoS One 2010, 5, e13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sefah K.; Shangguan D.; Xiong X.; O’Donoghue M. B.; Tan W. Nat. Protoc. 2010, 5, 1169–1185. [DOI] [PubMed] [Google Scholar]
- Adachi J.; Kumar C.; Zhang Y.; Olsen J. V; Mann M. Genome Biol. 2006, 7, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surinova S.; Schiess R.; Hüttenhain R.; Cerciello F.; Wollscheid B.; Aebersold R. J. Proteome Res. 2011, 10, 5–16. [DOI] [PubMed] [Google Scholar]
- Hanash S. M.; Pitteri S. J.; Faca V. M. Nature 2008, 452, 571–9. [DOI] [PubMed] [Google Scholar]
- Walsh N.; Larkin A.; Swan N.; Conlon K.; Dowling P.; McDermott R.; Clynes M. Cancer Lett. 2011, 306, 180–9. [DOI] [PubMed] [Google Scholar]
- Yildirim M. A.; Goh K.-I.; Cusick M. E.; Barabási A.-L.; Vidal M. Nat. Biotechnol. 2007, 25, 1119–26. [DOI] [PubMed] [Google Scholar]
- Cibiel A.; Dupont D. M.; Ducongé F. Pharmaceuticals 2011, 4, 1216–1235. [Google Scholar]
- American Cancer Society. Cancer Facts & Figures 2011; American Cancer Society: Atlanta, GA, 2011. [Google Scholar]
- Kosary C.Cancer of the ovary. In SEER Survival Monograph: Cancer Survival among Adults: US SEER Program, 1988–2001, Patient and Tumor Characteristics; Ries L. A. G., Young J. L., Keel G. E., Eisner M. P., Lin Y. D., Horner M.-J., Eds.; National Cancer Institute: Bethesda, MD, 2007. [Google Scholar]
- Anglesio M. S.; Carey M. S.; Köbel M.; Mackay H.; Huntsman D. G. Gynecol. Oncol. 2011, 121, 407–15. [DOI] [PubMed] [Google Scholar]
- Hedin L. E.; Illerg K.; Elofsson A. J. Proteome Res. 2011, 10, 3324–3331. [DOI] [PubMed] [Google Scholar]
- Tan S.; Tan H. T.; Chung M. C. M. Proteomics 2008, 8, 3924–32. [DOI] [PubMed] [Google Scholar]
- Hermann T. Science 2000, 287, 820–825. [DOI] [PubMed] [Google Scholar]
- Pang Z.; Al-Mahrouki A.; Berezovski M.; Krylov S. N. Electrophoresis 2006, 27, 1489–94. [DOI] [PubMed] [Google Scholar]
- Mallikaratchy P.; Tang Z.; Kwame S.; Meng L.; Shangguan D.; Tan W. Mol. Cell. Proteomics 2007, 6, 2230–8. [DOI] [PubMed] [Google Scholar]
- Orlando V.; Strutt H.; Paro R. Methods (San Diego, CA, U.S.) 1997, 11, 205–14. [DOI] [PubMed] [Google Scholar]
- McGhee J. D.; Von Hippel P. H. Biochemistry 1975, 14, 1281–1296. [DOI] [PubMed] [Google Scholar]
- Trepel J.; Mollapour M.; Giaccone G.; Neckers L. Nat. Rev. Cancer 2010, 10, 537–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T.-H.; Chao A.; Tsai C.-L.; Chang C.-L.; Chen S.-H.; Lee Y.-S.; Chen J.-K.; Lin Y.-J.; Chang P.-Y.; Wang C.-J.; Chao A.-S.; Chang S.-D.; Chang T.-C.; Lai C.-H.; Wang H.-S. Mol. Cell. Proteomics 2010, 9, 1873–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanata S. M.; Lopes M. H.; Mercadante A. F.; Hajj G. N. M.; Chiarini L. B.; Nomizo R.; Freitas A. R. O.; Cabral A. L. B.; Lee K. S.; Juliano M. a; de Oliveira E.; Jachieri S. G.; Burlingame A.; Huang L.; Linden R.; Brentani R. R.; Martins V. R. EMBO J. 2002, 21, 3307–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocks B. G.; Theriault T. P. Drug Discovery Today: Targets 2004, 3, 165–171. [Google Scholar]
- Berger S.; Siegert A.; Denkert C.; Köbel M.; Hauptmann S. Cancer Immunol. Immunother. 2001, 50, 328–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eustace B. K.; Sakurai T.; Stewart J. K.; Yimlamai D.; Unger C.; Zehetmeier C.; Lain B.; Torella C.; Henning S. W.; Beste G.; Scroggins B. T.; Neckers L.; Ilag L. L.; Jay D. G. Nat. Cell Biol. 2004, 6, 507–14. [DOI] [PubMed] [Google Scholar]
- Zueva E.; Rubio L. I.; Ducongé F.; Tavitian B. Int. J. Cancer 2011, 128, 797–804. [DOI] [PubMed] [Google Scholar]
- Shin B. K.; Wang H.; Yim A. M.; Le Naour F.; Brichory F.; Jang J. H.; Zhao R.; Puravs E.; Tra J.; Michael C. W.; Misek D. E.; Hanash S. M. J. Biol. Chem. 2003, 278, 7607–16. [DOI] [PubMed] [Google Scholar]
- Eustace B. K.; Jay D. G. Cell Cycle 2004, 1098–1100. [PubMed] [Google Scholar]
- Tsai C. L.; Tsai C.-N.; Lin C.-Y.; Chen H.-W.; Lee Y.-S.; Chao A.; Wang T.-H.; Wang H.-S.; Lai C.-H. Cell Rep. 2012, 2, 283–293. [DOI] [PubMed] [Google Scholar]
- Noma T.; Ikebukuro K.; Sode K.; Ohkubo T.; Sakasegawa Y.; Hachiya N.; Kaneko K. Biotechnol. Lett. 2006, 28, 1377–81. [DOI] [PubMed] [Google Scholar]
- Partridge J.; Flaherty P. J. Visualized Exp. 2009, e1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.