

Abstract
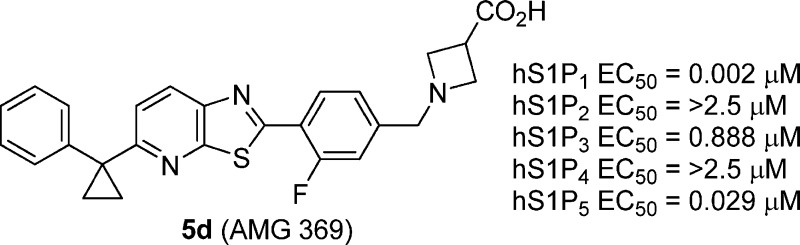
The optimization of a series of thiazolopyridine S1P1 agonists with limited activity at the S1P3 receptor is reported. These efforts resulted in the discovery of 1-(3-fluoro-4-(5-(1-phenylcyclopropyl)thiazolo-[5,4-b]pyridin-2-yl)benzyl)azetidine-3-carboxylic acid (5d, AMG 369), a potent dual S1P1/S1P5 agonist with limited activity at S1P3 and no activity at S1P2/S1P4. Dosed orally at 0.1 mg/kg, 5d is shown to reduce blood lymphocyte counts 24 h postdose and delay the onset and reduce the severity of experimental autoimmune encephalomyelitis in rat.
Keywords: Sphingosine-1-phosphate receptor, S1P1, lymphocyte, inflammation, multiple sclerosis
The lysophospholipid sphingosine-1-phosphate (1, S1P) has emerged as a versatile signaling molecule,1,2 capable of impacting a wide range of cellular functions, including proliferation and apoptosis, differentiation, and migration. The diverse activities of S1P are believed to be mediated through its interaction with the five S1P GPCRs (S1P1−5) encoded by the EDG (endothelial differentiation gene) family as well as possible additional intracellular targets.1−6 The discovery that the potent immunomodulator FTY720 (fingolimod, 2) is phosphorylated in vivo to the monophosphate ester FTY720P,7,8 an agonist of all S1P GPCRs except S1P2, sparked intense research into the functions of the individual S1P receptors (see Figure 1 for structures). From these studies, a consensus emerged that the immunomodulatory effects of FTY720 are primarily due to agonism and subsequent downregulation of the S1P1 receptor by FTY720P, which interrupts the normal pattern of lymphocyte migration and leads to a state of peripheral lymphocyte depletion.3−8 It was also established that, in rodents, agonism of the S1P3 receptor by FTY720P causes a reduction in heart rate9,10 similar to that observed in human subjects.11 These findings, combined with the impressive efficacy of FTY720 in clinical trials for the treatment of transplant rejection12 and multiple sclerosis,13,14 led to a wide-ranging search for S1P1 agonists with limited activity at the S1P3 receptor.15,16 Herein we report the discovery of a dual S1P1/S1P5 thiazolo[5,4-b]pyridine agonist (5d) and describe its in vivo pharmacological and pharmacokinetic characteristics.
Figure 1.

S1P, FTY720, benzothiazole 3a, and optimized thiazolo[5,4-b]pyridine 5d (AMG 369).
Previously, our research team produced a series of potent and selective benzothiazole S1P1 agonists.17 Compound 3a was representative of this series and exhibited double-digit nanomolar potency in an assay measuring receptor internalization (RI) of an hS1P1-GFP fusion protein in U2OS cells, and it exhibited limited activity at hS1P3 as determined by Ca2+ mobilization in hS1P3- and Gq/i5-transfected CHO-K1 cells (Table 1).18 Compound 3a also demonstrated on-mechanism activity in rat, with a single oral dose of 1 mg/kg producing a significant reduction of circulating blood lymphocytes 24 h postdose. However, 3a is characterized by both relatively high lipophilicity (CLogP = 3.9) and low topological polar surface area (tPSA = 53 Å2),19 and therefore, it resides in a region of physicochemical space with a higher probability of off-target toxicity (CLogP > 3, tPSA < 75 Å2).20 Therefore, modifications of 3a were undertaken with the goal of identifying S1P1 agonists with lower CLogP and higher tPSA. Single nitrogen for CH substitutions were identified as a means of lowering CLogP (−1.1 units) and raising tPSA (+12 Å2). The three possible aza-analogues of 3a (3b−3d) arising from nitrogen substitution at each benzothiazole C−H position were investigated (Table 1) and found to exhibit reduced potency (3−10-fold loss vs 3a) in the S1P1 receptor internalization assay. Aza-derivatives (4b−4d) of the isomeric 6-benzyl-benzothiazole 4a were also investigated, and in one case, the substitution of nitrogen for CH resulted in no loss in potency (4d, S1P1 RI EC50 = 0.199 μM) relative to the parent (4a, hS1P1 RI EC50 = 0.221 μM). Of the six aza-analogues, 3b, 3d, and 4d exhibited reasonable S1P1 potency (EC50 = 0.138−0.199 μM) and were selected for further optimization.
Table 1. SAR of Thiazolopyridine Analoguesa.
| compd | X | Y | Z | hS1P1 RI EC50, μM (% efficacy) | hS1P3 Ca2+ EC50, μM (% efficacy) |
|---|---|---|---|---|---|
| 3a | CH | CH | CH | 0.041 (102) | 1.21 (24) |
| 3b | N | CH | CH | 0.144 (106) | 10.1 (63) |
| 3c | CH | N | CH | 0.429 (115) | >25 |
| 3d | CH | CH | N | 0.138 (125) | 2.89 (36) |
| 4a | CH | CH | CH | 0.221 (84) | 3.47 (50) |
| 4b | N | CH | CH | 1.60 (75) | >25 |
| 4c | CH | N | CH | 3.98 (77) | 5.46 (64) |
| 4d | CH | CH | N | 0.199 (92) | 0.478 (94) |
Data represents an average of at least two determinations. % efficacy is reported relative to 1.00 μM 1-(4-(6-benzylbenzofuran-2-yl)-3-fluorobenzyl)azetidine-3-carboxylic acid and 0.200 μM S1P for hS1P1 RI and hS1P3 Ca2+ assays, respectively; >[highest concentration tested] is reported for compounds that do not achieve >10% of control activity. For statistical analysis, see the Supporting Information.
Given the high lipophilicity of the endogenous S1P1 ligand, S1P (1, CLogP = 5.5), we hypothesized that the reduced potency of the aza-analogues could be remedied by addition of small lipophilic groups, with the goal of not exceeding the lipophilicity of 3a (CLogP = 3.9). After extensive exploration of more than seventy analogues of 3b, 3d, and 4d bearing small lipophilic groups, it was found that the α-methylbenzyl analogues of 4d, the thiazolo[5,4-b]pyridines 5a−b (Table 2), showed a 5-fold improvement in S1P1 potency and a 2-fold loss of S1P3 activity relative to the parent 4d. Geminal dimethyl substitution (5c) resulted in comparable S1P1 potency and reduced S1P3 activity. Encouraged by these results, benzylic 1,1-disubstituted carbocycles were investigated (5d−g). Of this series, cyclopropane-containing 5d was found to uniquely exhibit single-digit nanomolar potency at the S1P1 receptor (EC50 = 0.002 μM), with much weaker activity at the S1P3 receptor (EC50 = 0.888 μM). The calculated physicochemical properties of 5d (ClogP = 3.2, tPSA = 65 Å2) were also favorable relative to 3a (ClogP = 3.9, tPSA = 53 Å2). Further testing of 5d in a Ca2+ mobilization format revealed potent agonism of the hS1P5 receptor (EC50 = 0.029 μM, 50% efficacy) and a lack of activity at hS1P2 and hS1P4 receptors at concentrations up to 2.5 μM. The S1P5 activity of 5d was not considered a liability, as it has been suggested that agonism of oligodendrocyte S1P5 receptors by FTY720P may contribute to the efficacy of FTY720 (2) in the treatment of multiple sclerosis.21
Table 2. Optimization of Thiazolo[5,4-b]pyridine Agonistsa.
| compd | Z | R1 | R2 | hS1P1 RI EC50, μM (% efficacy) | hS1P3 Ca2+ EC50, μM (% efficacy) |
|---|---|---|---|---|---|
| 4d | N | H | H | 0.199 (92) | 0.478 (94) |
| 5a | N | Me | H | 0.035 (99) | 0.877 (44) |
| 5b | N | H | Me | 0.038 (87) | 0.996 (45) |
| 5c | N | Me | Me | 0.033 (103) | 2.51 (64) |
| 5d | N | -(CH2)2- | 0.002 (98) | 0.888 (26) | |
| 5e | N | -(CH2)3- | 0.119 (124) | 1.31 (68) | |
| 5f | N | -(CH2)4- | 0.026 (76) | 12% @ 2.5 μM | |
| 5g | N | -(CH2)5- | 0.033 (108) | 0.836 (41) | |
| 6 | CH | -(CH2)2- | 0.038 (93) | 4.67 (56) | |
Data represents an average of at least two determinations. % efficacy is reported as in Table 1. For statistical analysis, see the Supporting Information
The greater than 10-fold difference in S1P1 potency between 5d and both the gem-dimethyl analogue 5c and des-aza cyclopropane analogue 6 (Table 2) suggests that the presence of both cyclopropane and pyridine endow 5d with either a unique conformation or unique interactions with the S1P1 receptor, or both. To further understand the conformational preferences of 5c, 5d, and 6, quantum mechanical calculations22 were performed on fragments corresponding to 5c, 5d, and 6 (Figure 2, A, B, and C, respectively) to characterize the rotational energy profile about the indicated NCCC or CCCC bonds. The profiles for the gem-dimethyl fragment A and cyclopropyl fragment B show striking differences. Fragment A exhibits a fairly shallow profile and global minima at 120 and 240° angles, whereas B exhibits a steep profile greatly favoring a 180° dihedral angle. The des-aza fragment C does not share the profile of B; rather, a slight preference for a 0° dihedral angle is observed. It was further established that, like B, the simple fragment 2-(1-phenylcyclopropyl)pyridine also shows a strong preference for a 180° NCCC dihedral angle (data not shown, see Supporting Information). The unique conformational profile of 2-(1-phenylcyclopropyl)pyridines has, to the best of our knowledge, not been previously described. It may be that the 5d-S1P1 complex that is conducive to efficient receptor internalization contains 5d with a 180° NCCC dihedral angle and, therefore, benefits entropically from ligand preorganization.
Figure 2.
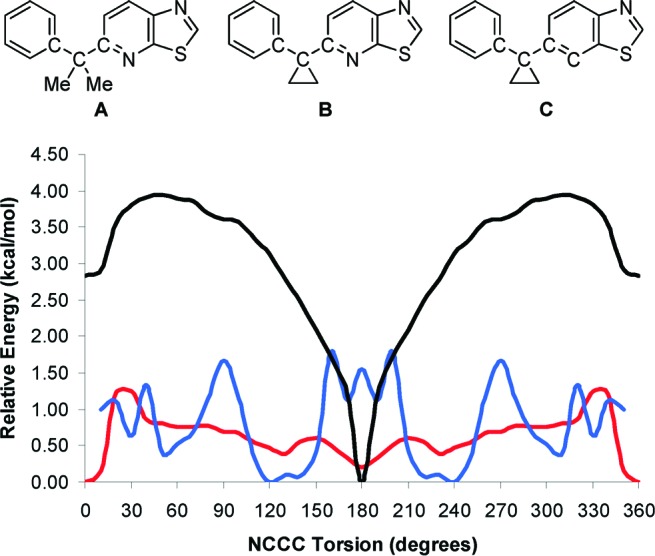
Rotational energy profile of fragments A (blue), B (black), and C (red) corresponding to 5c, 5d, and 6, respectively (B3LYP/6-31G* with Polarizable Continuum Model to simulate aqueous media). Minimization of fragment A at a 0° NCCC dihedral was not possible.
The remarkable in vitro potency of 5d translated to in vivo activity at doses that were more than an order of magnitude lower than had previously been achieved in our program (Figure 3a). Dosed orally at 0.01, 0.03, and 0.1 mg/kg to Lewis rats, 5d produced a dose-dependent reduction in circulating blood lymphocyte counts consistent with S1P1 agonism2 24 h postdose.23 Statistical significance (P < 0.05) was achieved at the 0.03 mg/kg (45% reduction in lymphocytes vs vehicle) and 0.1 mg/kg doses (65% reduction in lymphocytes vs vehicle). Since S1P1 agonists typically produce a maximal 70% reduction in lymphocyte counts,24 the 0.1 mg/kg dose of 5d in the rat provides a nearly maximal pharmacologic effect.25 Compound 5d was further profiled in a rat experimental autoimmune encephalomyelitis (EAE) model which has characteristics of the human disease multiple sclerosis (Figure 3b).26 In this EAE model, Lewis rats are immunized with guinea pig myelin basic protein and scored daily on a 0−6 scale reflecting the degree of disability observed (0 = no sign of disability), with the investigator assessing the clinical score blinded to the study group of the individual rats. The onset of disease typically occurs 10 days postimmunization, with complete resolution of disease at 20 days postimmunization. 5d dosed orally once daily at 20 μg per day (approximately 0.1 mg/kg) at the time of immunization resulted in delayed onset and significant reduction of disease score (Figure 3). A 10-fold lower dose of 5d (2 μg, approximately 0.01 mg/kg) did not significantly impact disease score in this model.
Figure 3.
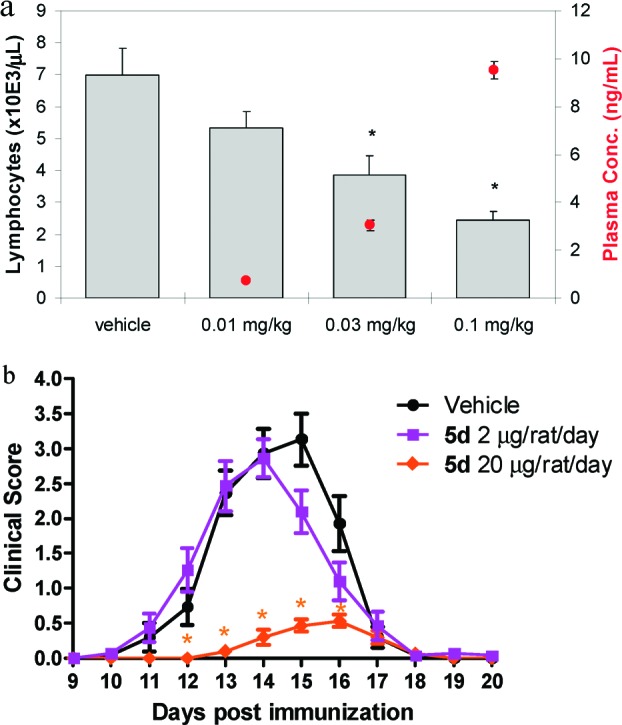
In vivo characterization of 5d. (a) 5d dosed orally reduces blood lymphocyte counts in female Lewis rats 24 h postdose (N = 5/group; bars represent average blood lymphocyte counts + SE; circles represent average plasma concentration ± SE; *P < 0.05 vs vehicle by ANOVA/Dunnett’s Multiple Comparison Test). (b) Efficacy of 5d dosed orally at 2 and 20 μg/rat/day (approximately 0.01 and 0.1 mg/kg, respectively) in delaying onset and reducing disease score in an experimental autoimmune encephalomyelitis (EAE) model in female Lewis rats (N = 15/group; *P < 0.05 vs vehicle by Wilcoxon exact 2-sided test). Vehicle = 20% captisol, pH 2.
Additional profiling established that 5d possesses acceptable characteristics for further development. In rat, nonhuman primate, and dog (Table 3), the compound had low clearance (4−21% of liver blood flow), a moderate steady state volume of distribution of total drug (Vss = 1.2−2.3 L/kg), a moderate to long mean residence time (5−17 h), and acceptable oral bioavailability (34−64%). Cardiovascular safety studies in telemetered rats established a no-effect level for heart rate and mean arterial pressure changes of 10 mg/kg (po), indicating a wide margin for S1P3-associated cardiovascular toxicity. In vitro studies established that 5d was not an inhibitor or inducer of human cytochrome P450 enzymes, was nonmutagenic (Ames and micronucleus negative), and did not significantly inhibit the hERG channel in an electrophysiology assay at the highest concentration tested (2.3 μM).27
Table 3. Pharmacokinetic Parameters for 5d.
| species | Cl (L/(h kg)) | Vss (L/kg) | T1/2 (h) | MRT (h) | % F |
|---|---|---|---|---|---|
| rata | 0.18 | 1.8 | 5.9 | 10 | 64 |
| canineb | 0.071 | 1.2 | 38 | 17 | 41 |
| NHPc | 0.50 | 2.3 | 24 | 4.7 | 34 |
Male Sprague−Dawley (iv: 1 mg/kg, DMSO, N = 3; po: 2 mg/kg, 20% captisol pH 4.0, N = 2).
Male beagle (iv: 1 mg/kg, 20% captisol pH 4.0, N = 3; po: 1 mg/kg, 20% captisol/1% pluronic F68/1% HPMC pH 2.1, N = 3).
NHP = nonhuman primate, male cynomolgus (iv: 1 mg/kg, 20% captisol pH 4.0, N = 3; po: 1 mg/kg, 20% captisol/1% pluronic F68/1% HPMC pH 2.1, N = 3).
The synthesis of 5d is detailed in Scheme 1. Aryl bromide 7, synthesized in one step from the readily available 4-bromo-3-fluorobenzaldehyde, was treated with n-BuLi to effect lithium-halogen exchange, and the resulting anion was allowed to react with 2,6-dichloro-3-isothiocyanatopyridine to give an intermediate thioamide. Treatment of the thioamide with sodium carbonate in DMF at elevated temperature effected cyclization to the thiazolo[5,4-b]pyridine 8. A Suzuki reaction between 8 and 1-phenylvinylboronic acid catalyzed by PdCl2{PtBu2-p-NMe2-Ph}228 provided 9. The cyclopropane was installed with dimethyloxosulfonium methylide under Corey−Chaykovsky conditions29 to give 10, and deprotection of the acetal revealed aldehyde 11. Reductive amination of 11 with methyl azetidine-3-carboxylate hydrochloride provided the penultimate intermediate 12. Saponification of 12 afforded 5d, which was isolated in high yield as its zwitterion.
Scheme 1. Synthesis of 5d.

(a) n-BuLi, 2,6-dichloro-3-isothiocyanatopyridine, THF, −78 °C; (b) Na2CO3, DMF, 90 °C, 83% (two steps); (c) bis(4-(di-tert-butylphosphino)-N,N-dimethylbenzenamine) palladium dichloride (2.5 mol %), 1-phenylvinylboronic acid, K2CO3, dioxane/water, 80 °C, 74%; (d) Me3SOI, tBuOK, DMSO/THF, 75%; (e) 5 N HCl, THF, 65 °C, 93%; (f) methyl azetidine-3-carboxylate hydrochloride, AcOH, DIPEA, NaBH3CN, MeOH/CH2Cl2, 75%; (g) NaOH/THF, then HCl and pH 6 sodium phosphate buffer, 86%.
In conclusion, a thiazolopyridine series of S1P1 agonists was developed with the goal of reducing the lipophilicity and increasing the topological polar surface area of an existing benzothiazole series. The moderate potency of the thiazolo[5,4-b]pyridine series was improved by addition of small lipophilic groups at the benzylic position. The cyclopropyl-containing derivative 5d had improved calculated physicochemical properties, exhibited single-digit nanomolar S1P1 potency with selectivity against S1P2, S1P3, and S1P4, and was capable of reducing blood lymphocyte counts and the severity of experimental autoimmune encephalomyelitis in rat at a dose of 0.1 mg/kg. Molecular modeling experiments revealed an unanticipated preference of the 5-(1-phenylcyclopropyl)thiazolo[5,4-b]pyridine fragment for a rotamer with an NCCC dihedral angle of 180°, which differentiates 5d from related less potent analogues. Additional in vitro and in vivo experiments established that 5d possesses acceptable properties for further development.
Acknowledgments
We thank Ronya Shatila and Beth Tominey for in vivo PKDM studies, Jay Larrow, Bobby Riahi, and Filisaty Vounatsos for large-scale synthesis (7 and methyl azetidine-3-carboxylate hydrochloride), Zheng Hua for chiral separation support, and Srinivasa Chekru, Mercedes Lobera, Yael Marantz, Sharon Shacham, Anurag Sharadendu, Xiang Yu, and Zhaoda Zhang (EPIX Pharmaceuticals).
Supporting Information Available
Statistics for in vitro data presented in Tables 1 and 2. Tabulated data for quantum mechanical calculations for fragments A−C and 2-(1-phenylcyclopropyl)pyridine, as well as a complete ref (22). Experimental procedures and characterization data for 3a−d, 4a−d, 5a−g, and 6. Details of in vitro and in vivo assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Hannun Y. A.; Obeid L. M. Principles of bioactive lipid signaling: lessons from sphingolipids. Nat. Rev. Mol. Cell. Biol. 2008, 9, 139–150. [DOI] [PubMed] [Google Scholar]
- Kihara A.; Mitsutake S.; Mizutani Y.; Igarashi Y. Metabolism and biological functions of two phosphorylated sphingolipids, sphingosine-1-phosphate and ceramide 1-phosphate. Prog. Lipid Res. 2007, 46, 126–144. [DOI] [PubMed] [Google Scholar]
- a Rosen H.; Gonzalez-Cabrera P. J.; Sanna M. G.; Brown S. Sphingosine-1-phosphate receptor signaling. Annu. Rev. Biochem. 2009, 78, 743–768. [DOI] [PubMed] [Google Scholar]
- Marsolias D.; Rosen H. Chemical modulators of sphingosine-1-phosphate receptors as barrier-oriented therapeutic molecules. Nat. Rev. Drug Discov. 2009, 8, 297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huwiler A.; Pfeilschifter J. New players on the center stage: sphingosine-1-phosphate and its receptors as drug targets. Biochem. Pharmacol. 2008, 75, 1893–1900. [DOI] [PubMed] [Google Scholar]
- Brinkmann V. Sphingosine-1-phosphate receptors in health and disease: mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol. Ther. 2007, 115, 84–105. [DOI] [PubMed] [Google Scholar]
- Mandala S.; Hajdu R.; Bergstrom J.; Quackenbush E.; Xie J.; Milligan J.; Thornton R.; Shei G.; Card D.; Keohane C.; Rosenbach M.; Hale J.; Lynch C. L.; Rupprecht K.; Parsons W.; Rosen H. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 2002, 296, 346–349. [DOI] [PubMed] [Google Scholar]
- Brinkmann V.; Davis M. D.; Heise C. E.; Albert R.; Cottens S.; Hof R.; Bruns C.; Prieschl E.; Baumruker T.; Hiestand P.; Foster C. A.; Zollinger M.; Lynch K. R. The immune modulator FTY720 targets sphingosine-1-phosphate receptors. J. Biol. Chem. 2002, 277, 21453–21457. [DOI] [PubMed] [Google Scholar]
- Forrest M.; Sun S.-Y.; Hajdu R.; Bergstrom J.; Card D.; Doherty G.; Hale J.; Keohane C.; Meyers C.; Milligan J.; Mills S.; Nomura N.; Rosen H.; Rosenbach M.; Shei G.-J.; Singer I. I..; Tian M.; West S.; White V.; Xie J.; Proia R. L.; Mandala S. Immune cell regulation and cardiovascular effects of sphingosine-1-phosphate receptor agonists in rodents are mediated via distinct receptor subytpes. J. Pharm. Exp. Ther. 2004, 309, 758–768. [DOI] [PubMed] [Google Scholar]
- Sanna M. G.; Liao J.; Jo E.; Alfonso C.; Ahn M.-Y.; Peterson M. S.; Webb B.; Lefebvre S.; Chun J.; Gray N.; Rosen H. Sphingosine-1-phosphate (S1P) receptor subtypes S1P1 and S1P3, respectively, regulate lymphocyte recirculation and heart rate. J. Biol. Chem. 2004, 279, 13839–13848. [DOI] [PubMed] [Google Scholar]
- Schmouder R.; Serra D.; Wang Y.; Kovarik J. M.; DiMarco J.; Hunt T. L.; Bastien M.-C. FTY720: Placebo-controlled study of the effect on cardiac rate and rhythm in healthy subjects. J. Clin. Pharmacol. 2006, 46, 895–904. [DOI] [PubMed] [Google Scholar]
- Salvadori M.; Budde K.; Charpentier B.; Klempnauer J.; Nashan B.; Pallardo L. M.; Eris J.; Schena F. P.; Eisenberger U.; Rostaing L.; Hmissi A.; Aradhye S. FTY720 versus MMF with Cyclosporine in de novo renal transplantation: a 1-year, randomized controlled trial in Europe and Australasia. Am. J. Transplant. 2006, 6, 2912–2921. [DOI] [PubMed] [Google Scholar]
- Kappos L.; Radue E.-W.; O’Connor P.; Polman C.; Hohlfeld R.; Calabresi P.; Selmaj K.; Agoropoulou C.; Leyk M.; Zhang-Auberson L.; Burtin P. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. New Engl. J. Med. 2010, 362, 387–401. [DOI] [PubMed] [Google Scholar]
- Cohen J. A.; Barkhof F.; Comi G.; Hartung H.-P.; Khatri B. O.; Montalban X.; Pelletier J.; Capra R.; Gallo P.; Izquierdo G.; Tiel-Wilck K.; de Vera A.; Jin J.; Stites T.; Wu S.; Aradhye S.; Kappos L. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. New Engl. J. Med. 2010, 362, 402–415. [DOI] [PubMed] [Google Scholar]
- a Buzard D. J.; Thatte J.; Lerner M.; Edwards J.; Jones R. M. Recent progress in the development of selective S1P1 receptor agonists for the treatment of inflammatory and autoimmune disorders. Expert Opin. Ther. Patents 2008, 18, 1141–1159. [Google Scholar]
- Cooke N.; Zécri F. Sphingosine-1-phosphate type 1 receptor modulators: recent advances and therapeutic potential. Annu. Rep. Med. Chem. 2007, 42, 245–263. [Google Scholar]
- Lanman B. A.; Cee V. J.; Cheruku S. R.; Frohn M.; Golden J.; Lin J.; Lobera M.; Marantz Y.; Muller K. M.; Neira S. C.; Pickrell A. J.; Rivenzon-Segal D.; Schutz N.; Sharadendu A.; Yu X.; Zhang Z.; Buys J.; Fiorino M.; Gore A.; Horner M.; Itano A.; McElvain M.; Middleton S.; Schrag M.; Vargas H. M.; Xu H.; Xu Y.; Zhang X.; Siu J.; Burli R. W.. Discovery of a potent, S1P3-sparing benzothiazole agonist of sphingosine-1-phosphate receptor 1 (S1P1). ACS Med. Chem. Lett., (DOI: 10.1021/ml00228m). [DOI] [PMC free article] [PubMed] [Google Scholar]
- During the course of our S1P1 agonist program, we found that receptor internalization (RI) of an hS1P1-GFP fusion protein in U2OS cells correlated much better to in vivo blood lymphocyte depletion activity than Ca2+ mobilization in hS1P1- and Gq/i5-transfected CHO-K1 cells. Therefore, receptor internalization was used for the main assessment of hS1P1 activity. Mechanistically, receptor internalization may provide a better representation of the receptor downregulation that is believed to be responsible for the inhibition of lymphocyte egress observed with S1P1 agonists. For more information, see ref (6).
- Hughes J. D.; Blagg J.; Price D. A.; Bailey S.; DeCrescenzo G. A.; Devraj R. V.; Ellsworth J.; Wager T.; Whiteley L.; Zhang Y. Physicochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [DOI] [PubMed] [Google Scholar]
- ChemBioDraw Ultra, v. 11.0; Cambridgesoft: 100 CambridgePark Drive, Cambridge, MA 02140. [Google Scholar]
- Brinkmann V. FTY720 (fingolimod) in multiple sclerosis: therapeutic effects in the immune and the central nervous system. Br. J. Pharmacol. 2009, 158, 1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calculations were run with density functional theory at the B3LYP/6-31G* level, and the aqueous medium effects were simulated by the Polarisable Continuum Model as included in the Gaussian 98 suite of programs:Frisch M. J.;et al. Gaussian 98, Revision A.5; Gaussian, Inc.: Pittsburgh, PA, 1998.
- The 24 h time point was ideal, as it reduced the effects of dosing on lymphocyte counts and generally produced a linear relationship between compound concentration in plasma and lymphocyte depletion.
- Hale J. J.; Lynch C. L.; Neway W.; Mills S. G.; Hajdu R.; Keohane C. A.; Rosenbach M. J.; Milligan J. A.; Shei G.-J.; Parent S. A.; Chrebet G.; Bergstrom J.; Card D.; Ferrer M.; Hodder P.; Strulovici B.; Rosen H.; Mandala S. A rational utilization of high-throughput screening affords selective, orally bioavailable 1-benzyl-3-carboxyazetidine sphingosine-1-phosphate-1 receptor agonists. J. Med. Chem. 2004, 47, 6662–6665. [DOI] [PubMed] [Google Scholar]
- This was confirmed in studies exploring higher doses of 5d in Lewis rats.
- Gold R.; Linington C.; Lassmann H. Understanding the pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain 2006, 129, 1953–1971. [DOI] [PubMed] [Google Scholar]
- hERG inhibition was assessed by a manual patch clamp assay (ChanTest, Cleveland, OH, USA: ). [Google Scholar]
- Guram A. S.; King A. O.; Allen J. G.; Wang X.; Schenkel L. B.; Chan J.; Bunel E. E.; Faul M. M.; Larsen R. D.; Martinelli M. J.; Reider P. J. New air-stable catalysts for general and efficient Suzuki−Miyaura cross-coupling reactions of heteroaryl chlorides. Org. Lett. 2006, 8, 1787–1789. [DOI] [PubMed] [Google Scholar]
- Corey E. J.; Chaykovsky M. Dimethyloxosulfonium methylide and dimethylsulfonium methylide. Formation and application to organic synthesis. J. Am. Chem. Soc. 1965, 87, 1353–1364. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.