

Abstract

A novel series of monocarbam compounds exhibiting promising antibacterial activity against multidrug resistant Gram-negative microorganisms is reported, along with the synthesis of one such molecule MC-1 (1). Also reported are structure−activity relationships associated with the in vitro and in vivo efficacy of 1 and related analogues in addition to the hydrolytic stability of such compounds and possible implications thereof.
Keywords: Monocarbam, monobactam, β-lactam, MC-1, siderophore-conjugate, antibiotic
Multidrug resistance among Gram-negative nonfermenters such as Pseudomonas aeruginosa is a significant public health problem.1 Particularly concerning is the growing resistance that these pathogens now exhibit to multiple classes of frontline antibiotics.2 While broad-spectrum antibiotics such as carbapenems and fluoroquinolones still maintain activity against most strains, there is a continual erosion of the effectiveness of members of these classes against multidrug-resistant (MDR) P. aeruginosa.3,4 Combining this fact with results of multiple surveillance studies suggesting that up to 50% of all hospital infections are MDR Gram-negative in nature makes the need for new agents capable of addressing this medical threat readily apparent.5,6 Few new introductions of antibiotics with significantly improved activity against such MDR strains have occurred over the past decade. Modifications of existing classes of antibiotics have proven unsuccessful in providing potent activity against MDR strains that possess multiple resistance mechanisms. Focus in this field has shifted to efforts at finding antimicrobials that enter MDR pathogens by novel mechanisms, thus avoiding many common efflux and low permeability mutations.
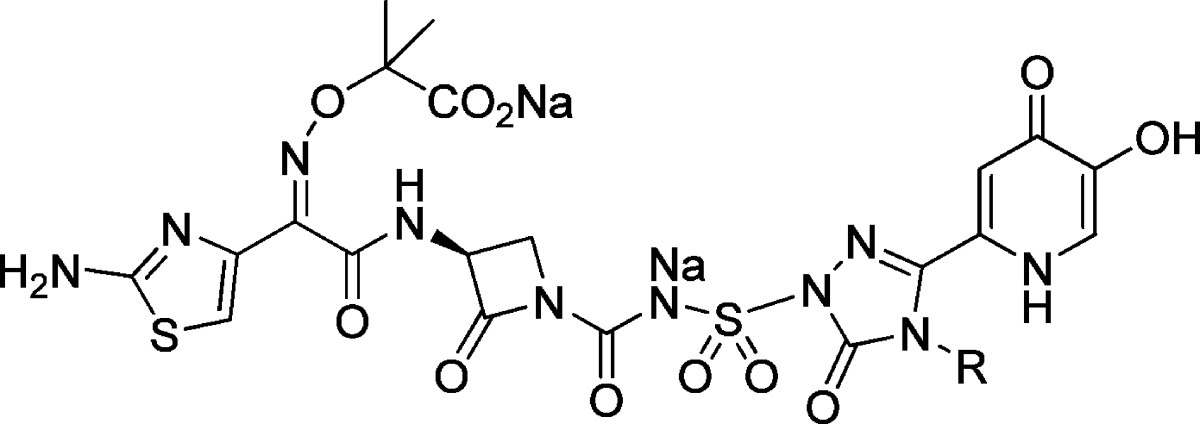
One strategy for circumventing the resistance exhibited by these pathogens involves exploiting the essential iron uptake pathway of these Gram-negative microorganisms by conjugating a siderophore to a β-lactam antibiotic.7−11 This approach was investigated by a number of groups in the 1980s, with siderophore conjugates pursued primarily within the cephalosporin and monobactam β-lactam subclasses.12−15 The Upjohn monocarbam U-78608 (2) is an example of the latter and was used as a comparator agent for these studies (Figure 1).16 Monocarbams are monocyclic β-lactams that are unique from other members of this class, such as aztreonam (3), in that they contain a carbon-centered activating group attached to the β-lactam nitrogen. Siderophore-conjugated monocarbams like 2 and pirazmonam (4)17 were not progressed beyond early development despite impressive in vitro activity. This was likely due to their projected high cost of goods, lack of Gram-positive activity, and insufficient market potential, which at the time was dominated by effective and less structurally complex, broad spectrum β-lactams. Today, however, because of the changing landscape of the Gram-negative arena driven largely by the antibiotic resistance described above, this siderophore conjugate approach has received renewed attention.18 Herein, we report on a novel series of siderophore-conjugated monocarbam analogues and describe the synthesis of one such molecule MC-1 (1), along with structure−activity relationships (SAR) associating physical property attributes with in vivo biological outcomes.
Figure 1.

MC-1 (1) and related monobactam antibiotics.
On the basis of the encouraging in vitro data reported for 2, we initiated a program with this compound as our lead. Early in vitro evaluation of 2 in our hands showed it to be a potent Gram-negative agent, exhibiting a minimum inhibitory concentration (MIC)90 of 2 μg/mL against 91 clinical isolates of P. aeruginosa as compared to >64 μg/mL for 3 and meropenem (Table 3). This robust anti-Pseudomonal activity translated into excellent efficacy in a murine in vivo systemic infection model [intraperitoneally infected (IP infection model), Table 3]. However, somewhat surprisingly, 2 did not perform well in a murine model of respiratory tract infection (RTI PD50 > 150 mg/kg). This was concerning since the lung is considered to be an essential organ in which to demonstrate efficacy for any nosocomial, Gram-negative agent. Reasons for the poor RTI performance exhibited by 2 were not initially apparent; however, subsequent investigation provided possible explanations.
Table 3. In Vitro and in Vivo Results for Study Compounds MC-1 (1) and 25 through 32 along with Comparator Agents Aztreonam (3), Meropenem, and U-78608 (2).
| MIC (μg/mL) |
||||||
|---|---|---|---|---|---|---|
| compd | K. pneumoniae 1000-02 | A. baumannii AB-3167 | P. aeruginosa 1091-05 | MIC90a (μg/mL) P. aeruginosa | IP PD50 (mg/kg) P. aeruginosa 1091-05 (95% CI) | RTI PD50 (mg/kg) P. aeruginosa 1091-05 (95% CI) |
| aztreonam (3) | >64 | 32 | 8 | >64 (n = 138) | >50 | >200 |
| meropenem | <1 | 0.25 | 0.5 | >64 (n = 138) | 1.30 (0.4−2.1) | 1.04 (0.6−1.4) |
| U-78608 (2) | 0.5 | 1 | 0.5 | 2 | 4 (2.4−5.7) | >150 |
| MC-1 (1) | 0.5 | 0.5 | 0.125 | 0.5 (n = 138) | <1 | 20.6 (8.7−32.5) |
| 25 | 0.25 | 1 | 0.125 | 1 | NT | 15.7 (8.45−23.0) |
| 26 | 0.5 | 0.5 | 0.125 | 1 | NT | 18.6 (8.94−28.3) |
| 27 | 0.5 | 1 | 0.125 | 1 | <1 | 32.7 (23.7−42.0) |
| 28 | 1 | 1 | 0.125 | 1 | NT | 25.0 (24.8−25.2) |
| 29 | 0.5 | 4 | 0.5 | 1 | NT | >70.8 |
| 30 | 0.5 | 2 | 0.5 | NT | 2.8 (2.1−3.4) | >100 |
| 31b | 0.5 | 2 | 0.25 | 0.5 (n = 51) | NT | 1.4 (0.9−1.9) c |
| 32 | 1 | 4 | 0.25 | 0.5 (n = 51) | NT | 21.3 (13.0−30.0) |
Ninety-one clinical isolates tested unless otherwise indicated.
PPB for 31, fu-0.51.
Lower inoculum was used [PD50 for 1 was 6.9 mg/kg (5.0−8.8) in this experiment]. NT, not tested.
The hydrolytic instability of these monocarbam antibiotics (discussed in more detail later) presented complications in determining certain end points in assays requiring extended aqueous incubations, such as plasma protein binding (PPB). However, efforts to measure PPB for 2 suggested moderately high binding despite the polar nature of this compound (fu-0.26 in mouse). As a result, we postulated that modulation of the physical properties around 2 might affect PPB, thereby increasing free drug concentrations available for activity in the lung. Because most infections take place within the extravascular space, it is the free concentrations within this compartment that are most relevant for efficacy. Total drug distribution into the lung was generally equivalent for all monocarbam analogues and similar to that observed for aztreonam (lung/plasma ratios of 0.2−0.3).19 While total drug concentrations in tissue homogenates relative to the plasma can provide some indication of penetration, these data are often misleading, since uniform distribution of drug concentrations is assumed. Concentrations observed in tissue homogenates reflect an average of intracellular and extracellular concentrations, as well as drug that is bound to protein. This value could grossly underestimate the actual free drug concentrations in the extravascular space. In the absence of active transport processes, equilibrium between free drug in the tissue and the plasma occurs passively at steady state. This has been demonstrated in microdialysis experiments using several β-lactam antibiotics.20 Cefpodoxime, for instance, is 25% bound to human plasma and demonstrates greater than 2-fold higher peak unbound concentrations in muscle tissue, relative to cefixime (65% PPB). Thus, a strategy to reduce PPB to increase free concentration in the tissue was pursued in an attempt to increase pulmonary efficacy.
Integral to this strategy, was the need to find a position on these molecules to manipulate physicochemical property space, while maintaining adequate in vitro potency. We considered a number of synthetically tractable positions on 2, before settling on the triazalone (methyl) side chain. On the basis of earlier reports, this was an unexplored part of the molecule that at the same time was distal to portions that had previously been optimized, such as the siderophore and aminothiazole-containing substructure (also present in 3). Efforts to incorporate new functionality at this position of the triazalone were facilitated by enablement of the oxadiazalone intermediate 10 (Scheme 1A). This compound was prepared in good overall yield from commercially available kojic acid (5), in six chemical steps. (R)-3-Aminopropane-1,2-diol (11) was introduced in a two-step sequence involving addition of the amine, followed by cyclization to form triazalone 12 (while Scheme 1A is specific for construction of triazalone 12, many other triazalones of interest were prepared from 10 in this or a related manner21). Triazalone 12 was then activated as previously described16 by per-silylation using methylsilyltrifluoroacetamide (MSTFA) in THF (Scheme 1B). Separately, monobactam precursor 16(22) was treated with chlorosulfonylisocyanate in dichloromethane, forming a sulfamoylchloride adduct; this intermediate was then mixed with the activated triazalone 12, resulting in construction of the monocarbam moiety and affording 17 as the product. Stepwise deprotection of 17 and reverse-phase chromatographic purification of 1 was followed by treatment of this material with 2 equiv of sodium bicarbonate in water. Final isolation of the disodium salt of 1 was then accomplished by lyophilization.23
Scheme 1. Representative Monocarbam Synthesis: Preparation of MC-1 (1),

(A) Preparation of the substituted triazalone intermediate 12. Reagents and conditions: (a) Benzylchloride, K2CO3, DMF. (b) CrO3, H2SO4, H2O/acetone, −5 °C to room temperature. (c) NH4OH, H2O, 80 °C. (d) Benzylchloride, K2CO3, DMF, 80 °C. (e) Hydrazine monohydrate, methanol, 65 °C. (f) Carbonyldiimidazole, THF. (g) Compound 11, THF, 60 °C. (h) Aqueous KOH, 100 °C.
(B) Construction of the monocarbam moiety followed by stepwise deprotection leading to 1 disodium salt. Reagents and conditions: (i) H2, 10% Pd/C, toluene/ethanol (assumed quant., used directly in 20% theoretical excess). (j) Toluene/ethanol, 5 h. (k) MSTFA, THF, 12, concentrate at 60 °C. (l) ClSO2NCO, 16, CH2Cl2, 0 °C. (m) Add 12 in fresh THF to 16, 0 °C to room temperature. (n) Pd black, H2, acetic acid, THF. (o) Trifluoroacetic acid, CH2Cl2, 0 °C. (p) NaHCO3 (2 equiv), H2O. (q) Freeze at −80 °C and lyophilize.
Early SARs for compounds prepared in this manner are shown in Table 1. The organism used for screening of MICs24,800 was a virulent (non-MDR) strain of P. aeruginosa (1091-05) also used for in vivo efficacy studies. The SAR data from this initial survey suggested that the binding site was quite forgiving regarding aspects of polarity or moderate size for this triazalone side chain. Larger groups than methyl were accommodated well (e.g., compounds 18 and 19) in addition to compounds with polar functionality such as a basic amine (20) or an alcohol (compounds 21 and 22). The only side chains studied that were not well tolerated were those containing very large groups (data not shown) or those possessing aromatic functionality (e.g., compounds 23 and 24). This SAR is consistent with recent crystallographic studies of 1 bound to penicillin binding protein 3 (PBP3), indicating a relatively large pocket in the protein where this side chain resides.25
Table 1. P. aeruginosa MICs for Initial Triazalone Substituted Monocarbam Analogues.
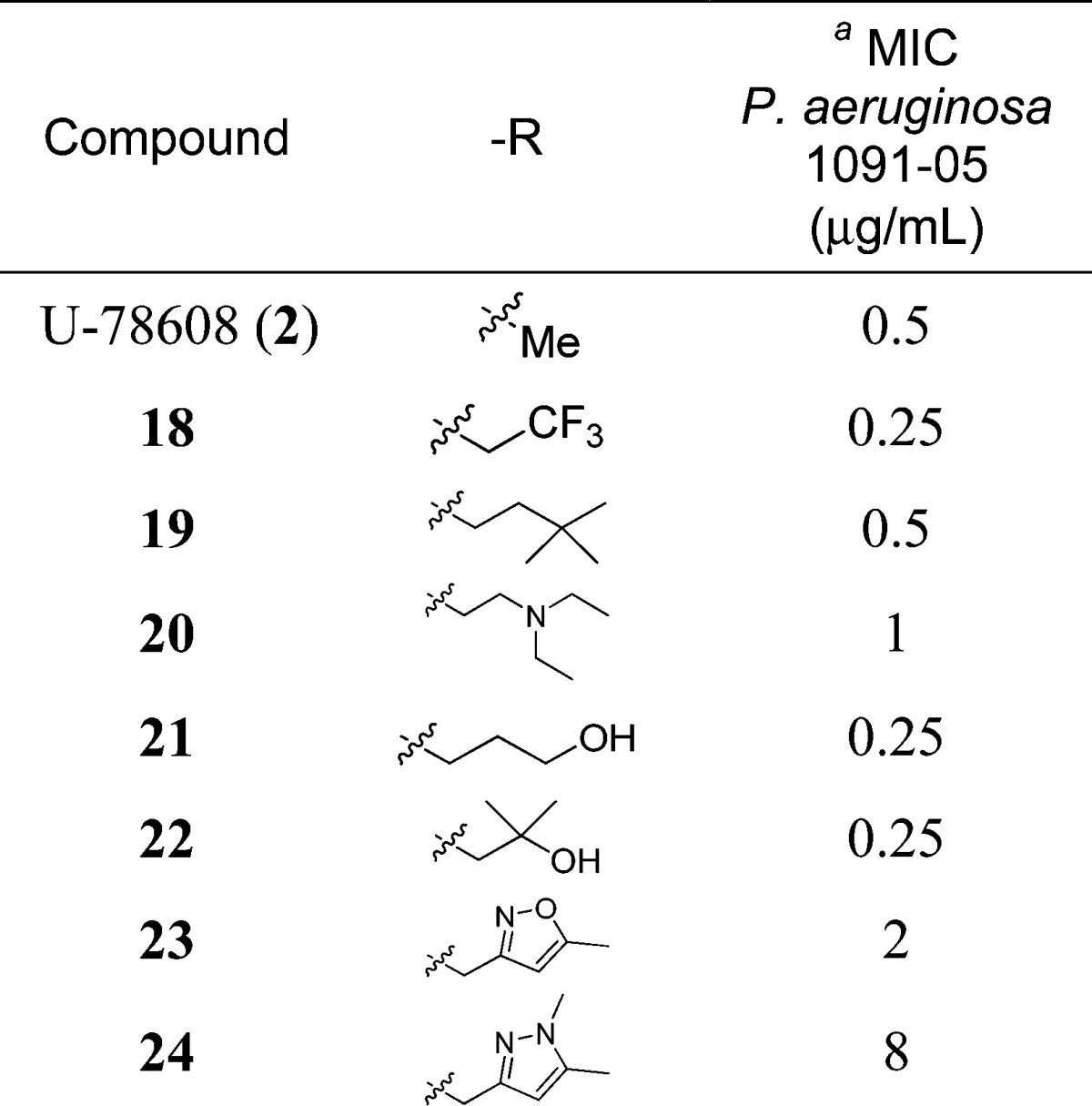 |
For additional MICs, see ref (27).
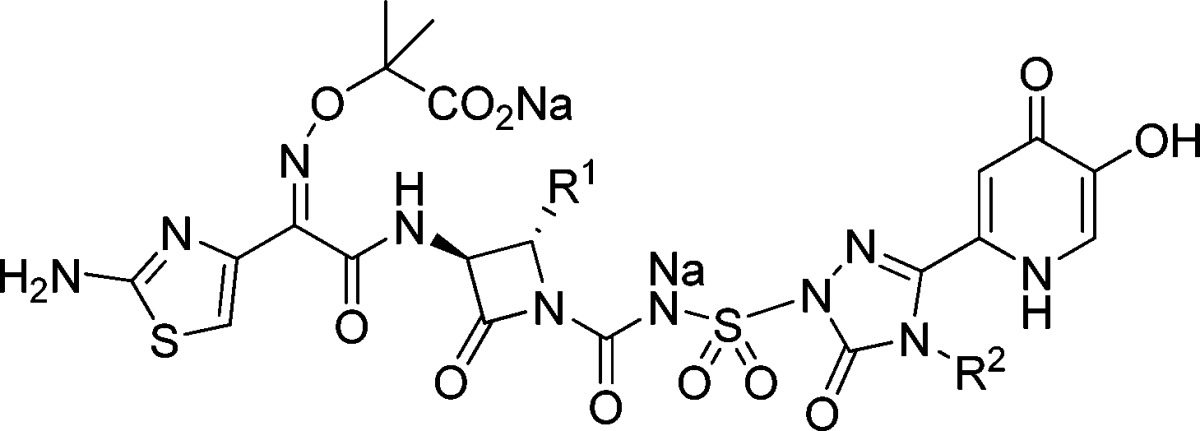
Encouraged by the flexibility in the SAR associated with this side chain, we selected a set of compounds for more extensive evaluation to address the question of triazalone side chain properties as a function of in vivo performance. Because essentially all compounds tested exhibiting good MICs against P. aeruginosa 1091-05 (including 2) also worked well in the IP infection model, we focused instead on RTI performance to differentiate compounds. The compounds selected are shown in Table 2. Compounds 1 and 25−28 all contained polar functionality on this side chain, including at least one hydrogen bond donor. However, the side chains of compounds 29 and 30 were more representative of the methyl group of 2 in terms of their hydrophobicity while still maintaining an overall size comparable to the other compounds being investigated. All of the compounds studied exhibited excellent MICs against an extended spectrum β-lactamase (ESBL)-producing strain of Klebsiella pneumoniae (1000-02) and a recent clinical isolate of Acinetobacter baumannii (AB-3167), in addition to P. aeruginosa (1091-05); furthermore, these compounds also displayed comparable MIC90 values against multiple clinical isolates of P. aeruginosa (Table 3).
Table 2. Additional Compounds Prepared To Study the Role of Physical Properties in RTI Efficacy.
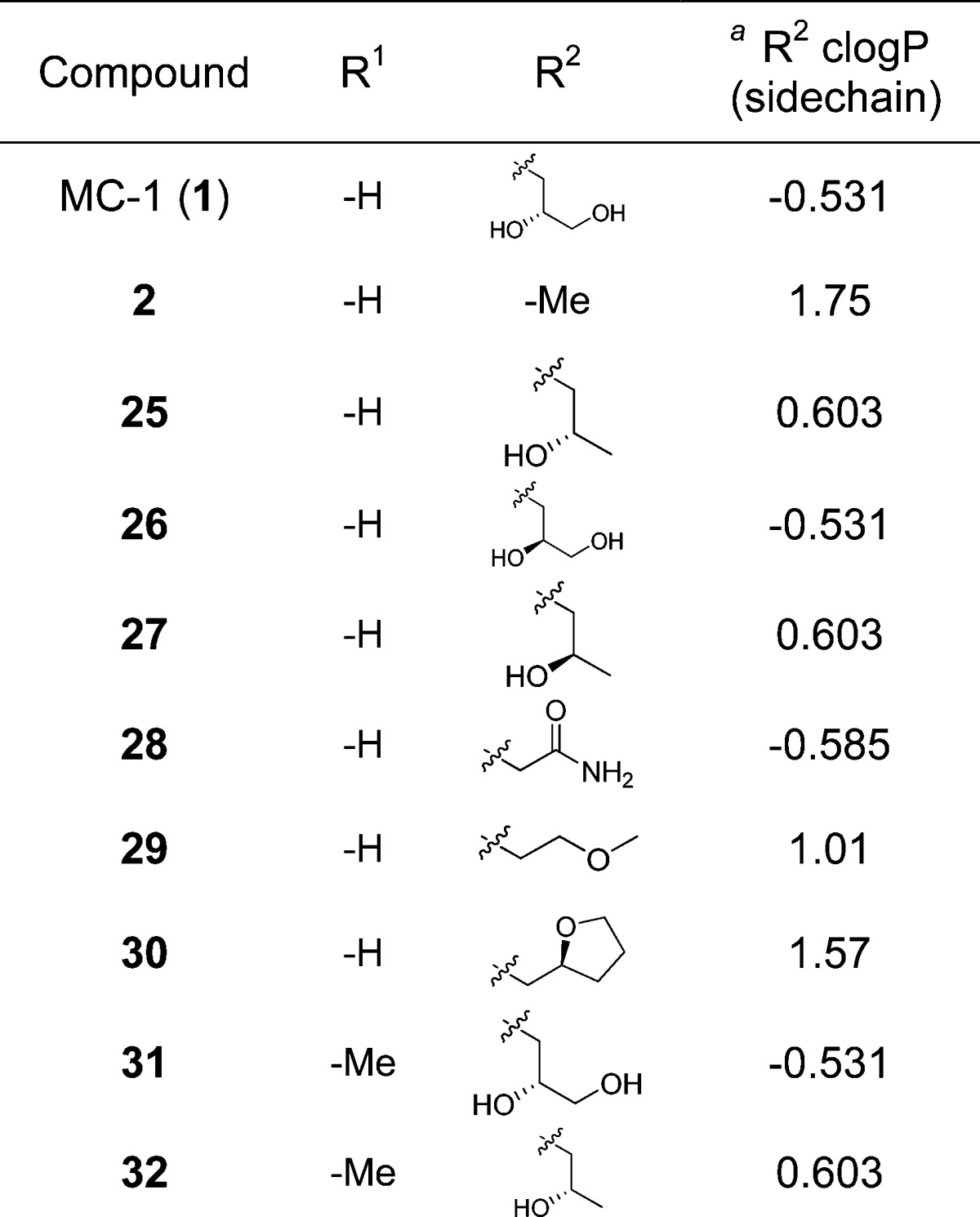 |
R2 side chain clogP values calculated using ChemDraw.
The results of this study confirmed that compounds containing more polar triazalone functionality were superior in terms of their RTI PD50 values. By contrast, compounds 29 and 30 behaved more like 2 in that they did not provide significant RTI protection within the same dose range. These data are consistent with free fraction determinations (PPB), which generally increased with the inclusion of a polar functionality [e.g., fu-0.46 for 1 vs 0.26 for 2 (mouse)]. Somewhat fortuitously, the increase in free fraction did not result in a proportional increase in drug clearance. This is likely due to the fact that drug excretion/clearance for these compounds is not exclusively mediated by glomerular filtration of unbound drug into the urine. Taken collectively, these findings were consistent with the observed RTI PD50 data, suggesting that improved tissue activity is achieved through higher free drug concentrations at the site of infection. Lack of differentiation and very low PD50 values in the IP model were likely a reflection of the high free drug concentrations obtained systemically for all analogues. Combining these results with studies of resistance frequency26 and additional MIC testing, 1 emerged as a compound of interest for further profiling.
As alluded to earlier, one complicating factor associated with studying 1 was its hydrolytic instability. The instability of monocarbams is largely a manifestation of the electron-withdrawing nature of the sulfonylurea activating group. In addition to complicating certain assays requiring longer-term incubations, the instability of the monocarbam moiety also impeded the synthetic scale-up of 1. Despite this obstacle, sufficient compound (1) was produced to conduct additional pharmacology27 (Table 4) and toxicology28 studies. The pH dependence associated with the hydrolytic ring-opening of the β-lactam moiety of 1 was measured, and the results are as follows: at pH 6.0, 1 was relatively stable; however, at pH 8.0, 50% degradation occurred over 100 h at 37 °C (see the Supporting Information, Figure 1). As a consequence, one strategy employed to improve the stability of 1 was to incorporate a methyl group at the C4 position of the β-lactam ring, such as that, which is present on 3. In the case of 3, the primary role for the C4-methyl is reported to be improved stability toward β-lactamases.29 However, other studies also reveal a role for inductive effects of C4-β-lactam substituents in activating (or deactivating) the β-lactam ring toward hydroxide ion.30 To test this hypothesis, compounds 31 and 32 were prepared by an analogous synthetic sequence as that described for 1 (Scheme 1). As can be seen from Table 3, these compounds exhibited comparable in vitro data and in vivo efficacy at least as good as that determined for 1 and 25.31 Compound 31 was evaluated for its hydrolytic stability alongside 1 and meropenem. At pH 6.0 and 37 °C, 31 showed similar stability; however, at pH 8.0, 31 exhibited measurably improved hydrolytic stability as compared to 1 and meropenem32 over the 100 h incubation (∼20% degradation as compared to ∼50% for 1 and meropenem; see the Supporting Information, Figure 1). Studies are ongoing to determine if the nature and magnitude of this stability improvement is sufficient to overcome the obstacles associated with developing a compound of this complexity.
Table 4. Additional MIC50/90 Testing for MC-1 (1) and Comparator Agents Aztreonam (3) and Meropenem.
| MIC50/MIC90 (μg/mL) |
|||
|---|---|---|---|
| organism (no. tested) | MC-1 (1) | aztreonam (3) | meropenem |
| Citrobacter freundii (21) | 0.5/2 | 0.25/64 | 0.06/0.25 |
| Enterobacter aerogenes (21) | 0.125/0.25 | 16/64 | 0.06/0.25 |
| Escherichia coli (79) a | 0.25/2 | 1/>64 | 0.06/0.06 |
| Klebsiella pneumoniae (180) b | 0.5/8 | 64/>64 | 0.06 c/32 c |
| Serratia marcescens (25) | 0.125/0.5 | 0.5/>64 | 0.06/>64 |
| P. aeruginosa (138) d | 0.125/0.5 | 16/>64 | 8/>64 |
| Stenotrophomonas maltophilia (30) | 1/2 | >64/>64 | >64/>64 |
| Acinetobacter spp. (31) | 16/>64 | 32/>64 | 1/32 |
Includes 34 ESBL, 11 CTX-M (cefotaxime active-Munich, ESBL subtype), and 34 recent clinical isolates.
Includes 109 ESBL, 22 K. pneumoniae carbapenemase (KPC), and 49 recent clinical isolates.
N = 98 strains, includes 59 ESBL, 21 KPC, and 18 recent clinical isolates.
Includes 17 cystic fibrosis isolates, 15 metallo-β-lactamase producers, and 106 recent clinical isolates.
In conclusion, we have described the synthesis of novel siderophore-conjugated monocarbams with excellent in vitro and in vivo activity. Such compounds hold promise for the treatment of serious infections caused by MDR pathogens such as P. aeruginosa. Compounds 1, 31, and related analogues exhibited superior protection in an in vivo model of RTI as compared to U-78608 (2). Our data from an analysis of physical property attributes suggest that the polarity and presence of hydrogen bond donors on the triazalone side chain of these monocarbam compounds dramatically affect in vivo outcomes, presumably due to higher free concentrations achieved in the lung. While the hydrolytic instability associated with 1 presents challenges to its development, the impressive in vitro and in vivo activity for this compound warrants further investigation. Efforts to increase stability by incorporating a C4 β-lactam methyl group on 1 (i.e., 31) have shown promise. Consequently, this and other strategies toward addressing this issue are being investigated in our laboratories and will be reported in due course.
Acknowledgments
We are grateful to Dr. Michael Barbachyn, Prof. Alvin Crumbliss (Duke University), Dr. Thomas Magee, Dr. Anthony Marfat, Prof. Marvin Miller (University of Notre Dame), and Prof. Ute Möllmann (Hans Knöll Institute) for many helpful conversations. We also acknowledge David Boyles, Dr. Sally Gut-Ruggeri, and Daniel Widlicka for helpful input on monocarbam syntheses; Glenn Wilcox and Ruchi Thombre for helpful information and for performing separations and isolations of monocarbam products; Jian Lin and Mark Niosi for PPB measurements; and Hongying Gao for bioanalytical analysis.
Glossary
Abbreviations
- MDR
multidrug-resistant
- MIC
minimum inhibitory concentration
- SAR
structure−activity relationships
- fu
fraction unbound
- IP
intraperitoneal
- RTI
respiratory tract infection
- PPB
plasma protein binding
- MSTFA
methylsilyltrifluoroacetamide
- PBP3
penicillin binding protein 3
- ESBL
extended spectrum β-lactamase
- CTX-M
cefotaxime active-Munich ESBL subtype
- KPC
Klebsiella pneumoniae carbapenemase.
This research was sponsored by Pfizer, Inc.
Supporting Information Available
In vivo efficacy assays, protein binding and hydrolytic stability protocols, and cumulative % of susceptibility data for 1, 3, and meropenem, in addition to experimental procedures and characterization data for compounds 1 and 6−32. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Briceno D. F.; Quinn J. P.; Villegas M. V. Treatment Options for Multidrug-Resistant Nonfermenters. Exp. Rev. Anti-Infect. Ther. 2010, 83303–315. [DOI] [PubMed] [Google Scholar]
- Giamarellou H.; Poulakou G. Multidrug-Resistant Gram-Negative Infections: What are the Treatment Options?. Drugs 2009, 69141879–1901. [DOI] [PubMed] [Google Scholar]
- Spellberg B.; Guidos R.; Gilbert D.; Bradley J.; Boucher H. W.; Scheld W. M.; Bartlett J. G.; Edwards J. Jr. The Epidemic of Antibiotic-Resistant Infections: A Call to Action for the Medical Community from the Infectious Diseases Society of America. Clin. Infect. Dis. 2008, 19781079–1081. [DOI] [PubMed] [Google Scholar]
- Boucher H. W.; Talbot G. H.; Bradley J. S.; Edwards J. Jr.; Gilbert D.; Rice L. B.; Scheld M.; Spellberg B.; Bartlett J. Bad Bugs, No Drugs: NO ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 4811–12. [DOI] [PubMed] [Google Scholar]
- Rossolini G. M.; Mantengoli E. Treatment and Control of Severe Infections Caused by Multiresistant Pseudomonas aeruginosa. Clin. Microbiol. 2005, 11Suppl. 417–32. [DOI] [PubMed] [Google Scholar]
- National Nosocomial Infections Surveillance (NNIS) System Report, Data Summary from January 1992 through June 2004, Issued October 2004. Am. J. Infect. Cont. 2004, 32 (8), 470−485. [DOI] [PubMed] [Google Scholar]
- Cornelis P. Iron Uptake and Metabolism in Pseudomonads. Appl. Microbiol. Biotechnol. 2010, 8661637–1645. [DOI] [PubMed] [Google Scholar]
- Girijavallabhan V.; Miller M. J.. Therapeutic Uses of Iron(III) Chelators and their Antimicrobial Conjugates. Iron Transport in Bacteria; American Society for Microbiology: Washington, DC, 2004; pp 413−433. [Google Scholar]
- Hider R. C.; Kong X. Chemistry and Biology of Siderophores. Nat. Prod. Rep. 2010, 27, 637–657. [DOI] [PubMed] [Google Scholar]
- Miethke M.; Marahiel M. A. Siderophore-Based Iron Acquisition and Pathogen Control Microbiol. Mol. Biol. Rev. 2007, 713413–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möllmann U.; Heinisch L.; Bauernfeind A.; Köhler T.; Ankel-Fuchs D. Siderophores as Drug Delivery Agents: Application of the “Trojan Horse” Strategy. Biometals 2009, 22, 615–624. [DOI] [PubMed] [Google Scholar]
- Ogino H.; Iwamatsu K.; Katano K.; Nakabayashi S.; Yoshida T.; Shibahara S.; Tsuruoka T.; Inouye S.; Kondo S. New Aminothiazolylglycylcephalosporins with a 1,5-dihydroxy-4-pyridone-2-carbonyl Group: II Synthesis and Anitibacterial Activity of MT0703 and its Diastereomers. J. Antibiot. 1990, 432189–198. [DOI] [PubMed] [Google Scholar]
- Dolence E. K.; Minnick A. A.; Lin C. E.; Miller M. J.; Payne S. M. Synthesis and Siderophore and Antibacterial Activity of N5-acetyl-N5-hydroxy-l-ornithine-Derived Siderophore-β-Lactam Conjugates: Iron-Transport-Mediated Drug Delivery. J. Med. Chem. 1991, 343968–978. [DOI] [PubMed] [Google Scholar]
- Katsu K.; Kitoh K.; Inoue M.; Mitsuhashi S. In Vitro Antibacterial Activity of E-0702, A New Semisynthetic Cephalosporin. Antimicrob. Agents Chemother. 1982, 22, 181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes R. B.; Koster W. H.; Bonner D. P. The New Monobactams: Chemistry and Biology. J. Clin. Pharmacol. 1988, 282113–119. [DOI] [PubMed] [Google Scholar]
- Barbachyn M. R.; Touminen T. C. Synthesis and Structure-Activity Relationships of Monocarbams Leading to U-78608. J. Antibiot. 1990, 4391199–1203. [DOI] [PubMed] [Google Scholar]
- Ohi N.; Aoki B.; Shinozaki T.; Moro K.; Noto T.; Nehashi T.; Okazaki H.; Matsunaga I. Semisynthetic beta-Lactam Antibiotics. I. Synthesis and Antibacterial Activity of New Ureidopenicillin Derivatives Having Catechol Moieties. J. Antibiot. 1986, 392230–241. [DOI] [PubMed] [Google Scholar]
- Page M. G. P.; Dantier C.; Desarbre E. In Vitro Properties of BAL30072, a Novel Siderophore Sulfactam with Activity against Multiresistant Gram-Negative Bacilli. Antimicrob. Agents Chemother. 2010, 5462291–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita Y.; Fugono T.; Imada A. Comparative Pharmacokinetics of Carumonam and Aztreonam in Mice, Rats, Rabbits, Dogs and Cynomolgus Monkeys. Antimicrob. Agents Chemother. 1986, 29, 127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P.; Muller M.; Derendorf H. Rational Dosing of Antibiotics: The Use of Plasma Concentrations Versus Tissue Concentrations. Int. J. Antimicrob. Agents 2002, 194285–290. [DOI] [PubMed] [Google Scholar]
- See also WO 2010070523.
- Barbachyn M. R.; Brickner S. J.; Thomas R. C. EP 281 289A.
- All final monocarbam products used for in vitro and in vivo experimentation were isolated as disodium salt lyophiles.
- CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard, 8th ed.; CLSI document M07-A8; CLSI: Wayne, PA, 2009. [Google Scholar]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing; Twentieth Informational Supplement; CLSI document M100-S20; CLSI: Wayne, PA, 2010. [Google Scholar]
- Han S.; Zaniewski R. P.; Marr E. S.; Lacey B. M.; Tomaras A. P.; Evdokimov A.; Miller R. J.; Shanmugasundaram V. Structural Basis for Effectiveness of Siderophore-Conjugated Monocarbams Against Clinically Relevant Strains of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 2010, 1075122002–22007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuscript in preparation. See also Lacey B. M.; Aschenbrenner L. A.; McPherson C. J.; Lemmon M. M.; Penzien J. B.; Fahnoe K. C.; Finegan S. M.; Tadakamalla B.; Huband M. D.; O'Donnell J. P.; Miller J. R.; Mueller J. P. Tomaras A. P. De-risking Clinically-Relevant Antibiotic Resistance Mechanisms to MC-1, a Siderophore-Monocarbam Conjugate with Broad-Spectrum Gram-Negative Activity, poster presentation, presented at the 2010. ICAAC Meeting, Boston, MA, paper no. C1-657.
- Manuscript in preparation. See also Huband M. D.; Gootz T. D.; Flanagan M. E.; Quinn J. P.; Mullins L. M.; Penzien J. B.; McCurdy S. P.; Lemmon M. M.; Finegan S. M.; O'Donnell J. P.; George D. M.; Tomaras A. P.; Tadakamalla B. Mueller J. P. In Vitro Antibacterial Activity of MC-1: A New Siderophore-Monocarbam Conjugate Versus Recent Gram-Negative Bacterial Clinical Isolates, poster presentation, presented at the 2010. ICAAC Meeting, Boston, MA, paper no. F1-2129.
- Manuscript in preparation.
- Bush K.; Freudenberger J. S.; Sykes R. B. Interaction of Azthreonam and Related Monobactams with β-Lactamases from Gram-Negative Bacteria. Antimicrob. Agents Chemother. 1982, 223414–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulchande J.; Martins L.; Moreira R.; Archer M.; Oliveira T. F.; Iley J. The Efficiency of C-4 Substituents in Activating the b-Lactam Scaffold Towards Serine Proteases and Hydroxide Ion. Org. Biomol. Chem. 2007, 5162617–2626. [DOI] [PubMed] [Google Scholar]
- The low inoculum used to study 31 in this experiment resulted in a correspondingly low PD50. By contrast, the PD50 generated for 32 was comparable to the other alcohol-containing compounds (1 and 25−27).
- Takeuchi Y.; Sunagawa M.; Isobe Y.; Hamazume Y.; Noguchi T. Stability of a 1β-methylcarbapenem antibiotic, Meropenem (SM-7338) in aqueous solution. Chem. Pharm. Bull. 1995, 434689–692. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.