

Abstract
The serotonin 5-HT3 receptor is a ligand-gated ion channel, which by virtue of its pentameric architecture, can be considered to be an intriguing example of intrinsically multivalent biological receptors. This paper describes a general design approach to the study of multivalency in this multimeric ion channel. Bivalent ligands for 5-HT3 receptor have been designed by linking an arylpiperazine moiety to probes showing different functional features. Both homobivalent and heterobivalent ligands have shown 5-HT3 receptor affinity in the nanomolar range, providing evidence for the viability of our design approach. Moreover, the high affinity shown by homobivalent ligands suggests that bivalency is a promising approach in 5-HT3 receptor modulation and provides the rational basis for applying the concepts of multivalency to the study of 5-HT3 receptor function.
Keywords: Serotonin receptors, 5-HT3, bivalent ligands, multivalency, molecular modeling
Serotonin 5-HT3 receptor (5-HT3R) belongs to the family of the ligand-gated ion channels (LGICs) and shows a pentameric structure similar to that of nicotinic acetylcholine (nACh), glycine, and type A γ-aminobutyric acid (GABAA) receptors.1,1b By virtue of its multimeric architecture, 5-HT3R represents an intriguing example of intrinsically multivalent biological receptors. Initially, a single 5-HT3R subunit (5-HT3A) was cloned, and subsequently, other 5-HT3R subunits (5-HT3B-5-HT3E) have been identified.1,1b The 5-HT3A cDNA forms functional homopentameric cell surface receptors, whereas 5-HT3B was reported to form functional 5-HT3Rs when expressed together with 5-HT3A but not when expressed alone.1,1b The three-dimensional structure of 5-HT3R has not yet been determined at an atomic level, but a large amount of site-directed mutagenesis experiments have been performed to gather information about structure and function,1−2 and the data obtained have been used in the building of 3D receptor homology models.3
The research activity performed by the pharmaceutical companies in the development of 5-HT3R antagonists has produced a large number of drug molecules showing remarkable efficacy in preventing acute chemotherapy-induced nausea and vomiting. Our interest in the development of 5-HT3R ligands started from the study of arylpiperazine derivatives related to quipazine (Scheme 1) and has since produced a considerable amount of information on the interaction of this class of ligands with their receptor.4,4b
Scheme 1. Development of Arylpiperazine-Based Bivalent 5-HT3R Ligands 1 and 2a,b.

In a previous paper, we described both the results of the exploration of the receptor pocket interacting with the c-edge (positions 3 and 4) of the quipazine quinoline nucleus by means of a systematic approach based on lipophilic probes (primarily alkyl chains of different length in compounds 34DSQ) and the development of tacrine-related heterobivalent ligand 1. This compound showed nanomolar potency both for 5-HT3R and for human AChE and represented the first example of a rationally designed high affinity 5-HT3R ligand showing nanomolar AChE inhibitory activity.5 More recently, we reported a noncovalent approach to dynamic multivalent complexes composed of a hydrophobic urea adamantyl-functionalized poly(propylene imine) dendrimer, a solubilizing oligo(ethylene glycol)-based guest molecule, and an arylpiperazine heterobivalent 5-HT3R ligand (2a,b).6 This work provided new perspectives for obtaining dynamic and adjustable noncovalent complexes with pharmacological potential. However, the stability of the supramolecular complexes in the biological environment should be further increased to allow for the in vivo evaluation of the construct. Thus, the exploration of multivalency in 5-HT3R function by means supramolecular constructs involves the preparation of new guest molecules showing an increased interaction strength with the host.
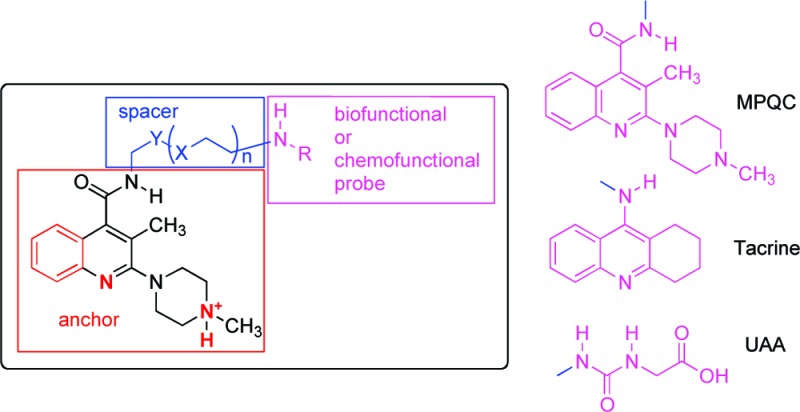
The rational design of tailored bivalent ligands requires a deep understanding of their interaction with the receptor. Therefore, in the present paper, we report on the results of a study focused on the characterization of the interaction of the 5-HT3R with bivalent ligands assembled by (a) an optimized arylpiperazine 5-HT3R ligand (playing the role of an anchor in interacting with the main binding site of the 5-HT3R), (b) a spacer showing different length, and (c) a probe showing different stereoelectronic and functional features. On the basis of their functional features, the probes can be classified into chemofunctional [e.g., the ureido acetic acid (UAA) moiety of heterobivalent guests 2a,b] or biofunctional [a group capable of interacting with receptors or enzymes, e.g., tacrine in heterobivalent ligand 1, or the same arylpiperazine anchor (MPQC) in the corresponding homobivalent ligands] (Figure 1).
Figure 1.
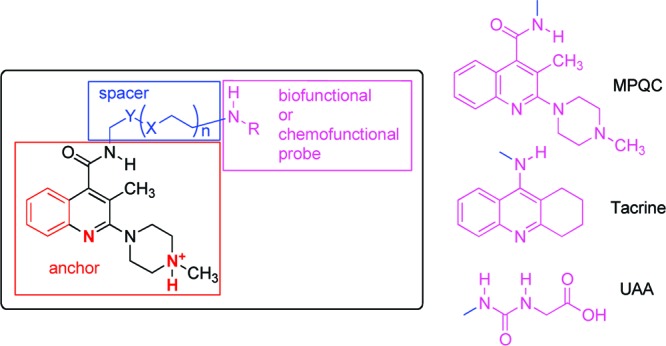
Design of arylpiperazine bivalent ligands 3–5. The elements in red are the pharmacophoric groups, which allow the arylpiperazine moiety (anchor) to interact with the 5-HT3R binding site; magenta indicates the probe responsible for the interaction with a biological target (biofunctional probe, MPQC and Tacrine) or for a particular chemical property (chemofunctional probe, UAA); blue indicates the spacer.
Homobivalent ligands 3a–f were synthesized as depicted in Scheme 2, while the synthesis of the remaining target compounds included in Table 1 is described in the Supporting Information. Key intermediates 9a–f were obtained from acyl chloride 7(5) following two different pathways. When short spacers are involved, compound 7 was reacted with the suitable diamine to give chlorinated homodimers 9a–d, while longer homodimers were obtained starting from amides 8e,f by deprotection and coupling with 7. The nucleophilic substitution with N-methylpiperazine completed the reaction sequence to target homodimers 3a–f.
Scheme 2. Synthesis of Homobivalent Ligands 3a–f.

Reagents: (i) H2N-CH2-Y-(X-CH2CH2)n-NH2, (C2H5)3N, CH2Cl2. (ii) H2N-CH2-Y-(X-CH2CH2)n-NH-COOC(CH3)3, CH2Cl2, (C2H5)3N. (iii) CF3COOH. (iv) CH2Cl2, (C2H5)3N. (v) N-methylpiperazine. Spacers: 3a and 9a: Y = (CH2)2, X = CH2, and n = 1; 3b and 9b: Y = (CH2)3, X = CH2, and n = 1; 3c and 9c: Y = CH2, X = CH2, and n = 2; 3d and 9d: Y = CH2, X = O, and n = 2; 3e, 8e, and 9e: Y = CH2, X = O, and n = 7; 3f, 8f, and 9f: Y = CH2, X = O, and n = 9.
Table 1. 5-HT3R Binding Affinities of Bivalent Ligands 3–5 and Reference Monovalent Ligands 6.
| compd | probea | spacer (met. equiv.)b | Ki (nM)c | RPd |
|---|---|---|---|---|
| 3a | MPQC | –(CH2)6– (6) | 28 ± 7.1 | 10 |
| 3b | MPQC | –(CH2)7– (7) | 2.7 ± 0.7 | 1 |
| 3c | MPQC | –(CH2)8– (8) | 67 ± 15 | 25 |
| 3d | MPQC | –CH2–CH2–O–CH2–CH2–O–CH2–CH2– (8) | 7.7 ± 1.5 | 3 |
| 3e | MPQC | –CH2–CH2–O–(CH2–CH2–O)6–CH2–CH2– (23) | 27 ± 8.2 | 10 |
| 3f | MPQC | –CH2–CH2–O–(CH2–CH2–O)8–CH2–CH2– (29) | 15 ± 2.4 | 5 |
| 4a | tacrine | –(CH2)6– (6) | 71 ± 22 | 13 |
| 4be | tacrine | –(CH2)7– (7) | 5.6f | 1 |
| 4c | tacrine | –(CH2)8– (8) | 69 ± 17 | 12 |
| 5be | UAA | –(CH2)7– (7) | 26g | 1 |
| 5de | UAA | –CH2–CH2–O–CH2–CH2–O–CH2–CH2– (8) | 141g | 5 |
| 5e | UAA | –CH2–CH2–O–(CH2–CH2–O)6–CH2–CH2– (23) | 106 ± 18 | 4 |
| 5f | UAA | –CH2–CH2–O–(CH2–CH2–O)8–CH2–CH2– (29) | 154 ± 21 | 6 |
| 6a | NH-Boc | –(CH2)6– (6) | 178 ± 32 | 10 |
| 6b | NH-Boc | –(CH2)7– (7) | 18 ± 3.1 | 1 |
| 6c | NH-Boc | –(CH2)8– (8) | 127 ± 23 | 7 |
| 6d | NH-Boc | –CH2–CH2–O–CH2–CH2–O–CH2–CH2– (8) | 132 ± 25 | 7 |
| 6e | NH-Boc | –CH2–CH2–O–(CH2–CH2–O)6–CH2–CH2– (23) | 89 ± 18 | 5 |
| 6f | NH-Boc | –CH2–CH2–O–(CH2–CH2–O)8–CH2–CH2– (29) | 71 ± 17 | 4 |
See Figure 1.
Methylene equivalents.
Each value is the mean ± SEM of three determinations and represents the concentration producing half the maximum inhibition of [3H]granisetron (final concentration, 1 nM) specific binding to rat cortical membranes.
Relative potency with respect to the compound bearing the heptamethylene spacer (expressed as the ratio between the Ki value of the compound and that of the analogue showing the same probe and the heptamethylene spacer).
Compounds 4b and 5b,d are cited in the introduction as 1 and 2a,b, respectively.
See ref (5).
See ref (6).
The affinity of compounds 3–6 for the 5-HT3R was measured by means of displacement studies performed with radiolabeled granisetron specifically bound to the 5-HT3R in rat cortical membranes7 (see the Supporting Information), and the results are summarized in Table 1.
The results suggest that the 5-HT3R is capable of accommodating bivalent ligands showing different spacers and probes, which modulate the affinity in a significant manner. Homobivalent ligands 3 are the most potent bivalent ligands in each subgroup characterized by the same spacer, and most notably, they are more potent than the corresponding reference monovalent ligands 6a–f bearing a t-butoxycarbonylamino probe. Moreover, the spacer's features appear to affect the interaction of the different probes. Thus, the heptamethylene tether appears to behave as the optimal spacer (see RP values in Table 1), and either its shortening to the hexamethylene or the elongation to the octamethylene counterparts decreases the 5-HT3R affinity of about 1 order of magnitude in compounds 3 and 4. The replacement of two methylene units in the octamethylene spacers of homobivalent ligand 3c with two oxygen atoms as in 3d produces an increase in potency of about 1 order of magnitude, whereas the spacer elongation leading to 3e,f generates a subtle affinity modulation. When long spacers are considered, a possible fluctuation in the extrareceptorial aqueous environment can be assumed, but the structure–affinity relationships in the compounds 3e,f bearing the longest spacers (23 or 29 methylene equivalents, respectively) appear to suggest that the probe could interact with different environments.
The homology model of the homomeric 5-HT3AR described in a previous paper3 was used as an instrument to check the receptor's ability to accommodate bivalent ligands. The protonated structures of homobivalent ligands with different length spacers (3a–d, see Table 1) were docked into the interface of the 5-HT3A-A dimer. In all cases, the main poses of the ligand's anchor moiety lie within the serotonin binding site and share similar features: The charged head of the piperazine moiety establishes a charged-reinforced H-bond interaction with the Glu129 side chain, the nearby amide NH is hydrogen-bonded to the hydroxyl side chain of Tyr153, and the ligand aromatic moiety establishes π-interactions with Trp183 and Trp90.8−9
On the contrary, the docking poses of the arylpiperazine probe can be clustered into two main groups. The first putative binding site for the probe is located below the known active site, close to the membrane (Figure 2d), and is characterized by both aromatic and polar residues: Tyr73, Tyr88, and Phe130, which last has been proved to be a key residue for receptor function,10 and Ser177, which establishes H-bond interaction with the piperazine ring.
Figure 2.

Docking of compounds 3a–c into the 5-HT3A-A receptor dimer: binding mode of the probe in a pincerlike manner (a), on the receptor surface (b), in the upper extracellular region (c), and close to the membrane (d).
The second putative binding site, instead, was found in the upper extracellular region of the dimer interface (in the opposite direction with respect to the membrane, Figure 2c) and is characterized by Asp189, which forms a charge-reinforced H-bond with the charged head of the piperazine moiety, and polar and aromatic residues, such as Tyr143 and Gln151, whose role in antagonist binding, particularly granisetron, was previously proven by single-point mutagenesis experiments.8b,11,12
Although all of the studied homobivalent ligands can dock into these two putative binding pockets, they show binding peculiarities worthy of note. The heptamethylene spacer ligand (3b), in its most frequent low energy poses, shows a stretched out spacer, and its probe is found to be lying into one of the two putative probe binding pockets (Figure 2c,d). These poses may indicate the presence of a secondary or allosteric binding site, as previously pointed out by Barbosa et al.,3 which could explain the greater affinity of this homobivalent ligand for the 5-HT3R. On the contrary, the homobivalent ligand with the shortest hexamethylene spacer (3a) prefers the docking pose where both of the cationic amines of the piperazine moieties interact with Glu129 in a pincerlike manner (Figure 2a), while the ligand with the longer octamethylene spacer (3c) prefers a pose where the probe extends outside the receptor surface (Figure 2b). The molecular dynamic simulation performed on this latter docking pose of 3c shows that the probe progressively moves from the receptor interface toward a polar surface area near loop C provoking a partial unfolding of the loop (mainly due to the formation of persistent H-bonds between the probe and the side chains of Ser227 and Glu225 on the receptor surface). This might be responsible for the misplacement of the anchor moiety, which moves away from Glu129 (a key residue for the agonist/antagonist binding), preventing the formation of the important charged-reinforced H-bond interaction, thus explaining the lower affinity of compound 3c, which is 25 times less active than 3b. Finally, it is worth noting that the docking poses of the homobivalent ligand 3d are different from those of 3c, which is characterized by the same spacer length in methylene equivalents. In the most populated low energy clusters, the orientation of compound 3d is very similar to that of the heptamethylene spacer ligand (3b); therefore, the probe does not extend outside the receptor surface but lies into one of the two putative probe binding pockets (Figures 2c,d). In fact, the replacement of two methylene units in the octamethylene spacers of homodimer 3c with two oxygen atoms in the homodimer 3d stabilizes the complex by favoring the interactions between the oxygen atoms in the spacer and the interface residues. These results could justify the significant increase in potency (about 1 order of magnitude) of the homobivalent ligand 3d with respect to compound 3c.
The results of the molecular dynamics simulations suggest the possibility that the probe of homobivalent ligands with longer spacers (e.g., 23 or 29 methylene equivalents) reaches other binding sites in the same receptor. To check this hypothesis, the compound with the longest spacer (3f, see Table 1) was manually docked into the 5-HT3R dimer: Although the spacer was forced to adopt an unrealistic all-extended conformation, the probe could not reach the adjacent binding interface. However, the probe of these long bivalent ligands may either intercept active sites of other receptors or dock into other probable binding pockets on the same receptor surface.
In the attempt to discover new possible binding sites, rigid docking of the probe was performed on the whole receptor surface. The results showed the presence of three main surface areas capable of accommodating the arylpiperazine probe (Figure 3).
Figure 3.

Docking of the arylpiperazine probe on the 5-HT3A receptor surface: charged surface of the putative sites in the extracellular receptor portion (a), above the loop C (b), and near the membrane (c).
The main pose of the probe is found in the extracellular portion of the receptor, near the channel entrance, opposite to the membrane (Figure 3a). Here, the probe occupies a pocket that shows features very similar to those of the serotonin site. In fact, the main residues involved in ligand binding are aromatic or negatively charged: Asn49 (whose carboxylic group is hydrogen-bonded to the cationic head of the piperazine ring), Glu98 (whose side chain forms a H-bond interaction with the ligand amino group), Tyr50, Phe99, and Trp102 (which stabilize the complex interacting with the aromatic group).
The second putative surface site shows different features. It is a polar pocket (Pro63, Ile187, Asn191, and Gln188) surrounded by a few positively charged residues: Arg 61, Arg196 (whose guanine group is hydrogen-bonded to the cationic head of the piperazine ring), and Lys62 (which forms a H-bond with the ligand carbonyl group, Figure 3b). This secondary site is localized in a surface region just above loop C, very near the active site; therefore, it may be exploited by bivalent ligands with a relatively short spacer, as in the case of compound 3c discussed above. In addition, as this site is positively charged, it may easily accommodate negative chemofunctional probes, such as the deprotonated ureido acetic moiety of compounds 5e,f. Finally, the least important pose shows the ligand probe lying in a surface pocket near the membrane formed by Ile242, Asn175, Asp172, Val173, Tyr167, and Gln174 (Figure 3c). Therefore, depending on the spacer length, a bivalent ligand probe may interact with one of the putative sites on the surface or reach other receptors and act as a biofunctional or chemofunctional ligand on the basis of its stereoelectronic and functional features.
In conclusion, the study of the interactions of the 5-HT3R with arylpiperazine ligands led to conceive a design approach to bivalent ligands in which an arylpiperazine moiety is linked by means of a spacer to a probe showing different functional (chemofunctional or biofunctional) features. The high 5-HT3R affinity shown by homobivalent and heterobivalent ligands provides evidence of the viability of our approach, which can be considered of broad applicability in the design of bivalent ligands tailored for specific applications. Moreover, the existence on the receptor surface of three potential accessory binding sites for the arylpiperazine moiety shown by the molecular modeling studies implies that multivalency in 5-HT3R could involve receptor domains different from the main binding site. The high affinity shown by homobivalent ligands 3a–f suggests that bivalency is a promising approach in 5-HT3R modulation. Because bivalency can be considered the first step of multivalency, the results obtained provide the rational basis for the application of the concepts of multivalency to the study of 5-HT3R function.
Acknowledgments
Prof. Stefania D'Agata D'Ottavi's careful reading of the manuscript is acknowledged.
Supporting Information Available
Experimental details for the synthesis and the characterization of compounds 3–5 and their intermediates and the computational methods and the experimental procedures used in binding assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Thanks are due to Italian MIUR for financial support.
Supplementary Material
References
- Thompson A. J.; Lummis S. C. R. 5-HT3 Receptors. Curr. Pharm. Des. 2006, 12, 3615–3630and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannon J.; Hoyer D. Molecular Biology of 5-HT Receptors. Behav. Brain Res. 2008, 195, 198–213. [DOI] [PubMed] [Google Scholar]
- Thompson A. J.; Lummis S. C. R. The 5-HT3 Receptor as a Therapeutic Target. Expert Opin. Ther. Targets 2007, 11, 527–540and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moura Barbosa A. J.; De Rienzo F.; Ramos M. J.; Menziani M. C. Computational Analisys of Ligand Recognition Sites of Homo- and Heteropentameric 5-HT3 Receptors. Eur. J. Med. Chem. 2010, 45, 4746–4760and references therein. [DOI] [PubMed] [Google Scholar]
- Cappelli A.; Anzini M.; Vomero S.; Mennuni L.; Makovec F.; Hamon M.; De Benedetti P. G.; Menziani M. C. The Interaction of 5-HT3 Receptor with Arylpiperazine, Tropane, an Quinuclidine Ligands. Curr. Top. Med. Chem. 2002, 2, 599–624. [DOI] [PubMed] [Google Scholar]
- Cappelli A.; Butini S.; Brizzi A.; Gemma S.; Valenti S.; Giuliani G.; Anzini M.; Mennuni L.; Campiani G.; Brizzi V.; Vomero S. The Interactions of the 5-HT3 Receptor with Quipazine-Like Arylpiperazine Ligands. The Journey Track at the End of the First Decade of the Third Millennium. Curr. Top. Med. Chem. 2010, 10, 504–526. [DOI] [PubMed] [Google Scholar]
- Cappelli A.; Gallelli A.; Manini M.; Anzini M.; Mennuni L.; Makovec F.; Menziani M. C.; Alcaro S.; Ortuso F.; Vomero S. Further Studies on the Interaction of the 5-hydroxytryptamine3 (5-HT3) Receptor with Arylpiperazine Ligands. Development of a New 5-HT3 Receptor Ligand Showing Potent Acetylcholinesterase Inhibitory Properties. J. Med. Chem. 2005, 48, 3564–3575. [DOI] [PubMed] [Google Scholar]
- Galeazzi S.; Hermans T. M.; Paolino M.; Anzini M.; Mennuni L.; Giordani A.; Caselli G.; Makovec F.; Meijer E. W.; Vomero S.; Cappelli A. Multivalent Supramolecular Dendrimer-Based Drugs. Biomacromolecules 2010, 11, 182–186. [DOI] [PubMed] [Google Scholar]
- Nelson D. R.; Thomas D. R. [3H]-BRL 43694 (Granisetron), a Specific Ligand for 5-HT3 Binding Sites in Rat Brain Cortical Membranes. Biochem. Pharmacol. 1989, 38, 1693–1695. [DOI] [PubMed] [Google Scholar]
- Beene D. L.; Brandt G. S.; Zhong W.; Zacharias N. M.; Lester H. A.; Dougherty D. A. Cation-pi Interactions in Ligand Recognition by Serotonergic (5-HT3A) and Nicotinic Acetylcholine Receptors: the Anomalous Binding Properties of Nicotine. Biochemistry 2002, 41, 10262–10269. [DOI] [PubMed] [Google Scholar]
- Price K. L.; Lummis S. C. R. The Role of Tyrosine Residues in the Extracellular Domain of the 5-Hydroxytryptamine3 Receptor. J. Biol. Chem. 2004, 279, 23294–23301. [DOI] [PubMed] [Google Scholar]
- Brejc K.; van Dijk W. J.; Klaassen R. V.; Schuurmans M.; van Der Oost J.; Smit A. B.; Sixma T. K. Crystal Structure of an ACh-Binding Protein Reveals the Ligand-Binding Domain of Nicotinic Receptors. Nature 2001, 411, 269–276. [DOI] [PubMed] [Google Scholar]
- Price K. L.; Bower K. S.; Thompson A. J.; Lester H. A.; Dougherty D. A.; Lummis S. C. R. A Hydrogen Bond in Loop A is Critical for the Binding and Function of the 5-HT3 Receptor. Biochemistry 2008, 47, 6370–6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataraman P.; Joshi P.; Venkatachalan S. P.; Muthalagi M.; Parihar H. S.; Kirschbaum K. S.; Schulte M. K. Functional Group Interactions of a 5-HT3R Antagonist. BMC Biochem. 2002, 3, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.