

Abstract

Multiple CRTH2 antagonists are currently evaluated in human clinical trials for asthma and chronic obstructive pulmonary disease (COPD). During our lead optimization for CRTH2 antagonists, an observation of an intramolecular hydrogen bond in ortho-phenylsulfonamido benzophenone derivatives led to the design and synthesis of conformationally constrained benzodiazepinones as potent CRTH2 antagonists. The benzodiazepinones are 2 orders of magnitude more potent than the original flexible bisaryl ethers in our binding assay. Selected benzodiazepinones, such as compound 6, were also potent in the human eosinophil shape change assay. Analysis of the rigid conformations of these benzodiazepinones and ortho-phenylsulfonamido benzophenones provided an explanation for the structure–activity relationship and revealed the possible bound conformations to CRTH2, which may be useful for building a pharmacophore model of CRTH2 antagonists.
Keywords: CRTH2, PGD2, antagonist, asthma, benzodiazepinones, pharmacophore
CRTH2 (chemoattractant receptor-homologous molecule expressed on Th2 cells), also known as DP2, is a G-protein-coupled receptor, which has received increasing attention as a promising new target for the treatment of asthma and other allergic diseases.1−5 CRTH2 is selectively expressed on Th2 cells, T cytotoxic type 2 (Tc2) cells, eosinophils, and basophils.6−8 Its endogenous ligand is prostaglandin D2 (PGD2), which plays a key role in mediating allergic reactions.9,10 Stimulation of CRTH2 by PGD2 mediates multiple inflammatory responses, such as chemotaxis of eosinophils, basophils, and Th2 cells, eosinophil activation and degranulation, cytokine production from Th2 cells, and leukotriene production by mast cells.11−16 Therefore, blockade of CRTH2 has the potential to be beneficial in the treatment of allergic diseases triggered by PGD2.
Multiple research groups have disclosed their efforts in identifying CRTH2 antagonists.17−23 We also reported the discovery of phenylacetic acid derivatives as CRTH2 and DP dual antagonists and the optimization that led to the identification of AMG 009 and AMG 853.24,25
During the optimization of our phenylacetic acid derivatives, such as compounds 1 and 3, the bisaryl ether linker was studied (Table 1). One of the linker replacements explored was carbonyl. The resulting ortho-phenylsulfonamido benzophenones (2 and 4) were significantly more potent than the corresponding bisaryl ethers (1 and 3), as assessed by 3H-PGD2 displacement from the CRTH2 receptors expressed on HEK 293 cells.26 The 1H NMR spectra of the ortho-phenylsulfonamido benzophenones indicate the existence of an intramolecular hydrogen bond between the sulfonamide NH and the carbonyl oxygen, because the proton signal of the sulfonamide NH is shifted downfield by 2–3 ppm in CDCl3 and by 1–1.5 ppm in DMSO-d6 as compared to the sulfonamide NH of the bisaryl ethers. It is likely that the intramolecular hydrogen bond in compounds 2 and 4 locks these molecules into a desired shape for binding to the CRTH2 receptor. Even though the methoxy next to the carbonyl may also contribute to the improvement of the potency, its contribution should be small based on the structure–activity relationship (SAR).24
Table 1.
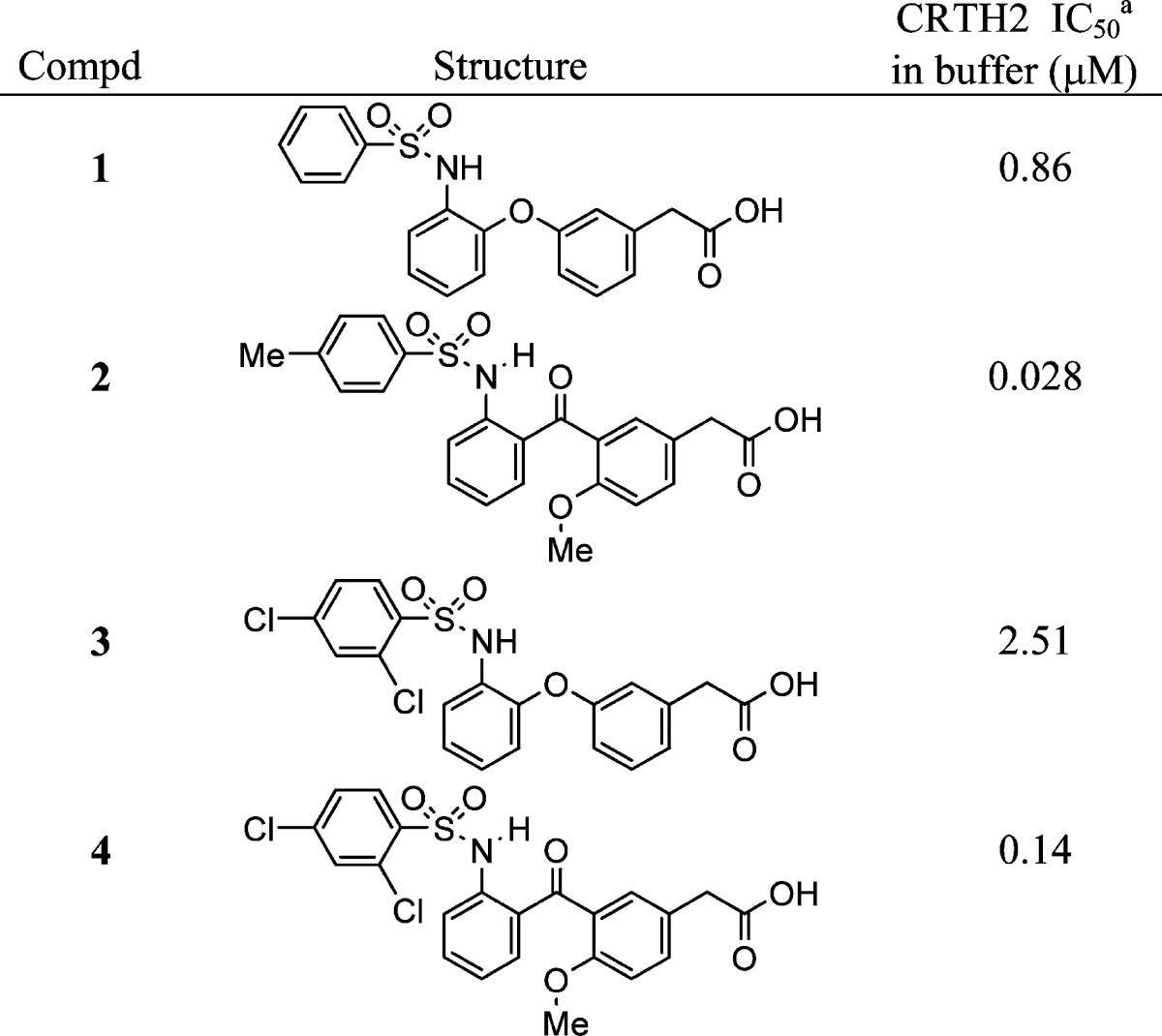 |
Displacement of 3H-PGD2 from the CRTH2 receptors expressed on HEK 293 cells. Assay run in buffer containing 0.5% bovine serum albumin. See ref (26) for assay protocol. Values are the means of three experiments, and the standard deviation is ±30%.
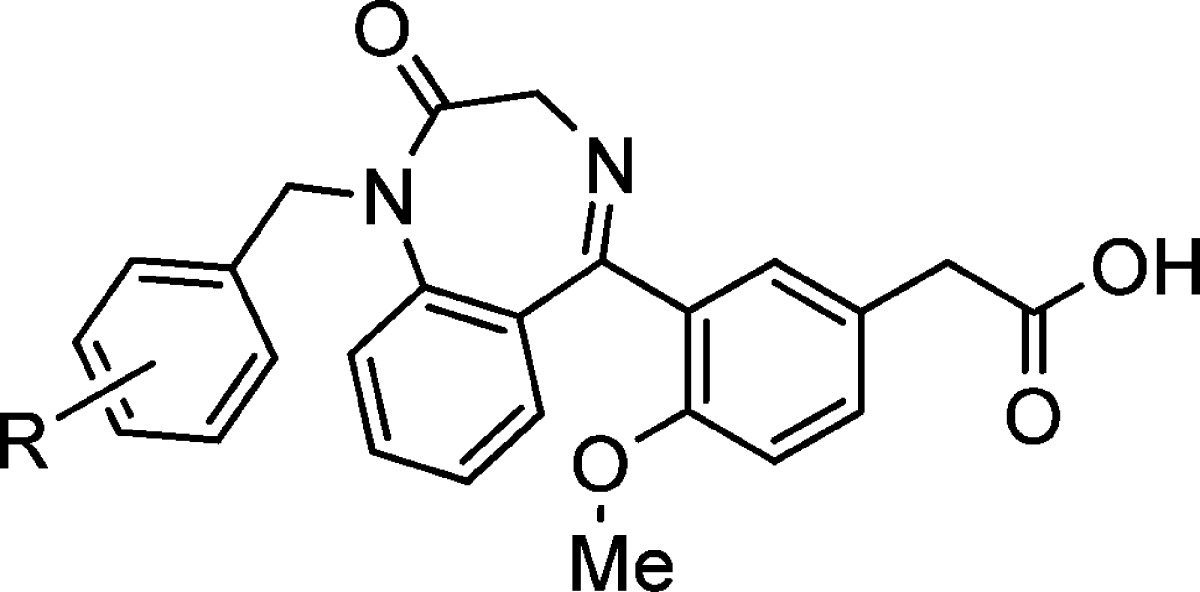
Constraining flexible molecules to adopt desired binding conformations is a very attractive tactic in lead optimization, because it can improve binding affinity without increasing molecule size. The observation of the intramolecular hydrogen bond and improved potency encouraged us to design constrained molecules based on the conformation locked by the intramolecular hydrogen bond. Benzodiazepinone derivatives were among the molecules designed (Figure 1), and the benzodiazepinones were conveniently synthesized from the same intermediate for the preparation of the ortho-phenylsulfonamido benzophenones (Schemes 1 and 2).
Figure 1.

Compound design based on intramolecular hydrogen bond.
Scheme 1.

Scheme 2.

Compounds 1 and 3 were prepared following our previously reported route.24,25 The syntheses of compounds 2 and 4 are shown in Scheme 1. Frieldel–Crafts reaction of 2-nitrobenzoic acid chloride with 4-methoxyphenylacetic acid methyl ester yielded the 2-nitrobenzophenone, which was reduced to the 2-aminobenzophenone using iron. Sulfonamide formation followed by ester hydrolysis afforded compounds 2 and 4.
The benzodiazepinone derivatives (5–13) were prepared from the same 2-aminobenzophenone intermediate in Scheme 1. Reaction of the 2-aminobenzophenone with glycine in pyridine gave the benzodiazepinone (Scheme 2), which was alkylated with various substituted benzyl bromides. Finally, the hydrolysis of the methyl esters provided compounds 5–13.
The rigid benzodiazepinone derivatives were found to be much more potent than the flexible compounds (Table 2). The benzodiazepinone with an unsubstituted benzyl group (5) had a CRTH2 binding IC50 of 5 nM in buffer and 60 nM in the presence of 50% plasma. Fluorine and chlorine at the para position of the benzyl group (6 and 8) further improved the potency, especially in the presence of plasma. Compound 6 had an IC50 of 9 nM in 50% plasma. More bulky substituents at the para position of the benzyl group, such as trifluoromethyl (12) and trifluoromethoxy (13), decreased the binding affinity. Ortho substitution was found to be detrimental to the potency, as indicated by compound 10. All of these benzodiazepinone derivatives (5–13) are selective for the CRTH2 receptor over the prostanoid D receptor (DP or DP1), the other GPCR for PGD2 (DP binding IC50 >10 μM).
Table 2.
| CRTH2 IC50a |
|||
|---|---|---|---|
| compd | R | in buffer (μM) | in plasma (μM) |
| 5 | H | 0.005 | 0.060 |
| 6 | 4-F | 0.003 | 0.009 |
| 7 | 3,4-F | 0.006 | 0.015 |
| 8 | 4-Cl | 0.007 | 0.022 |
| 9 | 3-Cl | 0.008 | 0.078 |
| 10 | 2,4-Cl | 0.029 | 0.24 |
| 11 | 4-Me | 0.015 | 0.104 |
| 12 | 4-CF3 | 0.030 | ND |
| 13 | 4-OCF3 | 0.070 | ND |
Displacement of 3H-PGD2 from the CRTH2 receptors expressed on HEK 293 cells. Assay run in buffer containing 0.5% bovine serum albumin or in 50% plasma. See ref (26) for assay protocol. Values are the means of three experiments, and the standard deviation is ±30%.
While constraining the molecule conformation in the form of benzodiazepinone resulted in dramatic improvement of the potency, in the form of quinazolinone, it decreased the binding potency (Figure 2). Quinazolinone 14 had a CRTH2 binding IC50 of 11 μM in buffer. This interesting SAR prompted us to analyze the rigid conformations to seek an explanation.
Figure 2.

Compound 14.
The strong binding of benzodiazepinone 6 to the CRTH2 receptor indicates that one of its low energy conformations may be close to the bound conformation to the CRTH2 receptor. This bound conformation should also overlap with one of the low energy conformations of ortho-phenylsulfonamido benzophenone 2 if they share a binding pocket. It was gratifying to see the calculated lowest energy conformation of benzodiazepinone 6 matches very well with that of benzophenone 2 (Figure 3).27 Therefore, these lowest energy conformations may be the bound conformations. Other low energy conformations of the two compounds do not overlap well, with the exception of the symmetric overlay (the mirror image, shown as an insert in Figure 3).
Figure 3.
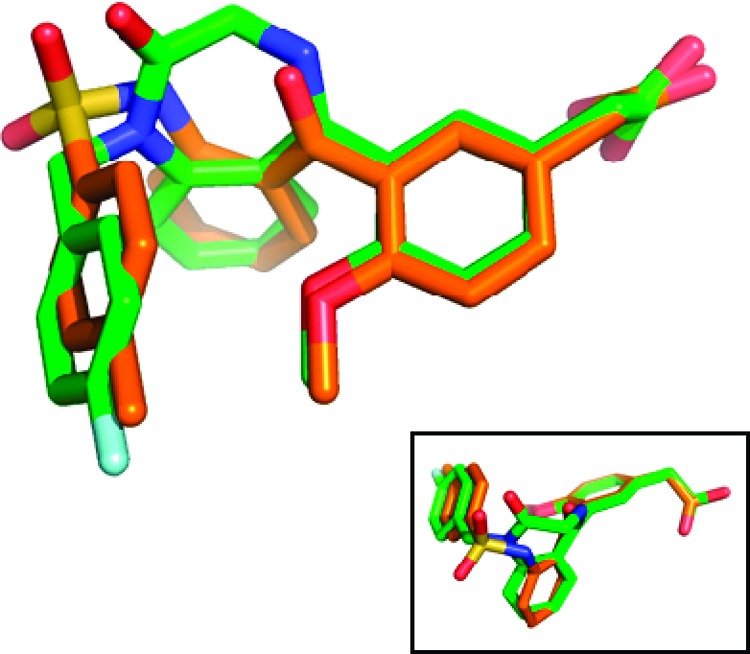
Overlap of benzodiazepinone 6 (green) and benzophenone 2 (orange). An alternate possible symmetric overlay is shown as an insert.
On the other hand, none of the low energy conformations of quinazolinone 14 matches those of benzophenone 2, which may explain why the binding potency of 14 is low. Figure 4 shows the overlap of the lowest energy conformations of compound 14 and 2. As expected, there are many low energy conformations for the flexible bisaryl ethers (1 and 3), and the closest overlay with the lowest energy conformations of 2 and 6 only occurs at a higher energy conformation (calculated to be about 3.5 kcal/mol from the minimal energy conformation). Therefore, the low binding affinity of the bisaryl ethers is likely due largely to the higher conformational energy of the CRTH2-bound conformation.
Figure 4.
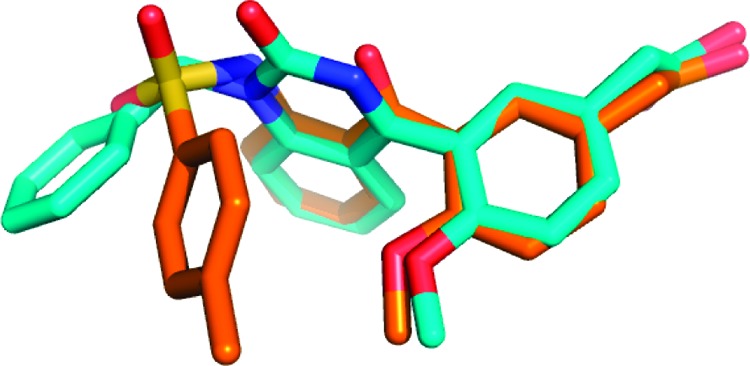
Overlap of quinazolinone 14 (blue) and benzophenone 2 (orange).
Compound 6 was also evaluated for its CRTH2 functional activity. It inhibited PGD2-mediated human eosinophil shape change with a Kb of 2 nM.28 In addition, this compound has favorable pharmacokinetics properties in mouse. When dosed orally at 15 mg/kg, the measured concentration of compound 6 in plasma was 5 μM at 1 h, 0.8 μM at 2 h, and 0.4 μM at 8 h. This compound could also achieve similar or greater exposures when dosed either subcutaneously or intraperitoneally.
In summary, we designed and synthesized benzodiazepinone derivatives as potent CRTH2 antagonists based on the observation of an intramolecular hydrogen bond in the ortho-phenylsulfonamido benzophenone derivatives. The benzodiazepinones are 2 orders of magnitude more potent than the corresponding bisaryl ethers. The SAR can be explained through analysis of the low energy conformations. The identification of the possible bound conformation could be useful for building a pharmacophore model of CRTH2 antagonists.
Acknowledgments
We thank Tod Martin and Dr. Alison Budelsky for the human eosinophil shape change assay data and useful suggestions.
Supporting Information Available
Detailed synthetic experimental procedures and characterization for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Schuligoi R.; Sturm E.; Luschnig P.; Konya V.; Philipose S.; Sedej M.; Waldhoer M.; Peskar B. A.; Heinemann A. CRTH2 and D-Type Prostanoid Receptor Antagonists as Novel Therapeutic Agents for Inflammatory Diseases. Pharmacology 2010, 85, 372–382. [DOI] [PubMed] [Google Scholar]
- Norman P. DP2 receptor antagonists in development. Expert Opin. Invest. Drugs 2010, 19, 947–961. [DOI] [PubMed] [Google Scholar]
- Ulven T.; Kostenis E. Novel CRTH2 antagonists: a review of patents from 2006 to 2009. Expert Opin. Ther. Patents 2010, 11, 1505–1530. [DOI] [PubMed] [Google Scholar]
- Medina J. C.; Liu J. PGD2 Antagonists. Annu. Rep. Med. Chem. 2006, 41, 221–235. [Google Scholar]
- Pettipher R.; Hansel T. T.; Armer R. Antagonism of the prostaglandin D2 receptors DP1 and CRTH2 as an approach to treat allergic diseases. Nature Rev. Drug Discovery 2007, 6, 313–325. [DOI] [PubMed] [Google Scholar]
- Nagata K.; Hirai H.; Tanaka K.; Ogawa K.; Aso T.; Sugamura K.; Nakamura M.; Takano S. CRTH2, an orphan receptor of T-helper-2-cells, is expressed on basophils and eosinophils and responds to mast cell-derived factor(s). FEBS Lett. 1999, 459, 195–199. [DOI] [PubMed] [Google Scholar]
- Nagata K.; Tanaka K.; Ogawa K.; Kemmotsu K.; Imai T.; Yoshie O.; Abe H.; Tada K.; Nakamura M.; Sugamura K.; Takano S. Selective expression of a novel surface molecule by human Th2 cells in vivo. J. Immunol. 1999, 162, 1278–1286. [PubMed] [Google Scholar]
- Cosmi L.; Annunziato F.; Galli M. I. G.; Maggi R. M. E.; Nagata K.; Romagnani S. CRTH2 is the most reliable marker for the detection of circulating human type 2 Th and type 2 T cytotoxic cells in health and disease. Eur. J. Immunol. 2000, 30, 2972–2979. [DOI] [PubMed] [Google Scholar]
- Mitsumori S. Recent Progress in Work on PGD2 Antagonists for Drugs Targeting Allergic Diseases. Curr. Pharm. Des. 2004, 10, 3533–3538. [DOI] [PubMed] [Google Scholar]
- Nagai H. Prostaglandin as a Target Molecule for Pharmacotherapy of Allergic Inflammatory Diseases. Allergol. Int. 2008, 57, 187–196. [DOI] [PubMed] [Google Scholar]
- Miadonna A.; Tedeschi A.; Brasca C.; Folco G. C.; Sala A.; Murphy R. C. Mediator release after endobronchial antigen challenge in patients with respiratory allergy. J. Allergy Clin. Immunol. 1990, 85, 906–913. [DOI] [PubMed] [Google Scholar]
- Turner N. C.; Fuller R. W.; Jackson D. M. Eicosanoid release in allergen-induced bronchoconstriction in dogs. Its relationship to airways hyperreactivity and pulmonary inflammation. J. Lipid Mediat. Cell Signal 1995, 11, 93–102. [DOI] [PubMed] [Google Scholar]
- Hirai H.; Tanaka K.; Yoshie O.; Ogawa K.; Kenmotsu K.; Takamori Y.; Ichimasa M.; Sugamura K.; Nakamura M.; Takano S.; Nagata K. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J. Exp. Med. 2001, 193, 255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto H.; Shichijo M.; Iino T.; Manabe Y.; Watanabe A.; Shimazaki M.; Gantner F.; Bacon K. B. An orally bioavailable small molecule antagonist of CRTH2, ramatroban (BAY u3405), inhibits prostaglandin D2-induced eosinophil migration in vitro. J. Pharmacol. Exp. Ther. 2003, 305, 347–352. [DOI] [PubMed] [Google Scholar]
- Monneret G.; Gravel S.; Diamond M.; Rokach J.; Powell W. S. Prostaglandin D2 is a potent chemoattractant for human eosinophils that acts via a novel DP receptor. Blood 2001, 98, 1942–1948. [DOI] [PubMed] [Google Scholar]
- Gosset P.; Bureau F.; Angeli V.; Pichavant M.; Faveeuw C.; Tonnel A. B.; Trottein F. Prostaglandin D2 affects the maturation of human monocyte-derived dendritic cells: Consequence on the polarization of naive Th cells. J. Immunol. 2003, 170, 4943–4952. [DOI] [PubMed] [Google Scholar]
- Tumey L. N.; Robarge M. J.; Gleason E.; Song J.; Murphy S. M.; Ekema G.; Doucette C.; Hanniford D.; Palmer M.; Pawlowski G.; Danzig J.; Loftus M.; Hunady K.; Sherf B.; Mays R. W.; Stricker-Krongrad A.; Brunden K. R.; Bennani Y. L.; Harrington J. J. 3-Indolyl sultams as selective CRTh2 antagonists. Bioorg. Med. Chem. Lett. 2010, 20, 3287–3290. [DOI] [PubMed] [Google Scholar]
- Grimstrup M.; Receveur J. M.; Rist O.; Frimurer T. M.; Nielsen P. A.; Mathiesen J. M.; Högberg T. Exploration of SAR features by modifications of thiazoleacetic acids as CRTH2 antagonists. Bioorg. Med. Chem. Lett. 2010, 20, 1638–1641. [DOI] [PubMed] [Google Scholar]
- Liu J.; Wang Y.; Sun Y.; Marshall D.; Miao S.; Tonn G.; Anders P.; Tocker J.; Tang H. L.; Medina J. Tetrahydroquinoline derivatives as CRTH2 antagonists. Bioorg. Med. Chem. Lett. 2009, 19, 6840–6844. [DOI] [PubMed] [Google Scholar]
- Stearns B. A.; Baccei C.; Bain G.; Broadhead A.; Clark R. C.; Coate H.; Evans J. F.; Fagan P.; Hutchinson J. H.; King C.; Lee C.; Lorrain D. S.; Prasit P.; Prodanovich P.; Santini A.; Scott J. M.; Stock N. S.; Truong Y. P. Novel tricyclic antagonists of the prostaglandin D2 receptor DP2 with efficacy in a murine model of allergic rhinitis. Bioorg. Med. Chem. Lett. 2009, 19, 4647–4651. [DOI] [PubMed] [Google Scholar]
- Ulven T.; Kostenis E. Minor Structural Modifications Convert the Dual TP/CRTH2 Antagonist Ramatroban into a Highly Selective and Potent CRTH2 Antagonist. J. Med. Chem. 2005, 48, 897–900. [DOI] [PubMed] [Google Scholar]
- Crosignani S.; Page P.; Missotten M.; Colovray V.; Cleva C.; Arrighi J. F.; Atherall J.; Macritchie J.; Martin T.; Humbert Y.; Gaudet M.; Pupowicz D.; Maio M.; Pittet P. A.; Golzio L.; Giachetti C.; Rocha C.; Bernardinelli G.; Filinchuk Y.; Scheer A.; Schwarz M. K.; Chollet A. Discovery of a New Class of Potent, Selective, and Orally Bioavailable CRTH2 (DP2) Receptor Antagonists for the Treatment of Allergic Inflammatory. Diseases. J. Med. Chem. 2008, 51, 2227–2243. [DOI] [PubMed] [Google Scholar]
- Armer R. E.; Ashton M. R.; Boyd E. A.; Brennan C. J.; Brookfield F. A.; Gazi L.; Gyles S. L.; Hay P. A.; Hunter M. G.; Middlemiss D.; Whittaker M.; Xue L.; Pettipher R. Indole-3-acetic Acid Antagonists of the Prostaglandin D2 Receptor CRTH2. J. Med. Chem. 2005, 48, 6174–6177. [DOI] [PubMed] [Google Scholar]
- Liu J.; Fu Z.; Wang Y.; Schmitt M.; Huang A.; Marshall D.; Tonn G.; Seitz L.; Sullivan T.; Tang H. L.; Collins T.; Medina J. Discovery and optimization of CRTH2 and DP dual antagonists. Bioorg. Med. Chem. Lett. 2009, 19, 6419–6423. [DOI] [PubMed] [Google Scholar]
- Liu J.; Li A.; Wang Y.; Johnson M.; Su Y.; Shen W.; Wang X.; Lively S.; Brown M.; Lai S.; Gonzalez Lopez De Turiso F.; Xu Q.; Van Lengerich B.; Schmitt M.; Fu Z.; Sun Y.; Lawlis S.; Seitz L.; Danao J.; Wait J.; Ye Q.; Tang L.; Grillo M.; Collins T.; Sullivan T.; Medina J.. Discovery of AMG 853, a CRTH2 and DP Dual Antagonist. ACS Med. Chem. Lett. 2011, 54, DOI: 10.1021/ml1002234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The CRTH2 radioligand binding assay was performed on HEK 293 cells stably expressing human CRTH2. To measure binding, [3H]-PGD2 was incubated together with HEK 293 (hCRTH2) cells in the presence of increasing concentrations of compounds. After washing, the amount of [3H]-PGD2 that remained bound to the cells was measured by scintillation counting, and the concentration of compounds required to achieve a 50% inhibition of [3H]-PGD2 binding (the IC50) was determined. The binding buffer contains either 0.5% bovine serum albumin (buffer binding) or 50% human plasma (plasma binding).
- The lowest energy conformations for compounds were generated using a stochastic search of bond torsions followed by energy minimization using the MMFF95 force field, as implemented in MOE (Chemical Computing Group, v2009.10). The lowest energy conformations were then manually superimposed.
- Human erythrocytes and granulocytes were enriched from normal donor peripheral blood by Isolymph (Gallard-Schlesinger Industries, Plainview, NY) gradient centrifugation. The erythrocytes were removed using ACK lysing buffer (Gibco, Carlsbad, CA). The mixed granulocyte population was preincubated with vehicle (0.05% DMSO) or antagonists for 10 min at room temperature prior to stimulation with PGD2 (0.003–600 nM at 1:3 dilution) (Cayman Chemical Co., Ann Arbor, MI) for 10 min at 37 °C. The cells were fixed using 1% final paraformaldehyde (Alpha Aesar, Ward Hill, MA) and were analyzed on a FACS caliber (BD Biosciences, San Jose, CA) flow cytometer. Leukocytes were gated on using forward/side scatter parameters. The FL2 positive cells (eosinophils) were then gated, and their geometric mean of the forward scatter was calculated. The geometric means were graphed using GraphPad Prism 5 (GraphPad Software Inc., La Jolla, CA), and IC50 values were calculated. The Kb values were calculated from their IC50 values using the equation A/(R – 1) where A is the concentration of the inhibitor used. The value R = X/Y where X is the IC50 value of PGD2 in the presence of the inhibitor and Y is the IC50 value of PGD2 alone.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.