

Abstract
We report the development and characterization of compound 22 (MK-5046), a potent, selective small molecule agonist of BRS-3 (bombesin receptor subtype-3). In pharmacological testing using diet-induced obese mice, compound 22 caused mechanism-based, dose-dependent reductions in food intake and body weight.
Keywords: MK-5046, bombesin receptor subtype-3 agonist, obesity
The mammalian bombesin G-protein-coupled receptor subfamily1 comprises three structurally related members, the receptors for neuromedin B (NMBR or BB1), gastrin-releasing peptide (GRPR or BB2),2 and bombesin receptor subtype-3 (BRS-3 or BB3).3 BRS-3 is an orphan receptor whose natural ligand is not known and which, despite its name, does not bind bombesin with high affinity. BRS-3 is expressed predominantly in the central nervous system (CNS). Mice lacking functional BRS-3 develop mild obesity and insulin resistance, suggesting a role for the receptor in the regulation of energy homeostasis.4 In addition, BRS-3 as well as the other mammalian family members are expressed by several human carcinoma cell lines, where they may play a role in growth regulation.3 Until recently, there have been limited reports of peptide5 or small-molecule6−9 pharmacological tools to further elucidate the role of the receptor and determine its potential as a drug target.
Figure 1.

Structures of leads 1−3.
Our laboratory has described the lead identification and initial optimization of the first reported series of small-molecule BRS-3 agonists with efficacy in body weight lowering.10−12 Exploratory medicinal chemistry efforts focused on lead 1. The potency at the human receptor was improved through the incorporation of branched substituents at the 4-position of the imidazole, while systemic and CNS exposure were increased through replacement of the benzoic acid moiety with pyridine. This work eventually led to the identification of 2 (Bag-1), the first key pharmacological tool in our program. Results of in vivo testing with this compound furnished a preclinical proof of concept for the BRS-3 mechanism: Administration of the BRS-3 agonist in established diet-induced obese (eDIO) mice caused a significant reduction of acute food intake and increase in fasting metabolic rate, and these effects were not evident in Brs3 null (KO) mice.13 Furthermore, subchronic dosing (100 mpk BID, 8 days) led to mechanism-based body weight lowering in eDIO mice, while it had no significant effects in KO mice.13
Undesirable features of 2 were its poor oral pharmacokinetics in preclinical species and a suboptimal off-target profile that included low micromolar binding to the human inward rectifying potassium ion channel (hERG, IC50 = 1.2 μM) as well as the diltiazem (DLZ) site of the rabbit calcium ion channel (IC50 = 0.5 μM).
The low unbound exposure of compound 2 was shown to be due primarily to high intrinsic clearance. A bile duct cannulated rat study was performed to examine the fate of the compound after in vivo dosing. For this purpose, a tritium radiolabel was incorporated in the 5-position of the pyridine ring. Biliary excretion of metabolized 2 accounted for ∼65% of the recovered radioactivity after both iv and po dosing, suggesting good absorption. There was extensive hydroxylation and concomitant loss of the radiolabel, suggesting oxidative metabolism of the pyridine ring. In addition, hydroxylation of both the 2-substituent (ethyl linker) and the 4-substituent (branched alkyl chain) of the imidazole was observed. Together, these oxidative processes were the cause for the poor pharmacokinetic profile of these early leads.
Table 1. Human BRS-3 Activity and Rat Liver Microsomal (RLM) Stability of 1−3.
| compd | hBRS-3a EC50 (nM) (% act)b | T1/2 (RLM incubation) (min) |
|---|---|---|
| 1 | 1900 ± 760 (74 ± 33%) | NDc |
| 2 | 47 ± 4 (99 ± 4%) | 1 |
| 3 | 66 ± 16 (97 ± 4%) | 3 |
Data expressed as means ± SDs (≥3 independent experiments).
% act represents the maximum activation of tested compound relative to that of the dY peptide {[d-Tyr6,β-Ala11,Phe13,Nle14]bombesin(6−14)}.
Not determined.
To advance the series, we searched for motifs that might furnish more metabolically stable structures for further structure−activity relationship (SAR) and identified analogue 3 bearing a 5-fluoropyridine. When we assessed the stability of this compound by incubation with rat liver microsomes, it had an improved half-life in comparison with 2. However, off-target activities remained problematic, with reduced selectivity vs the two key ion channels (hERG, Ki = 0.4 μM; DLZ, IC50 = 0.2 μM), as well as some activation of human PXR (IC50 = 3.5 μM with 44% maximal activation).
More information was garnered from early SAR focused on the imidazole substituent: In a survey of branched alkyl groups, use of a 2,2-dimethylpropane in place of the 2,2-dimethylbutane contained in compounds 2 and 3 resulted in ∼4-fold reduction in activity at human BRS-3. While a loss in potency and selectivity is not ideal, we considered it a small price to pay for increased metabolic stability and proceeded to incorporate the fluoropyridine and truncated substituent.
We next aimed to improve the selectivity for BRS-3 over off-target effects by introducing heteroatom substituents on the central ethylene linker to increase polarity and reduce the number of low energy, unfavorable configurations for the central ethyl linker region. We envisaged that such substitutions would also improve metabolic stability, given that the benzylic C−H would no longer be available for oxidation. Other attempts to further reduce metabolism in the pyridine region focused on incorporation of alternate heterocycles. Finally, we sought to further address the metabolism of the branched alkyl chain by incorporating metabolically more stable fluoroalkyl and cyclopropyl modifications.
Scheme 1.

To access the series of compounds with substituents attached to the carbon adjacent to the imidazole, 2-lithiated imidazoles were reacted with esters to generate ketone intermediates. For the purposes of the current series, reaction of 3,3-dimethylbutyraldehyde (4) with TosMIC generated 4-(2,2-dimethylpropyl)-1H-imidazole, which was subsequently protected as the dimethylsulfamoyl derivative 5. Lithiation of 5, followed by its reaction with methyl 4-bromophenylacetate, led to ketone 6, which was coupled to the stannane generated in situ from 2-bromo-5-fluoropyridine via palladium catalysis. Finally, the addition of methylmagnesium bromide, deprotection, and chiral separation afforded two enantiomeric alcohols. Compound 7 is the more potent isomer.
Scheme 2.

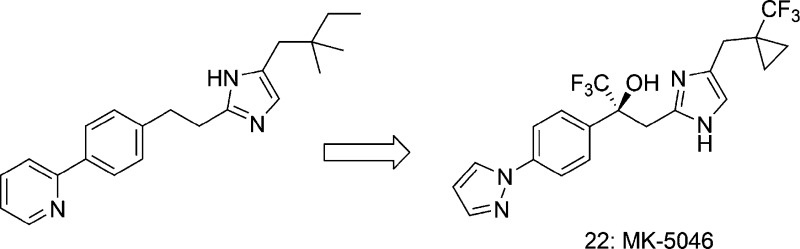
To access the series with substituents attached to the carbon distal from the imidazole, 1-trityl-2-methyl-4-(2,2-dimethylpropyl)imidazole, synthesized in three steps from 4,4-dimethylpentene, was lithiated in the α-position via treatment with n-butyllithium and subsequently reacted with methylaryl ketones 9 and 11a or the trifluorinated analogue 11b. As before, deprotection and chiral separation furnished agonists 14 and 15. Similarly, the use of the trityl-protected imidazole 20 (derived in five steps from 2-methylimidazole) led to compounds 21 and 22.14
Scheme 3.

Human BRS-3 binding potency and functional potency for the compounds are summarized in Table 2.15 The introduction of a tertiary alcohol on either carbon of the linker resulted in compounds that maintained similar activity to lead 3. Substitution distal from the imidazole (14 vs 7) gave slightly higher potency. Compounds 7 and 14 afforded improved off-target profiles relative to 3. In particular, binding to the hERG potassium channel was diminished (Ki = 5.9 and 4.2 μM for 7 and 14, respectively).
Table 2. Human BRS-3 Binding and Functional Potency for Compounds 3−22.
| compd | hBRS-3a IC50 (nM) | hBRS-3a EC50 (nM) (% act)b |
|---|---|---|
| 3 | 16 ± 6 | 66 ± 16 (97 ± 4%) |
| 7 | 82 ± 16 | 220 ± 22 (99 ± 4%) |
| 14 | 17 ± 5 | 67 ± 13 (99 ± 2%) |
| 15 | 5 ± 1 | 16 ± 3 (94 ± 5%) |
| 21 | 11 ± 2 | 14 ± 4 (94 ± 6%) |
| 22 | 27 ± 13 | 25 ± 3 (102 ± 6%) |
Data expressed as means ± SDs (≥3 independent experiments).
% act represents the maximum activation of tested compounds relative to that of dY peptide.
The initial leads 7 and 14 were assessed in rat pharmacokinetic studies. Although compound 7 had lower in vivo rat plasma clearance than 14 (14 vs 28 mL/min/kg), the unbound fraction (Fu) in plasma was considerably lower, suggesting higher intrinsic clearance. This was reinforced in studies assessing the stability of the compounds in incubations with rat liver microsomes. The half-life for compound 7 was significantly shorter than that for compound 14, indicating its propensity for higher metabolic intrinsic clearance (the two had similar unbound fractions under the conditions of the assay; Fu = 20%). On this basis, we focused our efforts on analogues of 14, leading to the identification of pyrazole 15 with improved human potency. The rat pharmacokinetic profile of 15 was notable for very high total clearance, which was largely a function of increased plasma unbound fraction as compared to the other compounds. To further reduce the potential for oxidative metabolism, our attention turned to the remaining region (common to compound 2) that was identified to be susceptible to oxidation in rats: the imidazole 4-substituent. After considerable efforts, trifluoromethyl-substituted methyl cyclopropane was incorporated to furnish compound 21, where both the unbound fraction in rat plasma and the plasma clearance were reduced.
Table 3. Rat Plasma Clearance,a Protein Binding,b and Stabilityc for Compounds 3−22.
| rat in vivo |
rat microsomal incubation |
|||
|---|---|---|---|---|
| compd | Clp (mL/min/kg) | plasma Fu (%) | T1/2 (min) | Fu (%) |
| 3 | 12 | ND | 3 | NDd |
| 7 | 14 | 0.1 | 1 | 20 |
| 14 | 28 | 0.4 | 5 | 20 |
| 15 | 110 | 6.8 | ND | ND |
| 21 | 52 | 3.2 | ND | ND |
| 22 | 20 | 1.0 | ND | ND |
Plasma clearance (Clp) calculated following 1 mg/kg iv dose.
Protein binding assessed using human plasma (200 μL) or microsomal proteins (0.2 mg/mL) and test compound (1 μM).
Microsomal stability assessed using liver microsomes (0.2 mg protein/mL) and test compound (1 μM).
Not determined.
The pharmacokinetic properties of 21 were also evaluated in the dog where the half-life was found to be unacceptably short (Table 4). Replacement of the remaining methyl group with a trifluoromethyl group gave 22. The compound exhibits overall improvements in dog pharmacokinetic properties relative to 21, while maintaining good potency and improved selectivity over off-target activities (hERG potassium channel binding Ki > 8 μM).
Table 4. Dog Pharmacokinetic Dataa for Pyrazole Analogues 21 and 22.
| compd | Clp (mL/min/kg) | T1/2 (h) | po AUCN [μM h/(mg/kg)] | Foral (%) |
|---|---|---|---|---|
| 21 | 10 | 0.3 | 1.4 | 29 |
| 22 | 5.6 | 1.4 | 3.7 | 52 |
Plasma clearance (Clp) and half-life (T1/2) calculated following 0.5 mg/kg iv dose. Normalized oral exposure (po AUCN) and oral bioavailability (Foral) calculated following 1 mg/kg po dose.
Compound 22 was evaluated in a screen of >100 receptors, ion channels, and enzymes. Eight off-target activities were identified with IC50 values in the range of 0.5−10 μM, but none were considered significant enough to preclude the use of the compound as a pharmacological tool or for clinical development.
The BRS-3 potency and pharmacokinetic parameters for 22 across a range of species, as well as activity of the compound in selected off-target assays are summarized in Table 5. Potency, selectivity, and pharmacokinetics were all significantly improved as compared to compound 3, and while the half-life remained relatively short, this was partially ameliorated by prolonged absorption, thereby allowing oral dosing paradigms in pharmacological studies.
Table 5. BRS-3 Potency, Selectivity,a and Pharmacokineticb Profile of 22 (MK-5046).
| human | mouse | rat | dog | rhesus | |
|---|---|---|---|---|---|
| BRS3 IC50 (nM) | 28 ± 13 | 5.4 ± 2 | 1.2 ± 0.4 | 6.5 ± 1.9 | 50 ± 15 |
| BRS3 Ki (nM) | 3.7 ± 0.5 | 1.6 ± 0.7 | 0.6 ± 0.2 | 9.9 ± 1.3 | 2.4 ± 1.3 |
| BRS3 EC50 (nM) | 14 ± 4 | 21 ± 0.9 | 2.2 ± 1 | 1.6 ± 0.8 | 6.9 ± 4 |
| % activationc | 100 ± 6 | 120 ± 10 | 110 ± 9 | 100 ± 58 | 110 ± 8 |
| NMBR IC50 (nM) | >10000 | ||||
| GRPR IC50 (nM) | >10000 | ||||
| hERG Ki (μM) | >8 | ||||
| DLZ IC50 (μM) | 1.9 | ||||
| hPXR EC50 (μM) | >25 | ||||
| po AUCN [μM h/(mg/kg)] | 0.1 | 1.2 | 3.7 | 0.8 | |
| Foral (%) | 9.6 | 64 | 52 | 18 | |
| Clp (mL/min/kg) | 32 | 20 | 5.6 | 8.8 | |
| Vdss (L/kg) | 2.2 | 0.98 | 0.45 | 0.44 | |
| T1/2 (h) | 1.1 | 0.7 | 1.4 | 1.0 |
Data expressed as means ± SDs (≥3 independent experiments) or single value.
Plasma clearance (Clp), volume of distribution (Vdss), and half-life (T1/2) calculated following 1 mg/kg iv dose in mouse and rat and 0.5 mg/kg iv dose in dog and rhesus. Oral bioavailability (Foral) calculated following 2 mg/kg in mouse and rat and 1 mg/kg in dog and rhesus.
% activation represents the maximum activation of tested compounds relative to that of dY-peptide for human, dog, and rhesus receptors and a reference small molecule BRS-3 agonist for mouse and rat receptors.
Compound 22 was assessed for acute efficacy in wild-type (WT) and Brs3 KO mice. In WT mice, the compound caused significant reductions in food intake and increase in fasting metabolic rate with no effect in the KO mice. After subchronic dosing in eDIO mice, compound 22 (25 mg/kg/day by sc infusion) caused reductions in body weight (8−9% as compared to vehicle-dosed animals) that were sustained for 14 days with no evidence of tachyphylaxis (manuscript submitted16).
In summary, a SAR about the core of lead compound 2 has resulted in the identification of a series of BRS-3 agonists with improved potency, selectivity, and oral exposure in preclinical species. On the basis of the in vitro and pharmacokinetic profiles, compound 22 (MK-5046) was chosen for further pharmacological study. In eDIO mice, sustained body weight loss was observed following 14 days of treatment with the compound. The data suggest that agonism of the BRS-3 may present a useful treatment for obesity, and compound 22 presents a suitable candidate for clinical investigation.
Acknowledgments
We thank the department of Laboratory Animal Resources for their assistance in animal dosing and sampling.
Supporting Information Available
Full experimental procedures and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Jensen R. T.; Battey J. F.; Spindel E. R.; Benya R. V. Mammalian bombesin receptors: Nomenclature, distribution, pharmacology, signaling, and functions in normal and disease states. Pharmacol. Rev. 2008, 60, 1–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battey J.; Wada E. Two distinct receptor subtypes for mammalian bombesin-like peptides. Trends Neurosci. 1991, 14, 524–528. [DOI] [PubMed] [Google Scholar]
- Fathi Z.; Corjay M. H.; Shapira H.; Wada E.; Benya R.; Jensen R.; Viallet J.; Sausville E. A.; Battey J. F. BRS3: A novel bombesin receptor subtype selectively expressed in testis and lung carcinoma cells. J. Biol. Chem. 1993, 268, 5979–5984. [PubMed] [Google Scholar]
- Ohki-Hamazaki H.; Watase K.; Yamamoto K.; Ogura H.; Yamano M.; Yamada K.; Maeno H.; Imaki J.; Kikuyama S.; Wada E.; Wada K. Mice lacking bombesin receptor subtype-3 develop metabolic defects and obesity. Nature 1997, 390, 165–169. [DOI] [PubMed] [Google Scholar]
- Boyle R. G.; Humphries J.; Mitchell T.; Showell G. A.; Apaya R.; Iijima H.; Shimada H.; Arai T.; Ueno H.; Usui Y.; Sakaki T.; Wada E.; Wada K. The design of a new potent and selective ligand for the orphan bombesin receptor subtype 3 (BRS3). J. Pept. Sci. 2005, 11, 136–141 and references therein. [DOI] [PubMed] [Google Scholar]
- Weber D.; Berger C.; Heinrich T.; Eickelmann P.; Antel J.; Kessler H. Systematic optimization of a lead-structure identities for a selective short peptide agonist for the human orphan receptor BRS-3. J. Pept. Sci. 2002, 8, 461–475. [DOI] [PubMed] [Google Scholar]
- Weber D.; Berger C.; Eickelmann P.; Antel J.; Kessler H. Design of Selective Peptidomimetic Agonists for the Human Orphan Receptor BRS-3. J. Med. Chem. 2003, 46, 1918–1930. [DOI] [PubMed] [Google Scholar]
- Carlton D. L.; Collin-Smith L. J.; Daniels A. J.; Deaton D. N.; Goetz A. S.; Laudeman C. P.; Littleton T. R.; Musso D. L.; Ott Morgan R. J.; Szewczyk J. R.; Zhang C. Discovery of small molecule agonists for the bombesin receptor subtype 3 (BRS-3) based on an omeprazole lead. Bioorg. Med. Chem. Lett. 2008, 18, 5451–5455. [DOI] [PubMed] [Google Scholar]
- Guo C.; Guzzo P. R.; Hadden M.; Sargent B.; Yet L.; Kan Y.; Palyha O.; Kelly T. M.; Guan X.-M.; Rosko K.; Gagen K.; Metzger J. M.; Dragovic J.; Lyons K.; Lin L. S.; Nargund R. P. Synthesis of 7-benzyl-5-(piperidin-1-yl)-6,7,8,9-tetrahydro-3H-pyrazolo[3,4-c]-[2,7]naphthyridin-1ylamine and its analogs as bombesin receptor subtype-3 agonists. Bioorg. Med. Chem. Lett. 2010, 20, 2785. [DOI] [PubMed] [Google Scholar]
- He S.; Dobbelaar P. H.; Liu J.; Jian T.; Sebhat I. K.; Lin L. S.; Goodman A.; Guo C.; Guzzo P. R.; Hadden M.; Henderson A. J.; Ruenz M.; Sargent B.; Yet L.; Kelly T. M.; Palyha O.; Kan Y.; Pan J.; Chen H.; Marsh D.; Shearman L. P.; Strack A. M.; Metzger J. M.; Feighner S. D.; Tan C.; Howard A. D.; Tamvakopoulos C.; Peng Q.; Guan X.-M.; Reitman M. L.; Patchett A. A.; Wyvratt M. J.; Nargund R. P. Discovery of substituted biphenyl imidazoles as potent bioavailable bombesin receptor subtype-3 agonists. Bioorg. Med. Chem. Lett. 2010, 20, 1913–1917. [DOI] [PubMed] [Google Scholar]
- Liu J.; He S.; Jian T.; Dobbelaar P. H.; Sebhat I. K.; Lin L. S.; Goodman A.; Guo C.; Guzzo P. R.; Hadden M.; Henderson A. J.; Pattamana K.; Ruenz M.; Sargent B.; Swenson B.; Yet L.; Tamvakopoulos C.; Peng Q.; Pan J.; Kan Y.; Palyha O.; Kelly T. M.; Guan X.-M.; Howard A. D.; Marsh D.; Metzger J.; Reitman M. L.; Wyvratt M. J.; Nargund R. P. Synthesis and SAR of derivatives based on 2-biarylethylimidazole as bombesin receptor subtype-3 (BRS-3) agonists for the treatment of obesity. Bioorg. Med. Chem. Lett. 2010, 20, 2074–2077. [DOI] [PubMed] [Google Scholar]
- Hadden M.; Goodman A.; Guo C.; Guzzo P. R.; Henderson A. J.; Pattamana K.; Ruenz M.; Sargent B.; Swenson B.; Yet L.; Liu J.; He S.; Sebhat I. K.; Lin L. S.; Tamvakopoulos C.; Peng Q.; Kan Y.; Palyha O.; Kelly T. M.; Guan X.-M.; Metzger J.; Reitman M. L.; Nargund R. P. Synthesis and SAR of heterocyclic carboxylic acid isosteres based on 2-biarylethylimidazole as bombesin receptor subtype-3 (BRS-3) agonists for the treatment of obesity. Bioorg. Med. Chem. Lett. 2010, 20, 2912–2915. [DOI] [PubMed] [Google Scholar]
- Guan X.-M.; Chen H.; Dobbelaar P. H.; Dong Y.; Fong T. M.; Gagen K.; Gorski J.; He S.; Howard A. D.; Jian T.; Jiang M.; Kan Y.; Kelly T. M.; Kosinski J.; Lin L. S.; Liu J.; Marsh D.; Metzger J. M.; Miller R.; Nargund R. P.; Palyha O.; Shearman L.; Shen Z.; Stearns R.; Strack A. M.; Stribling S.; Tang Y. S.; Wang S.-P.; White A.; Yu H.; Reitman M. L. Regulation of energy homeostasis by bombesin receptor subtype-3: Selective receptor agonists for the treatment of obesity. Cell Metab. 2010, 11, 101–112. [DOI] [PubMed] [Google Scholar]
- Absolute stereochemistry was determined via X-ray crystallography of the hydrochloride salt of an iodinated analogue ((2S)-1,1,1-trifluoro-3-(5-iodo-4-{[1-(trifluoromethyl)cyclopropyl]methyl}-1H-imidazol-2-yl)-2-[4-(1H-pyrazol-1-yl)phenyl]propan-2-ol). Crystals of this salt were grown by evaporation of an EtOAc/hexane solution over a period of approximately 1 week. Diffraction data were measured at room temperature. Examination of the intermolecular distances to the Cl ion as well as the electron density maps indicates the protonation by HCl to form the salt occurs at N23. With the presence of the two heavy atoms in the lattice, determination of the absolute configuration at the chiral C (C1, S) was unambiguous.
- Receptor binding was performed using membranes from CHO or HEK293 cells overexpressing the indicated receptor. A 30 pM concentration of [125I]-[d-Tyr6,β-Ala11,Phe13,Nle14]bombesin(6−14) (human assay) and 660 pM [3H]Bag-3 (rat and mouse assays) were used. For functional assays, agonist-induced mobilization of intracellular Ca2+ was measured in HEK293AEQ cells overexpressing BRS-3, using an aequorin bioluminescence assay.
- Guan X.-M. Manuscript submitted.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.