

Abstract
Deoxygenation of the diol groups in rings A and D of neomycin in combination with the introduction of an N1-(l)-HABA group in the 2-deoxystreptamine subunit (ring B) leads to a novel and potent antibiotic (1) with activity against strains of S. aureus carrying known aminoglycoside resistance determinants, as well as against an extended panel of Methicillin-resistant S. aureus isolates (n = 50). Antibiotic 1 displayed >64 fold improvement in MIC50 and MIC90 against this MRSA collection when compared to the clinically relevant aminoglycosides amikacin and gentamicin. The synthesis was achieved in six steps and 15% overall yield.
Keywords: Aminoglycoside, antibiotics, deoxygenation, MRSA, enzymatic modification, resistance, neomycin analogues
Staphylococcus aureus is a common commensal inhabitant of the human bacterial flora, colonizing approximately one third of the population’s nostrils.1,2 These Gram-positive facultative anaerobic cocci are responsible for conditions ranging from minor infections of skin and soft tissues to systemic illnesses such as pneumonia, meningitis, osteomyelitis, endocarditis, wound infections, bacteremia, and sepsis with high morbidity and mortality.1
The advent of antibiotics has had a major impact on the treatment of such infections for several decades. However, the appearance and dissemination of methicillin-resistant (or multidrug-resistant) S. aureus (MRSA) strains, which became prevalent in hospitals and later in community settings, with a current estimated asymptomatic reserve of 2.3 million people in the U.S. (0.8% of the population) and a potentially higher worldwide percentage,2 present a major hurdle for anti-infective therapy. Reliance on vancomycin as a last resort has generated selective pressure for the proliferation of vancomycin resistant S. aureus strains (VISA and VRSA).3 The current toll of MRSA infections in the U.S. surpasses that of HIV/AIDS, with approximately 100,000 yearly infections and 20,000 associated losses of lives.4,5
Aminoglycoside antibiotics are powerful broad-spectrum antibiotics which target a rRNA helix at the mRNA–tRNA decoding center of the bacterial 30S ribosomal subunit,6−8 affecting their bactericidal action by inducing translation inaccuracy and inhibition.9−12
The class of 4,5-disubstituted 2-deoxystreptamine aminoglycosides, including butirosin, paromomycin, and neomycin (Figure 1), share a common mode-of-action, ribosomal binding site, and powerful broad-spectrum antibacterial properties.6,7 Nevertheless, their use as anti-infectives is minimal due to their high susceptibility to multiple modifying enzymes (Figure 1).6,13−15
Figure 1.

Representative members of the 2-deoxystreptamine aminoglycoside classes characterized by 4,6-disubstitution (amikacin and gentamicin C1) or 4,5-disubstitution (neomycin B). Arrows indicate positions targeted by modifying enzymes prevalent in S. aureus (black, complete resistance; gray, medium resistance; white and crossed, evaded). See Supporting Information file for complete tabulation of aminoglycoside antibiotic structures.
The most prominent members of clinically used aminoglycosides belong to the 4,6-disubstituted 2-deoxystreptamine class, including gentamicin, tobramycin, amikacin, isepamicin, and arbekacin. However, their continued use has resulted in the emergence of S. aureus strains armored with a range of intracellular modifying enzymes,13−16 prevalently 4′-O-nucleotidyltransferase, ANT(4′)-I, 3′/5″-O-phosphotransferase, APH(3′/5″)-III, and the bifunctional 2″-O-phosphotransferase and 6′-N-acetyltransferase, APH(2″)/AAC(6′), whose respective target preferences are shown in Figure 1.13−16 As a result, semisynthetic members of the 4,6-disubstituted 2-deoxystreptamine class, such as arbekacin, were developed to overcome the inactivating action of a subset of the aforementioned enzymes and the different isoforms prevalent in other pathogens.16,17 In this regard, it should be noted that no new aminoglycoside antibiotic has been introduced since the early 1980s.6,17.
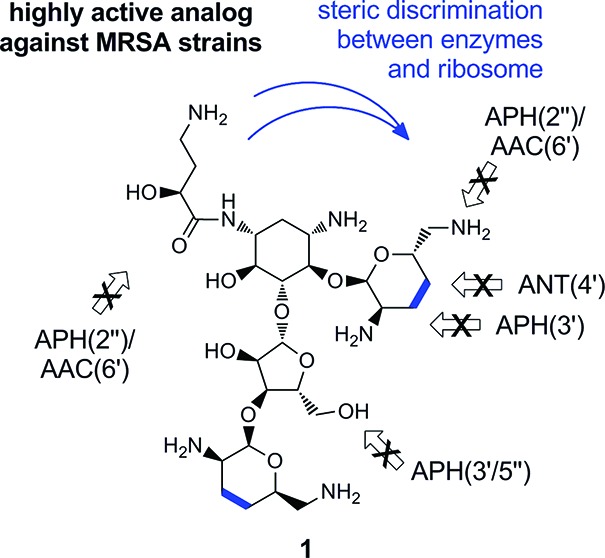
Deoxygenation methodologies have been an active area of investigation in aminoglycoside antibiotic semisynthesis with the hope to evade the action of resistance enzymes, in particular the ring-A 3′,4′-diol of the 2-deoxystreptamine classes.18−23 Among the subclasses, 4,5-disubstituted congeners have remained comparatively difficult to access in semisynthetic efforts, mainly due to lengthy protocols involving extensive protecting and functional group manipulations.18−24 Classic examples of N1-substitution have been performed on early semiprotected intermediates relying on selective reactivity among the secondary amines.6,19,24 Alternative strategies have also led to the synthesis of amphiphilic aminoglycosides with excellent antibacterial profiles.23 Herein we report the synthesis of a highly potent aminoglycoside (1) derived from neomycin B with antibacterial activity against strains of S. aureus expressing modifying enzymes and an extensive clinical MRSA collection. The key design features consisted of the implementation of a known but seldom used deoxygenation protocol and N1-amide substitution, exemplified by structure 1.
The synthetic effort began with the optimization of the Garegg–Samuelsson reaction, a modification of the classic Tipson–Cohen olefination by diol deoxygenation.25,26 The substrates were paromomycin and neomycin B intermediates 2 and 5, suitably protected with per-N-benzyloxycarbonyl groups and whose primary alcohols were capped by silylation and tritylation, respectively (Scheme 1). The resulting intermediates retained six secondary free alcohols, four of which featured trans-diol configurations. Engaging these intermediates with triphenylphosphine, imidazole, and triiodoimidazole in a refluxing 3:1 mixture of toluene and acetonitrile led to their reliable tetradeoxygenation, affording 3 and 6 in good yield and purity (Scheme 1).25−27
Scheme 1. Application of the Garegg–Samuelsson Modification of the Tipson–Cohen Reaction for Tetradeoxygenation in the Paromomycin and Neomycin Series.

Reagents: (a) TBSOTf, 2,6-lutidine, DCM, 0 °C; (b) PPh3, imidazole, triiodoimidazole, toluene, MeCN, reflux; (c) HF·py, py; (d) H2, Pd(OH)2/C, AcOH/H2O (4:1); (e) TrCl, cat. DMAP, py, 70 °C; (f) H2, Pd(OH)2/C, 1 N HCl, MeOH.
Under these conditions, the solitary alcohols at positions 6 and 2″ in intermediates 2 or 5 were unreactive toward iodination, and a common minor byproduct sporadically detected was comprised of vinyliodide intermediates.25−27 The silyl groups at positions 6′ and 5″ were removed with hydrogen fluoride to afford 3, followed by standard hydrogenation using Pearlman’s catalyst in a slightly acidified solution to yield 3′,4′,3‴,4‴-tetradeoxy-paromomycin (4). The analogous 5″-O-trityl neomycin intermediate 6 was globally deprotected by acid and Pearlman’s hydrogenation to afford N1-HABA-3′,4′,3‴,4‴-tetradeoxy-neomycin 7. The final analogues were purified by benchtop silica gel column chromatography with solvent mixtures of chloroform, methanol, and ammonia liquor, providing pure final products as free bases, which were thereafter transformed to acetate salts for convenient spectroscopic analysis and handling.28
In the case of butirosin, it has been shown that the (l)-α-hydroxy-γ-aminobutyric amide (l-HABA) chain naturally found on the antibiotic provides an enhancement and broadening of the antibacterial spectrum compared to its simple congener ribostamycin.6,17 This intricate side-chain has a close fit within the distorted H44 helix behind the bases A1492 and A1493 involved in mRNA decoding.10,29 Presumably, most aminoglycoside modifying enzymes have not evolved active sites able to recognize this unique modification in butirosin.6,16,17 Hence, semisynthetic introduction of a N1-amide moiety in the 4,6-disubstituted 2-deoxystreptamine class has provided the aforementioned benchmark drugs, such as amikacin, arbekacin, and isepamicin (Figure 1).6,17
Herein, we took advantage of the simplified reactivity of intermediates 3 and 6 to effect the chemoselective deprotection of N1 via N1,O6-oxazolidinones.6,19,29,30 Notably, the cyclic carbamate system can be selectively hydrolyzed in the presence of carboxybenzylamino groups with mild aqueous base, liberating N1 for amide coupling. Our procedure of choice was to expose intermediates 3 and 6 to a solution of LiOH in DMF for 24 h to liberate the C3-amino groups, in comparable yield to two-step procedures (Scheme 2).19−24,29,30 These intermediates were subsequently acylated under standard peptide coupling conditions with γ-N-Cbz protected l-HABA to afford 8 and 10 (Scheme 2). Global deprotection was effected using Pearlman’s hydrogenation under acidic conditions, thereby affording the novel antibiotics N1-HABA-3′,4′,3‴,4‴-tetradeoxy-paromomycin (9), and N1-HABA-3′,4′,3‴,4‴-tetradeoxy-neomycin (1), in overall yields unmatched by previous efforts with these frameworks.18−24
Scheme 2. Chemoselective N1-Deprotection of Tetradeoxygenated Paromomycin and Neomycin Intermediates via N1,O6-Oxazolidinones, for Subsequent Introduction of N1-HABA Groups.

Reagents: (a) LiOH, H2O/DMF, RT; (b) EDC, DIPEA, γ-N-Cbz-l-HABA; (c) H2, Pd(OH)2/C, AcOH/H2O (4:1); (d) DDC, Et3N, γ-N-Cbz-l-HABA; (e) H2, Pd(OH)2/C, 1 N HCl, MeOH.
The novel tetradeoxy analogues, 4, 7, 9, and 1 (Table 1), were tested against a panel of susceptible and aminoglycoside resistant S. aureus strains and compared to a series of naturally occurring aminoglycosides and the clinically relevant antibiotics gentamicin and amikacin.31,32
Table 1. Minimum Inhibitory Concentration of Tetradeoxygenated Antibiotic Analogues and Controls (μg/mL)a.
| S. aureus strain | Amk | Gent | NeoB | Par | H-Par | Ribo | Btr | 4 | 9 | 7 | 1 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ATCC 29213 | 4 | 0.5 | 0.5 | 2 | 2 | 8 | 8 | 2 | 1 | 0.5 | 0.5 |
| APH(3′/5″)-III | 8 | 0.5 | >64 | >64 | 32 | >64 | >32 | ND | ND | 32 | 1 |
| ANT(4′)-I | 64 | 0.5 | >64 | >64 | >64 | >64 | >32 | 4 | 8 | 0.5 | 1 |
| APH(2″)/AAC(6′) | 64 | >64 | >64 | >64 | >32 | >64 | >32 | >32 | 16 | >64 | 2 |
Abbreviations: Amk, amikacin; Gent, gentamicin C complex; NeoB, neomycin B; Par, paromomycin; H-Par, N1-HABA paromomycin; Ribo, ribostamycin; Btr, butirosin (N1-HABA ribostamycin); ATCC, American Type Colony Collection; APH, aminoglycoside phosphotransferase; ANT, aminoglycoside nucleotidyltransferase; AAC, aminoglycoside N-acetyltransferase; ND, not determined.
The novel tetradeoxygenated paromomycin analogues 4 and 9 were found to be 2–4-fold less active than their neomycin analogues 7 and 1 (Table 1). This loss of activity may be attributed to both loss of charge at position 6′ (e.g., paromomycin vs neomycin B) and slight disruption of the particular l-idose sugar motif of ring D (e.g., neomycin B vs ribostamicin).33
For both the paromomycin (4 vs 7) and neomycin (9 vs 1) analogues, a 2–4-fold increase in activity against wild-type S. aureus is observed in the presence of the N1-HABA substitution. Effects of similar magnitude have been observed for the naturally derived butirosin and ribostamicin pair.6,17
Although tetradeoxy-neomycin analogue 7 effectively frustrates the action of ANT(4′)-I, it appears to be a target of the two additional aminoglycoside-resistance determinants of S. aureus, namely APH(3′/5″)-III, likely acting on the 5″-hydroxyl group,34,35 and the 6′N-acetyltransferase activity of the bifunctional enzyme APH(2″)/AAC(6′).13−16 In contrast, tetradeoxy analogue 1 was active against all three strains tested (Table 1).
In the light of these results, N1-HABA-3′,4′,3‴,4‴-tetradeoxy-neomycin (1), was tested against a collection of 50 MRSA clinical isolates (Table 2). The MIC50 and MIC90 values obtained for 1 showed a >64 fold increase in activity compared to the currently clinically employed amikacin and gentamicin.
Table 2. MRSA Collection MIC50/90 (μg/mL).
| n = 50 | Amk | Gent | 1 |
|---|---|---|---|
| MIC50 | 32 | >32 | 0.25 |
| MIC90 | >64 | >32 | 0.5 |
In conclusion, we have demonstrated a six step synthesis of N1-HABA-3′,4′,3‴,4‴-tetradeoxy-neomycin (1), and its activity against an extended panel of clinical MRSA strains. Our semisynthetic strategy relied on a novel application of the chemoselective Garegg–Samuelsson deoxygenation reaction to aminoglycosides for the first time, allowing the combination of N1-modification via oxazolidinones for efficient access to promising antibiotic analogues in excellent overall yields (6 steps for 1, 15% overall yield). We are optimistic that carefully designed antibiotics, such as 1, can help in the fight against multidrug resistant S. aureus and warrant further investigations of structure–activity-relationships and therapeutic index.
Acknowledgments
We acknowledge Achaogen Inc., San Francisco, for financial assistance, Fonds de Recherche en Santé du Québec (FRSQ) for a fellowship to J.P.M., and continued support from the Natural Sciences and Engineering Research Council of Canada (NSERC).
Supporting Information Available
General methods, experimental procedures, HPLC purity reports, and 1H NMR and 13C NMR data. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Klevens R. M.; Morrison M. A.; Nadle J.; Petit S.; Gershman K.; Ray S.; Harrison L. H.; Lynfield R.; Dumyati G.; Townes J. M.; Craig A. S.; Zell E. R.; Fosheim G. E.; McDougal L. K.; Carey R. B.; Fridkin S. K. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 2007, 298, 1763–71. [DOI] [PubMed] [Google Scholar]
- Kuehnert M. J.; Kruszon-Moran D.; Hill H. A.; McQuillan G.; McAllister S. K.; Fosheim G.; McDougal L. K.; Chaitram J.; Jensen B.; Fridkin S. K.; Killgore G.; Tenover F. C. Prevalence of Staphylococcus aureus nasal colonization in the United States, 2001–2002. J. Infect. Dis. 2006, 193, 172–9. [DOI] [PubMed] [Google Scholar]
- Fridkin S. K.; Hageman J.; McDougal L. K.; Mohammed J.; Jarvis W. R.; Perl T. M.; Tenover F. C. Epidemiological and microbiological characterization of infections caused by Staphylococcus aureus with reduced susceptibility to vancomycin, United States, 1997–2001. Clin. Infect. Dis. 2003, 36, 429–39See also: VISA/VRSA—CDC Infection Control in Healthcare; http://www.cdc.gov/ncidod/ dhqp/ar_visavrsa.html. [DOI] [PubMed] [Google Scholar]
- Boucher H. W.; Corey G. R. Epidemiology of methicillin-resistant Staphylococcus aureus. Clin. Infect. Dis. 2008, 46, S344–9. [DOI] [PubMed] [Google Scholar]
- Boucher H. W.; Talbot G. H.; Bradley J. S.; Edwards J. E.; Gilbert D.; Rice L. B.; Scheld M.; Spellberg B.; Bartlett J. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [DOI] [PubMed] [Google Scholar]
- Arya D. P.Aminoglycoside Antibiotics: From Chemical Biology to Drug Discovery, 1st ed.; Wiley-Interscience: 2007. [Google Scholar]
- Walsh C.Antibiotics: Actions, Origins, Resistance; ASM Press: Washington, DC, 2003. [Google Scholar]
- Carter A. P.; Clemons W. M.; Brodersen D. E.; Morgan-Warren R. J.; Wimberly B. T.; Ramakrishnan V. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature 2000, 407, 340–8. [DOI] [PubMed] [Google Scholar]
- Ogle J. M.; Ramakrishnan V. Structural insights into translational fidelity. Annu. Rev. Biochem. 2005, 74, 129–77. [DOI] [PubMed] [Google Scholar]
- Francois B.; Russell R. J.; Murray J. B.; Aboul-ela F.; Masquida B.; Vicens Q.; Westhof E. Crystal structures of complexes between aminoglycosides and decoding A site oligonucleotides: role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acids Res. 2005, 33, 5677–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromadski K. B.; Rodnina M. V. Kinetic determinants of high-fidelity tRNA discrimination on the ribosome. Mol. Cell 2004, 13, 191–200. [DOI] [PubMed] [Google Scholar]
- Wilson D. N. The A-Z of bacterial translation inhibitors. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 393–433. [DOI] [PubMed] [Google Scholar]
- Magnet S.; Blanchard J. S. Molecular insights into aminoglycoside action and resistance. Chem. Rev. 2005, 105, 477–98. [DOI] [PubMed] [Google Scholar]
- Kim H. B.; Jang H. C.; Nam H. J.; Lee Y. S.; Kim B. S.; Park W. B.; Lee K. D.; Choi Y. J.; Park S. W.; Oh M. D.; Kim E. C.; Choe K. W. In vitro activities of 28 antimicrobial agents against Staphylococcus aureus isolates from tertiary-care hospitals in Korea: a nationwide survey. Antimicrob. Agents Chemother. 2004, 48, 1124–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz F.-J.; Fluit A. C.; Gondolf M.; Beyrau R.; Lindenlauf E.; Verhoef J.; Heinz H.-P.; Jones M. E. The prevalence of aminoglycoside resistance and corresponding resistance genes in clinical isolates of staphylococci from 19 european hospitals. J. Antimicrob. Chemother. 1999, 43, 253–259. [PubMed] [Google Scholar]
- Shaw K. J.; Rather P. N.; Hare R. S.; Miller G. H. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol. Rev. 1993, 57, 138–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price K. E. The potential for discovery and development of improved aminoglycosides. Am. J. Med. 1986, 80, 182–9. [DOI] [PubMed] [Google Scholar]
- Battistini C.; Franceschi G.; Zarini F.; Cassinelli G.; Arcamone F.. Semi-synthetic aminoglycoside antibiotics. 4. 3′,4′-dideoxyparomomycin and analogs. J. Antibiot. (Tokyo) [DOI] [PubMed] [Google Scholar]
- Torii T.; Tsuchiya T.; Umezawa S. Syntheses of 5-O-[2-O- and 3-O-(6-amino-6-deoxy-β-l-idopyranosyl)-β-d-ribofuranosyl]-1-N-[(S)-4-amino-2-hydroxybutanoyl]-3′-deoxyparomamine. J. Antibiot. 1982, 35, 58–61. [DOI] [PubMed] [Google Scholar]
- Nishimura T.; Tsuchiya T.; Umezawa S.; Umezawa H. A synthesis of 3′,4′-dideoxykanamycin B. Bull. Chem. Soc. Jpn. 1977, 50, 1580–3. [Google Scholar]
- Matsuno T.; Yoneta T.; Fukatsu S.; Umemura E. An improved synthesis of 3′,4′-dideoxykanamycin B. Carbohydr. Res. 1982, 109, 271–5. [Google Scholar]
- Rai R.; Chen H. N.; Chang H.; Chang C. W. T. Novel method for the synthesis of 3′,4′-dideoxygenated pyranmycin and kanamycin compounds, and studies of their antibacterial activity against aminoglycoside-resistant bacteria. J. Carbohydr. Chem. 2005, 24, 131–43. [Google Scholar]; 1982, 35, 98 - 101.
- Hanessian S.; Pachamuthu K.; Szychowski J.; Giguère A.; Swayze E. E.; Migawa M. T.; François B.; Kondo J.; Westhof E. Structure-based design, synthesis and A-site rRNA co-crystal complexes of novel amphiphilic aminoglycoside antibiotics with new binding modes—a synergistic hydrophobic effect against resistant bacteria. Bioorg. Med. Chem. Lett. 2010, 20, 7097–101. [DOI] [PubMed] [Google Scholar]
- Naito T.; Nakagawa S.. Paromomycin antibiotic derivatives. U.S. Patent 3,897,412, July 29, 1975.
- Garegg P. J.; Samuelsson B. Conversion of vicinal diols into olefins using triphenylphosphine and triiodoimidazole. Synthesis 1979, 10813–4. [Google Scholar]
- Garegg P. J.; Johansson R.; Samuelsson B. Introduction of 3,4-unsaturation in 2-amino-2-deoxy-d-glucopyranosides. J. Carbohydr. Chem. 1984, 32189–95. [Google Scholar]
- Garegg P. J.; Johansson R.; Ortega C.; Samuelsson B. Novel reagent system for converting a hydroxy-group into an iodo-group in carbohydrates with inversion of configuration—3. J. Chem. Soc., Perkin Trans. 1 1982, 681–3. [Google Scholar]
- See the Supporting Information for protocol details and HPLC purity reports.
- Kondo J.; Pachamuthu K.; Francois B.; Szychowski J.; Hanessian S.; Westhof E. Crystal structure of the bacterial ribosomal decoding site complexed with a synthetic doubly functionalized paromomycin derivative: a new specific binding mode to an a-minor motif enhances in vitro antibacterial activity. ChemMedChem 2007, 2, 1631–8. [DOI] [PubMed] [Google Scholar]
- Paulsen H.; Jansen R. Units of oligosaccharides. Part XXVI. Synthesis of a modified sisomicin with d-ribofuranose component. Carbohydr. Res. 1981, 92, 305–9. [Google Scholar]
- CLSI. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard, 7th ed. CLSI document M7-A7; Clinical and Laboratory Standards Institute: Wayne, PA, 2006. [Google Scholar]
- CLSI. Performance standards for antimicrobial susceptibility testing; 17th informational supplement. CLSI document M100-S18; Clinical and Laboratory Standards Institute: Wayne, PA, 2008. [Google Scholar]
- Haskell T. H.; Hanessian S. The configuration of paromose. J. Org. Chem. 1963, 28, 2598–604. [Google Scholar]
- Fong D. H.; Berghuis A. M. Structural basis of APH(3′)-IIIa-mediated resistance to N1-substituted aminoglycoside antibiotics. Antimicrob. Agents Chemother. 2009, 53, 3049–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong D. H.; Berghuis A. M. Substrate promiscuity of an aminoglycoside antibiotic resistance enzyme via target mimicry. EMBO J. 2002, 21, 2323–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.