

Abstract

A focused multiply N-methylated library of a cyclic hexapeptidic somatostatin analogue: MK678 cyclo(−MeAYwKVF−) was generated, which resulted in the unexpected observation of an efficacious tetra-N-methylated analogue, cyclo(−MeAYMewMeKVMeF−) with a potent inhibitory action on sensory neuropeptide release in vitro and on acute neurogenic inflammatory response in vivo. The analogue shows selectivity toward somatostatin receptor subtype 2 (sst2). Extensive 2D NMR spectroscopy and molecular dynamics simulation revealed the solution conformation of the analogue, which can be adopted as a lead for the further structure−activity relationship studies targeting neurogenic inflammation.
Keywords: Somatostatin, cyclic peptide, multiple N-methylation, sst, N-methylated peptide, neurogenic inflammation
Neurogenic inflammation refers to vasodilation and plasma protein extravasation (edema formation) induced by pro-inflammatory sensory neuropeptides such as calcitonin gene-related peptide (CGRP) and substance P (SP) released from the activated sensory nerve terminals in the innervated area.1 These processes are followed by the activation of inflammatory cells later. The classical nonsteroidal anti-inflammatory drugs cannot inhibit the neurogenic component, which plays an important role in the pathological mechanisms of several inflammatory diseases, for example, rheumatoid arthritis, allergic contact dermatitis, psoriasis, asthma, and inflammatory bowel diseases. Corticosteroids are only effective in very high doses in which they produce severe side effects.2 SP and CGRP are also involved in neuropathic pain, which is rather resistant to conventional analgesics. Therefore, agents acting at the level of the sensory nerve terminals themselves to inhibit the release of these pro-inflammatory/pro-nociceptive sensory neuropeptides are desired for the development of a new group of anti-inflammatory and analgesic drugs.
Somatostatin (SST), a naturally occurring neuropeptide, generally induces inhibitory activity in the central nervous system and acts as a neurotransmitter effecting the locomotor activity and cognitive function.3 In the peripheral nervous system, it is found in sensory and sympathetic neurons where it exerts down-regulation of nociception and neurogenic components of inflammatory processes.4,5 SST counteracts the effects of SP in neurogenic inflammation due to the inhibition of pro-inflammatory neuropeptide release or opposing receptor actions. SST acting on its own receptors inhibits neurokinin 1 receptor (NK1)-mediated SP actions on vasodilatation in arterioles, as well as on plasma protein extravasation from postcapillary venules, accumulation, and cytokine production of immune cells and other stimulated inflammatory cells.
Besides the locally released pro-inflammatory sensory neuropeptides, SST is also released from the peripheral terminals of the capsaicin-sensitive peptidergic population of primary sensory neurons in response to activation.4−7 It gets into the systemic circulation and inhibits inflammatory and nociceptive processes in rat and mouse experimental models presumably through SST receptor subtypes 1 and/or 4 (sst1/ sst4; SRIF1 receptor group), while inhibition of hormone secretion is mediated by the sst2, sst3, and sst5 subtypes (SRIF2 receptor group).
SST, because of its broad range of physiological activity, has a great potential as a therapeutic molecule. However, a major drawback of metabolic instability, that is, half-life of <3 min in circulation and lack of selectivity toward the different receptor subtypes (sst1−5), led to the development of hundreds of synthetic analogues of SST.8 One of these analogues, seglitide (MK678) (Figure 1), that was found to be selective toward the SST receptor subtype 2 (sst2) and showed improved control of postprandial hyperglycemia in diabetic animals when administered in combination with insulin attracted our attention.9
Figure 1.

SST (SRIF-14 and -28) and seglitide (MK678). The pharmacophore of SST and seglitide (MK678) is highlighted in red.
Multiple N-methylation has been shown to increase receptor selectivity and metabolic stability and to improve oral bioavailability of peptides.10,11 The potential of N-methylation on peptidic backbone long has been recognized and harnessed to modulate the physicochemical properties of peptides, like introducing cis-peptide bond12 and as β-sheet breaker.13 We have previously shown that multiple N-methylation of the peptidic backbone in cyclic peptides induces remarkable physicochemical properties, that is, increased metabolic stability and enhanced absorption through the intestinal wall with an overall increase in oral bioavailability.11
Thus, we synthesized a focused library of multiply N-methylated seglitide, where only the externally oriented (or solvent exposed) amide bonds were selectively N-methylated. This biased library was synthesized based on our previous experience with cyclic RGD hexapeptides14 and SST analogues, where N-methylation of the solvent exposed amide bonds (which are not involved in any interaction with the receptor) retained the bioactive conformation, showing significant biological activity.
The focused library of N-methylated seglitide is comprised of additional N-methylations at d-Trp8, Lys9, and Phe11 (the numbering is based on the original sequence of SRIF-14) (Table 1). These residues were selected, as N-methylation of these amide bonds is expected to retain the conformation of the cyclic peptide. The N-methylation of the amino acids Fmoc-l-Ala and Fmoc-l-Phe was carried out in solution by Freidinger's method.15 To obtain N-methylated derivatives containing N-Me-Lys(Boc) and N-Me-Trp(Boc), an enhanced Mitsunobu method was employed.16 The linear peptides were synthesized on solid support using standard SPPS (solid phase peptide synthesis) employing HOBt (1-hydroxybenzotriazole) and TBTU [2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate] as coupling reagents and DIEA (N,N-diisopropylethylamine) as base on chlorotritylchloride polystyrene resin (TCP-resin). For the coupling of the amino acids on N-methylated peptides, HOAt (1-hydroxy-7-aza-benzotriazole) and HATU [2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate] were employed with collidine as the base. The cyclization of the linear peptides was performed with HATU and HOBt with collidine as a base under high dilution conditions in solution to avoid any cyclodimerization and oligomerization.10
Table 1. Synthesized N-Methylated SST Analogues.
| code | peptide |
|---|---|
| MK678 | c(−MeAYwKVF−) |
| 1 | c(−MeAYMewKVF−) |
| 2 | c(−MeAYwKVMeF−) |
| 3 | c(−MeAYwMeKVF−) |
| 4 | c(−MeAYMewMeKVF−) |
| 5 | c(−MeAYwMeKVMeF−) |
| 6 | c(−MeAYMewKVMeF−) |
| 7 | c(−MeAYMewMeKVMeF−) |
The activities of the above-described SST analogues were investigated by means of an in vitro pharmacological assay on the release of the sensory neuropeptide CGRP from the peripheral terminals of sensory nerves of the isolated rat trachea. The experimental technique employed provides a sensitive detection of drug effects at the level of peripheral nerve endings.4,5 Electrical field stimulation (EFS) parameters to induce CGRP release were chosen to be selective for nerves. Rats were exsanguinated in deep anesthesia, and the whole trachea was removed and cleaned of fat adhering connective tissues. Tracheae from two rats were placed into the same organ bath (1.8 mL) to achieve a sufficient amount of peptide release and perfused (1 mL/min) with pH- (7.2) controlled oxygenized Krebs solution for 60 min (equilibration period) at 37 °C. After discontinuation of the flow, the solution was changed three times for 8 min to produce prestimulated, stimulated, poststimulated fractions. In the second 8 min period, EFS (40 V, 0.1 ms, 10 Hz for 120 s; 1200 pulses) was performed to elicit neurotransmitter release. The concentration of CGRP was determined from 200 μL samples of the incubation medium by a selective and specific radioimmunoassay (RIA) technique as described previously in detail.17 Peptide outflow was expressed as the released amount per wet tissue weight. The detection limit of the assay was 0.2 fmol/tube.
The peptides were added into the incubation medium at the beginning of the second and third 8 min fraction in 500 nM concentration. Each compound was tested in separate experiments; only one peptide was applied to the same tracheae. In each group, five experiments were performed to provide n = 5 data points per group (10 tracheae per group). Results were evaluated by the nonparametric Mann−Whitney U test (unpaired comparisons), and p < 0.05 was considered significant.
In control experiments, EFS (1200 pulses) evoked an approximately 5- and 3.5-fold increase in the outflow of CGRP in the stimulated and poststimulated fractions, respectively, as compared to the basal release. This was significantly inhibited in the presence of 500 nM 3 (di-N-methylated), 4 (tri-N-methylated), and 7 (tetra-N-methylated) (Figure 2A).
Figure 2.

(A) Graphical representation of the effect of the SST analogues on EFS (40 V, 0.1 ms, 10 Hz for 120 s; 1200 pulses)-induced CGRP release from sensory fibers of the isolated rat tracheae. Each column represents the mean + SEM CGRP concentration measured in the incubation medium of the prestimulated, stimulated, and poststimulated 8 min fractions of n = 5 experiments (5 × 2 tracheae/group). Analogues were added to the incubation medium (500 nM) at the beginning of the second and third fractions. *P < 0.05, and **P < 0.01 vs respective fraction of the control experiment (Mann−Whitney U test). (B) Graphical representation of the effect of the SST analogues on 1% mustard oil-induced acute neurogenic plasma protein extravasation in the dorsal skin of the rat paw. Each column represents the mean + SEM Evans blue accumulation per g wet skin of n = 6−10 experiments. Analogues were injected ip (100 μg/kg) 10 min before the induction of the inflammation. *P < 0.05 vs solvent-treated control experiment (Mann−Whitney U test).
Compound 7 induced the greatest, about 70%, inhibitory action in the stimulated period and completely abolished CGRP outflow in the poststimulated fraction. The inhibitory actions were seen for these three analogues (3, 4, and 7) on the total EFS-induced CGRP release (Table 2).
Table 2. Effect of the SST Analogue MK678 Peptide and Its Derivatives on EFS-Evoked Total CGRP Release (Percentage Release in the Stimulated and Poststimulated Fractions Together as Compared to the Respective Basal, Nonstimulated, Spontaneous Outflow)a.
| treatment (500 nM) | % CGRP release (comparison to prestimulated fraction) |
|---|---|
| control (Krebs) | 405 ± 58 |
| MK678 | 294 ± 33 |
| 1 | 360 ± 40 |
| 2 | 355 ± 45 |
| 3 | 182 ± 54* |
| 4 | 153 ± 30* |
| 5 | 343 ± 17 |
| 6 | 268 ± 16 |
| 7 | 100 ± 40** |
Values are means ± SEM of n = 5 experiments per group. *P < 0.05, and **P < 0.01, vs respective fraction of the control experiment determined with the nonparametric Mann−Whitney U test.
Because CGRP is an important mediator of neurogenic inflammation, the effectiveness of compounds in decreasing its electrically evoked release in vitro may be seen as an indicator for effectiveness to inhibit neurogenic inflammatory reactions in vivo. Because the analogues 3, 4, and 7 exerted significant inhibitory actions on CGRP release in vitro, they were examined on 1% mustard oil-induced neurogenic plasma protein extravasation in the rat paw skin. This concentration of mustard oil has been shown to selectively stimulate capsaicin-sensitive nerves and induce a purely neurogenic acute inflammatory reaction. Each compound (100 μg/kg) was administered ip 10 min before the induction of the inflammation. For comparison, the solvent was administered instead of the analogues. This study was undertaken in blocks with 14−15 rats per occasion. The whole set of data was obtained during 6 days. There were two or three solvent-treated rats every day, and the remaining 12 animals were randomized to receive each treatment.
Intraperitoneal injection of 100 μg/kg 7 induced the greatest, about 40% inhibitory action on 1% mustard oil-evoked acute neurogenic plasma protein extravasation in the rat paw skin (Figure 2B). However, 3 and 4 did not inhibit significantly this neurogenic inflammatory response.
The comparison of the receptor binding affinity (Table 3) of the synthesized N-methylated and the native SST toward sst1−5 revealed interesting facts. The native SST shows no receptor selectivity, whereas seglitide (MK678) shows high affinity and selectivity toward sst2. Out of the different N-methylated analogues, the di-N-methylated analogues 1, 2, and 3 exhibit a similar pattern in affinity and selectivity toward sst2. However, this can only be observed in case of two tri-N-methylated analogues 4 and 5, whereas in case of 6, there is a sudden loss in affinity toward sst2. This can be explained on the basis of pattern of N-methylation in the di-N-methylated analogues. In the case of these cyclic hexapeptide analogues of SST, N-methylation at Lys9 either retains or increases the affinity of the resulting analogue by reinforcing the bioactive conformation of the cyclic peptide by two additional γ-turns.11 However, in case of 4 and 5, N-methylations at d-Trp8 and Phe11, respectively, result in subtle loss of bioactive conformation by disrupting the stabilizing γ-turns, but this loss is compensated by the N-methylation at Lys9, showing a better affinity and selectivity profile. On the contrary, this is not the case for 6, where both of the γ-turns are destabilized by N-methylations at d-Trp8 and Phe11, resulting in a lower affinity, which could not be compensated by additional N-methylation at Lys9 as observed in the tetra-N-methylated analogue 7.
Table 3. IC50 Values (nM) of MK678 and the Synthesized N-Methylated Analogues Toward the Different SST Receptor Subtypes Using in Vitro Receptor Autroradiography18.
| code | peptide | sst1 | sst2 | sst3 | sst4 | sst5 |
|---|---|---|---|---|---|---|
| SST-28 | 2.0 (3) | 2.4 (3) | 1.1 (3) | 2.2 (3) | 2.1 (3) | |
| MK678 | c(−MeAYwKVF−) | >1000 (2) | 1.6 (2) | 105 (2) | >1000 (2) | 81 (2) |
| 1 | c(−MeAYMewKVF−) | >1000 (2) | 5.5 (2) | 205 (2) | >1000 (2) | 126 (2) |
| 2 | c(−MeAYwKVMeF−) | >1000 (2) | 4.4 (2) | 98 (2) | >1000 (2) | 53 (2) |
| 3 | c(−MeAYwMeKVF−) | >1000 (2) | 4.2 (2) | 308 (2) | >1000 (2) | 247 (2) |
| 4 | c(−MeAYMewMeKVF−) | >1000 (2) | 3.1 (2) | 172 (2) | >1000 (2) | 196 (2) |
| 5 | c(−MeAYwMeKVMeF−) | >1000 (2) | 4.1 (2) | 175 (2) | 373 (2) | 137 (2) |
| 6 | c(−MeAYMewKVMeF−) | >1000 (2) | 45 (2) | 82 (2) | >1000 (2) | 76 (2) |
| 7 | c(−MeAYMewMeKVMeF−) | >1000 (2) | 44 (2) | 200 (2) | >1000 (2) | 263 (2) |
As 7 was found to be highly potent in inhibiting CGRP release, in vitro and in vivo (inspite of having the lowest IC50 value toward sst2), the solution conformation was determined employing extensive 2D NMR studies and extended MD (molecular dynamics) simulations. The conformation of 7 is shown in Figure 3. A very strong ROE between N-Me-Phe11 Hα and N-Me-Ala6 Hα clearly demonstrates that the peptide bond between NMe-Phe11 and NMe-Ala6 is in cis-conformation. No further ROEs were observed between other α-protons of neighboring residues, which indicate that all other peptide bonds are in trans-conformation. Although most of the amide bonds are N-methylated, two β-turns about N-Me-d-Trp8, N-Me-Lys9 and N-Me-Phe11, N-Me-Ala6 exhibit a characteristic intramolecular hydrogen bond pattern via the two HN atoms.
Figure 3.
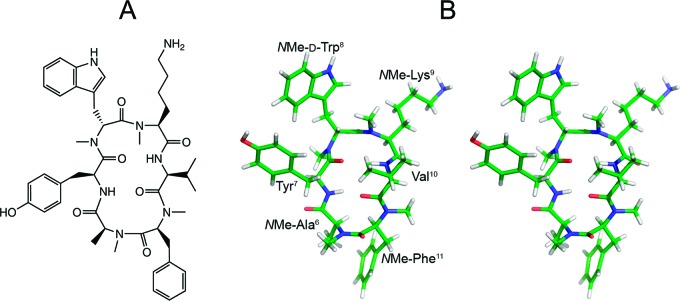
(A) Structural formula of 7. (B) Stereoview of the solution structure of 7 as determined by NMR spectroscopy and MD calculations. The conformation shown here was extracted by cluster analysis as the most representative frame of the MD calculation.
According to the backbone dihedral angles of NMe-d-Trp8 (Φ = 92°, Ψ = −99°) and N-Me-Lys9 (Φ = −119°, Ψ = 5°), the structure obtained from restrained MD simulation possesses a β-turn (type II′) centered at these two residues.19 A distance of 2.2 Å between the Val10 amide proton and the Tyr7 carbonyl oxygen, a hydrogen bond angle of 129°, and a weakly negative temperature gradient of Val10 HN (−0.3 ppb/K in DMSO) indicate that the hydrogen bond within this turn is rather strong. A second β-turn (type VIa) possesses dihedral angles of N-Me-Phe11 (Φ = −44°, Ψ = 122°) and N-Me-Ala6 (Φ = −119°, Ψ = 77°) and a cis configuration of the peptide bond between N-Me-Phe11 and N-Me-Ala6. A distance of 1.8 Å between the Tyr7 amide proton and the Val10 carbonyl oxygen, a hydrogen bond angle of 151°, and a weakly negative temperature gradient of Tyr7 HN (−1.3 ppb/K in DMSO) indicate that the hydrogen bond within this β-turn is even stronger than the hydrogen bond of the type II′ β-turn.
The Tyr7 HN-Val10 O′ and Val10 HN-Tyr7 O′ hydrogen bonds orient the Tyr7 and Val10 residues in a geometry that is usually found for individual hydrogen-bonded residues in neighbored antiparallel β-strands. The backbone dihedral angles of Tyr7 (Φ = −116°, Ψ = 110°) and Val10 (Φ = −163°, Ψ = 102°) confirm the similarity to this secondary structure element, as they are close to the corresponding backbone dihedral angles in parallel and antiparallel β-sheets.
Besides hydrogen bonding, hydrophobic clustering of the Val10 γ-methyl groups with the N-Me-Lys9N-methyl group and of the N-Me-Ala6 β-methyl group to the π-sufrace of the neighbored N-Me-Phe11 phenyl ring seem to stabilize the peptide backbone conformation (Figure 3).
The high spatial requirements of the four N-methyl groups and the two strong hydrogen bonds between the Tyr7 and the Val10 residues indicate that the backbone of 7 should be in a well-defined and rather inflexible conformation, as observed during the extended MD simulation. This suggests that its backbone conformation in solution is virtually identical to the conformation in the receptor bound state (detailed description in the Supporting Information). Even weak conformational changes of the backbone during the binding process might be thermodynamically highly disfavored and explain for the comparably weak affinity to sst2, as compared to other less constrained analogues that might adapt to the sst2 binding pocket more easily.
In conclusion, we show here that a designed multiple N-methylation of a SST analogue could modulate the pharmacological property of the N-methylated analogues in an unprecedented manner. Neurogenic inflammation has been previously reported to be inhibited via binding of SST and its analogues to SST receptors sst1 and sst4.20 However, the N-methylated analogues, which we describe here, show a significant decrease of CGRP release in vitro and acute neurogenic inflammation in vivo and reveal no significant binding to sst1/4 but preferentially to sst2. Therefore, it is encouraging to note that multiple N-methylation not only could prove to be an approach to modulate the pharmacokinetic property of peptides but also could add one more dimension to the peptides by showing novel pharmacological properties. We are currently investigating the detail conformational behavior of all of the N-methylated analogues to have a better understanding of the structure−activity relationship.
Acknowledgments
J.G.B. thanks the TUM Graduate School for support.
Supporting Information Available
Synthetic procedure and analytical data of MK678 and 1−7 with detailed NMR characterization and structure calculation of 7. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
J.C. conceived and performed the design, synthesis of peptides, and wrote the manuscript. B.L. synthesized peptides and edited the manuscript. J.G.B. performed NMR spectroscopy, structure calculation, and wrote the manuscript. Z.H. handled animals and edited the manuscript. E.P., J.S., A.H., and J.M. handled animals. J.C.R. contributed sst results. G.K. conceived animal experiments and wrote the manuscript. H.K. supervised the project and edited the manuscript.
We acknowledge DFG for their generous grant.
Supplementary Material
References
- Maggi C. A. Tachykinins and calcitonin gene-related peptide (CGRP) as co-transmitters released from peripheral endings of sensory nerves. Prog. Neurobiol. 1995, 45, 1–98. [DOI] [PubMed] [Google Scholar]
- Scholzen T.; Armstrong C. A.; Bunnett N. W.; Luger T. A.; Olerud J. E.; Ansel J. C. Neuropeptides in the skin: Interactions between the neuroendocrine and the skin immune systems. Exp. Dermatol. 1998, 7, 81–96. [DOI] [PubMed] [Google Scholar]
- Reisine T.; Bell G. I. Molecular properties of somatostatin receptors. Neuroscience 1995, 67, 777–790. [DOI] [PubMed] [Google Scholar]
- Szolcsányi J.; Helyes Z.; Oroszi G.; Németh J.; Pintér E. Release of somatostatin and its role in the mediation of the anti-inflammatory effect induced by antidromic stimulation of sensory fibres of rat sciatic nerve. Br. J. Pharmacol. 1998, 123, 936–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szolcsányi J.; Pintér E.; Helyes Z.; Oroszi G.; Németh J. Systemic anti-inflammatory effect induced by counter-irritation through a local release of somatostatin from nociceptors. Br. J. Pharmacol. 1998, 125, 916–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bölcskei K.; Helyes Z.; Szabó A.; Sándor K.; Elekes K.; Németh J.; Almási R.; Pintér E.; Petho G.; Szolcsányi J. Investigation of the role of TRPV1 receptors in acute and chronic nociceptive processes using gene-deficient mice. Pain 2005, 117, 368–376. [DOI] [PubMed] [Google Scholar]
- Helyes Z.; Elekes K.; Németh J.; Pozsgai G.; Sándor K.; Kereskai L.; Börzsei R.; Pintér E.; Szabó A.; Szolcsányi J. Role of transient receptor potential vanilloid 1 receptors in endotoxin-induced airway inflammation in the mouse. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L1173–L1181. [DOI] [PubMed] [Google Scholar]
- Ovadia O.; Greenberg S.; Laufer B.; Gilon C.; Hoffman A.; Kessler H. Improvement of drug-like properties of peptides: the somatostatin paradigm. Expert Opin. Drug Discovery 2010, 5, 655–671. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; Saperstein R.; Nutt R. F.; Freidinger R. M.; Brady S. F.; Curley P.; Perlow D. S.; Paldeva W. J.; Colton C. D.; Zacchei A. G.; Tocco D. J.; Hoff D. R.; Vandlen R. L.; Gerich J. E.; Hall L.; Mandarino L.; Cordes E. H.; Anderson P. S.; Hirschmann R. A super active cyclic hexapeptide analog of somatostatin. Life Sci. 1984, 34, 1371–1378. [DOI] [PubMed] [Google Scholar]
- Chatterjee J.; Gilon C.; Hoffman A.; Kessler H. N-methylation of peptides: A new perspective in medicinal chemistry. Acc. Chem. Res. 2008, 41, 1331–1342. [DOI] [PubMed] [Google Scholar]
- Biron E.; Chatterjee J.; Ovadia O.; Langenegger D.; Brueggen J.; Hoyer D.; Schmid H. A.; Jelinek R.; Gilon C.; Hoffman A.; Kessler H. Improving oral bioavailability of peptides by multiple N-methylation: Somatostatin analogues. Angew. Chem., Int. Ed. 2008, 47, 2595–2599. [DOI] [PubMed] [Google Scholar]
- Chatterjee J.; Mierke D.; Kessler H. N-methylated cyclic pentaalanine peptides as template structures. J. Am. Chem. Soc. 2006, 128, 15164–15172. [DOI] [PubMed] [Google Scholar]
- Yan L. M.; Tatarek-Nossol M.; Velkova A.; Kazantzis A.; Kapurniotu A. Design of a mimic of nonamyloidogenic and bioactive human islet amyloid polypeptide (IAPP) as nanomolar affinity inhibitor of IAPP cytotoxic fibrillogenesis. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 2046–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee J.; Ovadia O.; Zahn G.; Marinelli L.; Hoffman A.; Gilon C.; Kessler H. Multiple N-methylation by a designed approach enhances receptor selectivity. J. Med. Chem. 2007, 50, 5878–5881. [DOI] [PubMed] [Google Scholar]
- Freidinger R. M.; Hinkle J. S.; Perlow D. S.; Arison B. H. Synthesis of 9-fluorenylmethyloxycarbonyl-protected N-alkyl amino-acids by reduction of oxazolidinones. J. Org. Chem. 1983, 48, 77–81. [Google Scholar]
- Biron E.; Chatterjee J.; Kessler H. Optimized selective N-methylation of peptides on solid support. J. Pept. Sci. 2006, 12, 213–219. [DOI] [PubMed] [Google Scholar]
- Helyes Z.; Németh J.; Pintér E.; Szolcsányi J. Inhibition by nociceptin of neurogenic inflammation and the release of SP and CGRP from sensory nerve terminals. Br. J. Pharmacol. 1997, 121, 613–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reubi J. C.; Schär J.-C.; Waser B.; Wenger S.; Heppeler A.; Schmitt J. S.; Mäcke H. R. Affinity profiles for human somatostatin receptor subtypes SST1−SST5 of somatostatin radiotracers selected for scintigraphic and radiotherapeutic use. Eur. J. Nucl. Med. 2000, 27, 273–282. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; Freidlinger R. M.; Perlow D. S.; Paleveda W. J. Jr.; Holly F. W.; Strachan R. G.; Nutt R. F.; Arison B. H.; Homnick C.; Randall W. C.; Glitzer M. S.; Saperstein R.; Hirschmann R. A potent cyclic hexapeptide analogue of somatostatin. Nature 1981, 292, 55–58. [DOI] [PubMed] [Google Scholar]
- Pintér E.; Helyes Z.; Szolcsányi J. Inhibitory effect of somatostatin on inflammation and nociception. Pharmacol. Ther. 2006, 112, 440–456. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.