

Abstract

A series of N-pyridyl benzamide KCNQ2/Q3 potassium channel openers were identified and found to be active in animal models of epilepsy and pain. The best compound 12 [ICA-027243, N-(6-chloro-pyridin-3-yl)-3,4-difluoro-benzamide] has an EC50 of 0.38 μM and is selective for KCNQ2/Q3 channels. This compound was active in several rodent models of epilepsy and pain but upon repeated dosing had a number of unacceptable toxicities that prevented further development. On the basis of the structure−activity relationships developed around 12, a second compound, 51, [N-(2-chloro-pyrimidin-5-yl)-3,4-difluoro-benzamide, ICA-069673], was prepared and advanced into a phase 1 clinical study. Herein, we describe the structure−activity relationships that led to the identification of compound 12 and to the corresponding pyrimidine 51.
Keywords: Potassium channel, KCNQ2/Q3, epilepsy
KCNQ2−5 (Kv7.2−7.5) voltage-dependent potassium channels are expressed at high levels in the brain, including regions linked to seizure disorders, such as the cortex, hippocampus, and thalamus. They represent the molecular correlate of the neuronal M current (IM), a noninactivating, slowly deactivating subthreshold K+ current that opposes depolarizing current and serves to stabilize membrane potential and control neuronal excitability.1,1b
Given that KCNQ2/Q3 channels lead to benign familial neonatal convulsions, a rare form of neonatal epilepsy in humans,2−2d it is hypothesized that KCNQ2/Q3-based M currents play an important role in the control of neuronal excitability and epileptiform activity. Benign familial neonatal convulsions are characterized by generalized seizures in early life, which disappear within weeks or months of birth but may return later in life. Moreover, disruption of the KCNQ2 gene in mice results in hypersensitivity to the chemoconvulsant pentylenetetrazole (PTZ),3 decreased seizure threshold to electric shock-induced convulsions,4 and spontaneous seizures.5 This evidence suggests that openers or activators of KCNQ channels should decrease neuronal excitability, making them attractive drug targets for the treatment of epilepsy and related disorders of neuronal excitability.6−6d
A screening program to identify KCNQ agonists resulted in the identification of 3,4 dichloro-N-(pyridine-3-yl)benzamide (1) as a weak agonist (EC50 = 6 μM, Figure 1, 86Rb+ assay, see the Supporting Information). The corresponding 2-pyridyl [2, 3,4 dichloro-N-(pyridine-2-yl)benzamide] and 4-pyridyl [3, 3,4 dichloro-N-(pyridine-4-yl)benzamide] isomers as well as the benzene derivative (4) were inactive (EC50 > 10 μM). The subsequent chemistry effort focused on the N-(pyridine-3-yl)benzamide core. A large set of derivatives were prepared (>400), which included modifications to the amide (reduction and alkylation), substitutions on both aromatic rings as well as replacement of the substituted benzene component with heterocycles. The KCNQ agonist activity was only seen with compounds based directly upon the core shown in Scheme 1. The majority of the compounds had little or no KCNQ agonist activity (EC50 values > 10 μM).
Figure 1.

Benzanilide analogues.
Scheme 1. General Synthesis of 2-Chlorpyridine Benzanildes.

Reagents and conditions: (a) Nucleophile, MeOH. (b) Pd/C 10% H2, DCM. (c) DPPA, TEA, t-butanol. (d) TFA, DCM. (e) (CO)2Cl2, DCM, DMF (cat) 0° to room temperature. (f) DIEA, DCM, room temperature.
Tables 1 and 2 contain the key pyridine derivatives prepared, their KCNQ2/Q3 activity, and KCNQ1 activity when tested. KCNQ17−8b is found in cardiac tissue and therefore becomes a critical counter screen for early leads. In addition, Table 3 provides KCNQ3/Q5 data, which illustrates that compounds can be identified that are slightly more potent against KCNQ2/Q3 versus KCNQ3/Q5.9 The 2-substituted-5-amino pyridines illustrated in Table 1 were prepared by reacting the corresponding 2-chloro-5-nitropyridine with the appropriate nucleophile followed by catalytic reduction. The methyl and trifluoromethyl amino pyridines were prepared from the corresponding nicotinic acids by Curtius reaction with diphenylphosphorylazide followed by hydrolysis of the resulting tert-butyl-carbamate.
Table 1. KCNQ2/Q3 Activity of N-(6-Substituted Pyridin-3-yl)-3,4-diflourobenzamide.
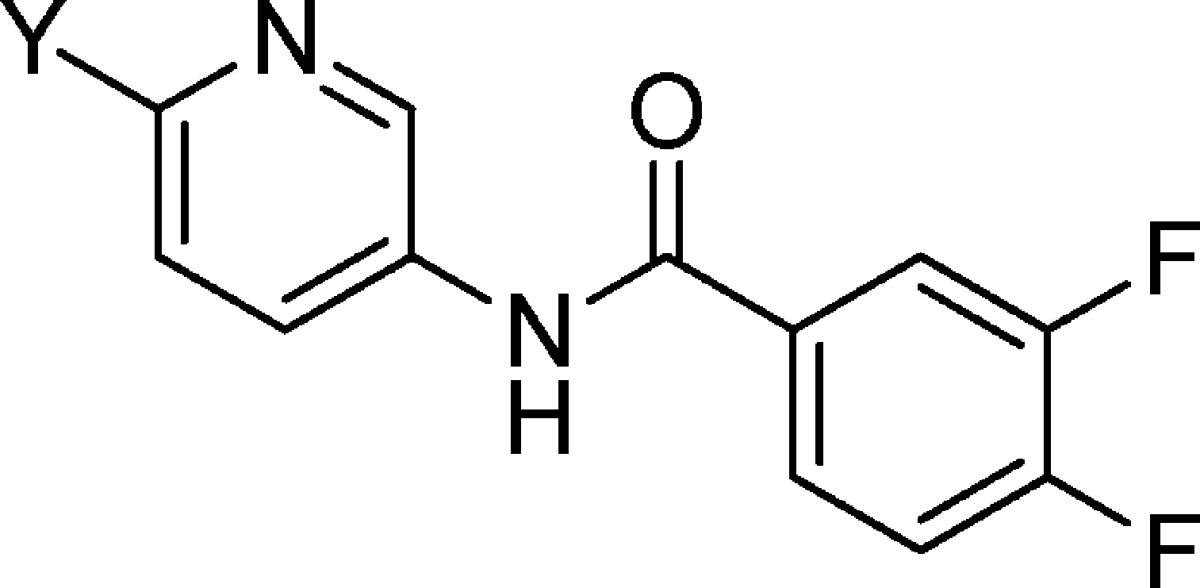
| compd no. | Y-substituent | KCNQ2/Q3 EC50 (μM) |
|---|---|---|
| 5 | F | 4.8 |
| 6 | CH3 | 6.8 |
| 7 | SCH3 | >10 |
| 8 | CF3 | >10 |
| 9 | CN | 10 |
| 10 | OH | >10 |
| 11 | OCH3 | >10 |
| 12 | Cl | 0.38 |
| 13 | NH-cyclopropyl | 2.7 |
| 14 | 1-pyrrolidine | 0.48 |
| 15 | 1-morpholine | 7.8 |
Table 2. KCNQ2/Q3 and KCNQ1 Activity of N-(6-Chloropyridin-3-yl) Benzamidesa.
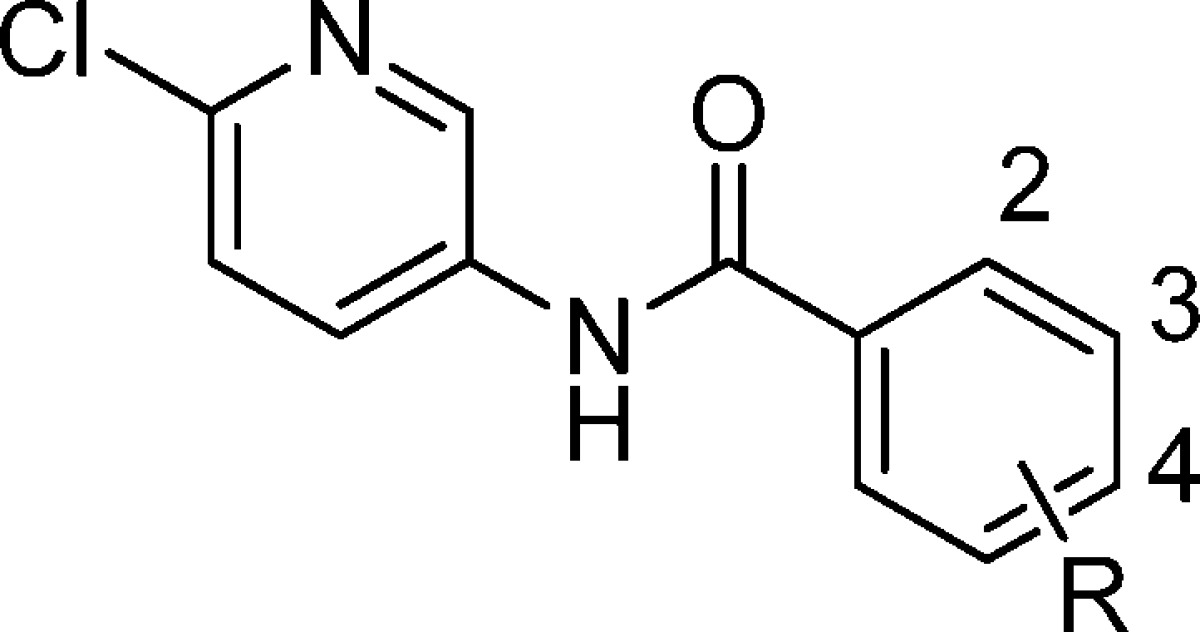
| compd no. | R-substituent | KCNQ2/Q3 EC50 (μM) | KCNQ1 IC50 (μM) |
|---|---|---|---|
| 12 | 3,4-F2 | 0.38 | 41 |
| 16 | 4-Cl | 0.38 | 15 |
| 17 | H | 1.6 | >100 |
| 18 | 2-F | >10 | NT |
| 19 | 3-F | 1.1 | 77 |
| 20 | 4-F | 0.98 | 66 |
| 21 | 3-CF3 | 0.39 | 5.8 |
| 22 | 4-CF3 | 0.32 | 2.9 |
| 23 | 4-Ph | 0.26 | >30 |
| 24 | 3-OCH3 | 5.5 | NT |
| 25 | 3,4 (CH3)2 | 0.69 | >100 |
| 26 | 3,4-Cl2 | 0.67 | 17 |
| 27 | 3-Cl | 0.27 | 41 |
| 28 | 3-F, 4-Cl | 0.08 | 15 |
| 29 | 3-CF3, 4-F | 0.42 | 3.4 |
| 30 | 4-SO2NH2 | >10 | NT |
| 31 | 3-F, 4-CF3 | 0.37 | 3.4 |
| 32 | 3-Cl, 4-F | 0.19 | 22 |
| 33 | 3-F, 4-CH3 | 0.28 | 33 |
| 34 | 3-CN | >10 | NT |
| 35 | 2,3-F2 | >10 | NT |
| 36 | 4-OC6H5 | 0.01 | 1.9 |
| 37 | 3-F, 4-CN | 1.1 | 51 |
| 38 | 3-F, 4-OCH3 | 3.2 | >100 |
| 39 | 3-F, 4-OEt | 0.57 | >100 |
| 40 | 3-F, 4-OiPr | 0.17 | >100 |
| 41 | 3-F, 4-OC6H5 | 0.03 | 1.3 |
| 42 | 3-F, 4-(4F−C6H5S) | 0.08 | >100 |
| 43 | 3-F, 4-(4F−C6H5SO2) | 0.07 | >100 |
| 44 | 3-F, 4-(4F−C6H5CH2S) | 1.6 | >100 |
| 45 | 3-F, 4-(4F−C6H5CH2SO2) | 0.16 | >100 |
| 46 | 3-F, 4-SiPr | 0.11 | 5.8 |
| 47 | 3-F, 4-SiBu | 0.14 | >100 |
| 48 | 3-F, 4-SO2iBu | 0.26 | 54 |
| 49 | 3-F, 4-NHCH2CH2C6H5 | 0.18 | >100 |
NT, not tested.
Table 3. KCNQ Activity of 2-(Chloropyrimidin-5-yl)benzamides.
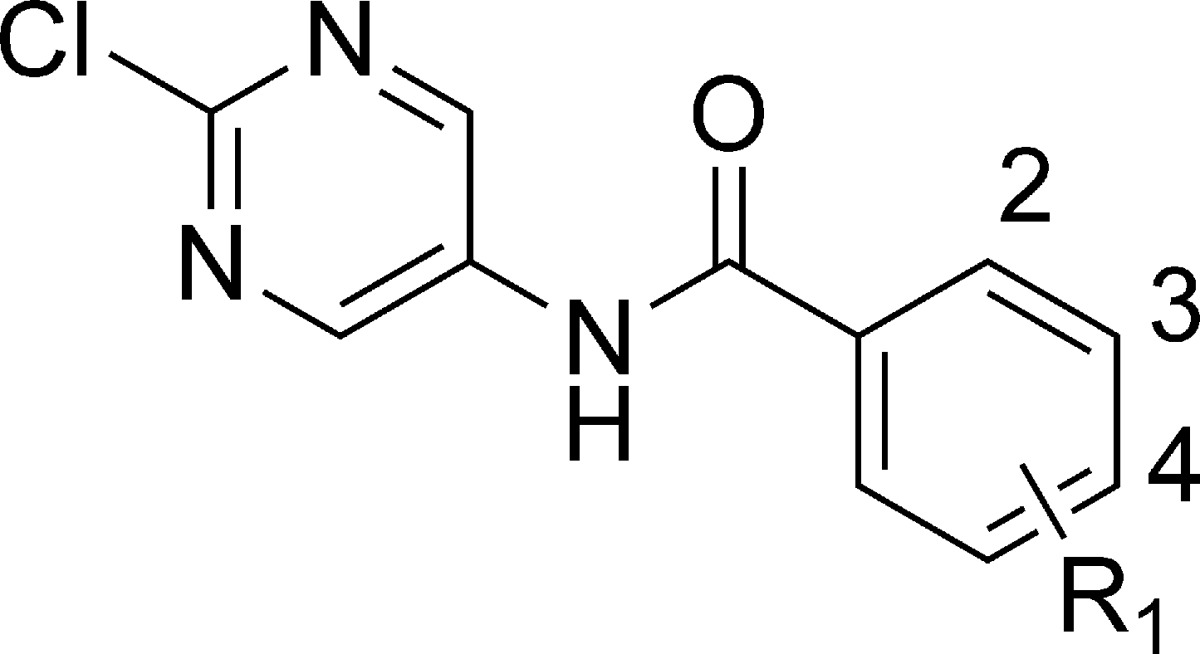
| compd no. | R1 | KCNQ2/Q3 EC50(μM) | KCNQ3/Q5 EC50 (μM) | KCNQ1 IC50 (μM) |
|---|---|---|---|---|
| 50 | 3-F | 7.2 | 23 | >100 |
| 51 | 3,4-F2 | 0.69 | 14.3 | >100 |
| 52 | 3-Cl | 1.1 | 14 | 78 |
| 53 | 3-CF3 | 4.3 | 28 | 35 |
| 54 | 3-F,4-Cl | 0.41 | 9 | 49 |
| 55 | 3-F,4-CH3 | 0.60 | 10 | 93 |
The general synthesis of the pyridine compounds involved the reaction of a 2-substituted-5-aminopyridine with substituted benzoic acid or acid chloride (Scheme 1). The yields ranged from 20 to 60%. Typically, if the acid chlorides were not commercially available, they were made from the corresponding benzoic acids but used directly in the next step without purification. Compounds 1−5 were commercially available. All of the final compounds (Tables 1−3) were >97% pure as determined by LC/MS and NMR.
On the basis of the initial results with the pyridine isomers, we focused on identifying the optimal substituent at the 6-position (Y) of the N-(pyridine-3-yl) benzamide core, while holding the benzene portion constant (R1, R2 = F, Table 1). The best Y-substituent was chlorine (12, EC50 = 0.38 μM). All of the other substituents at this position resulted in compounds that were at least 10-fold less active. An exception is compound 14 (EC50 = 0.48 μM). However, there was some weak activity with fluoro derivative (5) and the methyl derivative (6) as well as with the cyclopropylamine and morpholine analogues (13 and 15).
Next, we turned our attention to the various substituents on the benzene portion and how changes modulated KCNQ2/Q3 agonist activity as well as KCNQ1 antagonist activity (Table 2). In general, substituents at the R2-position were not well tolerated,10 and R3-substituted compounds varied in their activity with 3-chloro and trifluoromethyl containing compounds (27 and 21) having comparable activity to 12. However, the 3-cyano and 3-methoxy compounds (34 and 24) were weakly active. The most potent compound identified was the 3-fluoro-4-choro derivative 28 with a KCNQ2/Q3 EC50 = 0.08 μM, but 28 was also weakly active versus KCNQ1.
In addition to halogen and alkyl substituents at R3 and R4, we examined a series of heteroatom substituents (O, S, and N) at these positions. From the large number of compounds prepared, those compounds containing a fluorine substituent on the R3-position and various heteroatom substituents at the R4-position of the benzene ring (compounds 37−49) are highlighted here for comparison. In general, these compounds had KCNQ2/Q3 activity ranging from nanomolar to micromolar and varied considerably in their KCNQ1 (IC50) activity as well (Table 2). For example, the R4O-phenyl derivatives 36 and 41 have EC50 values of 0.01 and 0.03 μM, respectively, while the best R4O-alkyl derivative 40 has an EC50 of 0.17 μM. However, the corresponding KCNQ1 IC50 values for 36 and 41 are < 2 μM, while the KCNQ1 activity for compound 40 is > 100 μM.
In the pyridine series, the compound with the best overall in vitro and in vivo profile was 12.11,12 This compound was advanced into 28 day toxicity studies in rat and cynomolgus monkeys, but upon repeated dosing, the compound induced nonhemolytic anemia in both species. It was determined that a metabolite derived from 2-chloro-5-aminopyridine was the cause of the toxicity.13 Therefore, a search for a structurally diverse analogue with a similar in vitro profile minus the toxicity was initiated.
The first set of analogues investigated were the corresponding pyrimidine analogues (Table 3). The compounds were prepared starting with the 2-amino-5-nitropyrimidine (Scheme 2), which was converted to the 2-chloro analogue by treatment with t-butyl nitrite followed by cupric chloride. The nitro compound was reduced using iron in acetic acid to avoid reduction of the 2-choro substituent, and the resulting 2-choro-5-aminopyrimidine was reacted with various acid chlorides to provide the desired compounds (Table 3). As shown in the table, compound 51 was the most potent pyrimidine analogue and had an improved activity profile versus KCNQ1. For example, comparing the pyrimidines and the pyridines (i.e., 50 vs 19, 51 vs 12, and 55 vs 33), the pyrimidines overall were slightly less active on KCNQ2/Q3 channels but more selective for KNCQ2/Q3 over KCNQ1.
Scheme 2. Synthesis of 2-Chloropyrimidine-Substituted Benzanildes.

Reagents and conditions: (a) CuCl2, t-BuONO, MeCN, 65−80 °C. (b) Fe, AcOH, EtOH, H2O, 90−100 °C. (c) R1COCl, pyridine, DCM.
In fact, 51 was found to be 20-fold selective for KCNQ2/Q3 over KCNQ3/Q5 (Table 3) and had no measurable activity against a panel of cardiac ion channels (IC50 values > 30 μM for hERG, Nav1.5, L type channels, and KCNQ1) as well as no activity on GABA(A) gated channels at 10 μM.9 This compound was tested in the rat maximal electroshock (MES) and the PTZ anticonvulsant assays and had oral ED50 values of 1.5 and <1 mg/kg, respectively, which is slightly improved over the corresponding pyridine analogue 12 (compound 12 rat MES ED50 = 1.5 mg/kg, rat PTZ ED50 = 2.2 mg/kg). Following a 2 mg/kg iv administration (rat) of 51, the t1/2 was 1.2 h, clearance was 1039 mL/h/kg, the volume of distribution based on the terminal phase (Vz) was 1804 mL/kg, and the compound was well absorbed (63% bioavailable, Tmax = 0.5 h). This compound was advanced to IND enabling GLP safety studies and ultimately into a phase 1 clinical trials.
In summary, we have described the identification and structure−activity relationships for a series of N-pyridyl benzamide KCNQ2/Q3 potassium channel openers. The lead compounds 12 and 51 were orally active in several animal models of epilepsy and had sufficient in vitro selectivity and pharmacokinetic properties to advance to rat toxicity studies. Compound 51 was advanced into a phase 1 clinical trial, the results of which will be reported elsewhere.
Acknowledgments
The helpful comments on the manuscript by Doug Krafte are appreciated, as is the assistance of Steve Werness for performing the in vivo pharmacokinetic studies.
Supporting Information Available
General methods, experimental details, analytical data, and in vitro and in vivo assay protocols. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∥ Johnson and Johnson, San Diego, CA.
Author Present Address
⊥ CEAMED, Tenerife, Spain.
Supplementary Material
References
- Brown D. A.; Adams P. R. Muscarinic suppression of novel voltage-sensitive K+ current in a vertebrae neuron. Nature 1980, 283, 673–676. [DOI] [PubMed] [Google Scholar]
- Wang H. S.; Pan Z.; Brown B. S.; Wymore R. S.; Cohen I. S.; Dixon J. E.; McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: Molecular correlates of the M-channel. Science 1998, 282, 1890–1893. [DOI] [PubMed] [Google Scholar]
- Biervert C.; Schroeder B. C.; Kubisch C.; Berkovic S. F.; Propping P.; Jentasch T. J.; Steinlein. O. K. A potassium channel mutation in neonatal human epilepsy. Science 1998, 279, 403–406. [DOI] [PubMed] [Google Scholar]
- Charlier C.; Singh N. A.; Ryan S. G.; Lewis T. B.; Reus B. E.; Leach R. J.; Leppert M. A pore mutation in the novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat. Genet. 1998, 18, 53–55. [DOI] [PubMed] [Google Scholar]
- Sing N. A.; Chalier C.; Stauffer D.; DuPont B. R.; Leach R. J.; Melsi R.; Ronen G. M.; Bjerre I.; Quattlebaum T.; Murphy J.V. A novel potassium channel gene, KCNQ2 is mutated in inherited epilepsy of newborns. Nat. Genet. 1998, 18, 25–29. [DOI] [PubMed] [Google Scholar]
- Lerche H.; Biervert C.; Alekov A. K.; Schleithoff L.; Lindner M.; Klinger W.; Bretschneider F.; Mitrovic N.; Jurkat-Rott K.; Bode H. A reduced K+ current due to a novel mutation in KCNQ2 causes neonatal convulsions. Ann. Neurol. 1999, 46, 305–312. [DOI] [PubMed] [Google Scholar]
- Watanabe H.; Nagata E.; Kosaki A.; Nakurmura M.; Yokoyama M; Tanaka K.; Sasai H. Disruption of epilepsy KCNQ2 gene results in neural hyperexcitability. J. Neurochem. 2000, 75, 28–33. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Beyeer B. J.; Otto J. F.; O'Brien T. P.; Letss V. A.; White H. S.; Frankel W. N. Spontaneous deletion of epilepsy gene orthologs in a mutant mouse with a low electroconvulsive threshold. Mol. Genet. 2003, 12, 975–984. [DOI] [PubMed] [Google Scholar]
- Peters H. C.; Pongs O.; Storm J. F.; Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat. Neurosci. 2005, 8, 51–60. [DOI] [PubMed] [Google Scholar]
- Wickenden A. D.; Roeloffs R.; McNaughton-Smith G.; Rigdon G. C. KCNQ potassium channels: Drug targets for the treatment of epilepsy and pain. Expert Opin. Ther. Targets 2004, 14, 457–469. [Google Scholar]
- McNaughton-Smith G.; Wickenden A. D.. Compounds that activate KCNQ(2−5) family of potassium ion channels. In Voltage-Gated Ion Channels as Drug Targets; Triggle D., Ed.; Wiley-VCH: Weinheim, 2006; Chapter 7.6, pp 355−380. [Google Scholar]
- Munro G.; Dalby-Brown W. Kv7 (KCNQ) channel modulators and neuropathic pain. J. Med. Chem. 2007, 50, 2576–2582. [DOI] [PubMed] [Google Scholar]
- Gribkoff V. K. The therapeutic potential of neuronal Kv7 (KCNQ) channel modulators: An update. Expert Opin. Ther. Targets 2008, 12, 565–581. [DOI] [PubMed] [Google Scholar]
- Jespersen T.; Grunnet M.; Olesen S. P. The KCNQ1 potassium channel: From gene to physiological function. Physiology (Bethesda) 2005, 20, 408–416. [DOI] [PubMed] [Google Scholar]
- The following references demonstrate the presence of a conserved tryptophan in KCNQ2−5 that is not present in KCNQ1 and may account for the agonist activity seen vs KCNQ1 block. Lange W.; Geissendrfer J.; Schenzer A.; Grotzinger J.; Seebohm G.; Friedrich T.; Schwake M. Refinement of the binding site and mode of action of the anticonvulsant retigabine on KCNQ K+ channels. Mol. Pharmacol. 2009, 75, 272–280. [DOI] [PubMed] [Google Scholar]
- Padilla K.; Wickenden A. D.; Gerlach A. C.; McKormack C. The KCNQ2/Q3 selective channel opner ICA-27243 binds to a novel voltage-sensor domain site. Neurosci. Lett. 2009, 465, 138–142. [DOI] [PubMed] [Google Scholar]
- Activating KCNQ3/Q5 channels may produce vascular side effects:Ng F. L.; Davis A. J.; Jepps T. A.; Harhun M. I.; Yeung S. Y.; Wan A.; Reddy M.; Melville D.; Nardi A.; Khong T. K.; Greenwood I. A. Expression and function of the K+ channel KCNQ genes in human arteries. Br. J. Pharmacol. 2011, 162, 42–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- In addition to the 2-fluoro compound 18, the 2,3- and 2,4-difluoro analogues were prepared and found to have EC50 values >10 μM.
- Roeloffs R.; Wickenden A. D.; Crean C.; Werness S.; McNaughton-Smith G.; Staples J.; McNamara J. O.; Ghodadra N.; Rigdon G. C. In vivo profile of ICA-27243, a potent and selective KCNQ2/Q3 activator in rodent anticonvulsant models. J. Pharmacol. Exp. Ther. 2008, 3263818–828. [DOI] [PubMed] [Google Scholar]
- Wickenden A.; Krajewski J.; London B.; Wagoner P. K.; Wilson W. A.; Clark S.; Roeloffs R.; McNaughton-Smith G.; Rigdon G. C. ICA-27243: A novel, selective KCNQ2/Q3 potassium channel activator. Mol. Pharmacol. 2008, 73, 977–986. [DOI] [PubMed] [Google Scholar]
- A 14 day toxicity study conducted was conducted in rats dosed orally with compound 12 or the metabolite 2-chloro-5-aminopyridine. Nonhemolytic anemia was observed after dosing with both compounds.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.