

Abstract

Products 4 and 5, obtained by conjugation of doxorubicin with nitric oxide (NO) donor nitrooxy and phenylsulfonyl furoxan moieties, respectively, accumulate in doxorubicin-resistant human colon cancer cells (HT29-dx), inducing high cytotoxicity. This behavior parallels the ability of the compounds to generate NO, detected as nitrite, in these cells. Preliminary immunoblotting studies suggest that the mechanism that underlies the cytotoxic effect could involve inhibition of cellular drug efflux due to nitration of tyrosine residues of the MRP3 protein pump.
Keywords: Multidrug resistance, doxorubicin, nitric oxide, P-glycoprotein
Multidrug resistance (MDR) is one of the major reasons for the failure of cancer chemotherapy. Among the mechanisms that underlie this phenomenon, we know the most about those that increase the capacity of the cancer cells to efflux anticancer drugs, thus limiting their cellular accumulation with a consequent reduction of toxicity; this form of MDR is one of the most studied in cancer cell models.1,2 Indeed, cancer cells are able to overexpress ATP binding cassette (ABC) transporter proteins, which are the products of a family of 49 genes. ABC B1, better known as P-glycoprotein (P-gp) or MDR1, and ABC C1−6, better known as MDR-associated proteins MRP1−6, are the most representative pumps involved in MDR.3 One of the strategies followed to reverse MDR is the coadministration of an anticancer agent with an inhibitor of these efflux pumps. Elacridar, tariquidar, and laniquidar are examples of third-generation inhibitors that have been studied in clinical trials in association with a number of antitumor drugs.4,5 The identification of new MDR-reversing agents selectively targeting drug-resistance cells is a field of active investigation.6
In doxorubicin (DOXO)-resistant human epithelial colon cell line HT29-dx, there is a relationship between the reduced endogenous nitric oxide (NO) production and the resistance of these cells to the antibiotic.7 Classic NO donors, such as S-nitrosopenicillamine (SNAP), sodium nitroprusside (SNP), and S-nitrosoglutathione (GSNO), were able to reduce the efflux of DOXO from HT29-dx cells.7 This effect was neither inhibited by ODQ (1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one), a well-known inhibitor of the soluble guanylate cyclase (sGC), or by the protein-G kinase inhibitor 8-bromoguanosine-3′,5′-cyclic monophosphorothioate (Rp-8-Br-cGMPS), nor was it mimicked by 8-bromoguanosine-3′,5′-cyclic monophosphate (8-Br-cGMP), thus indicating independence from guanosine-3′,5′-cyclic monophosphate (cGMP). Experiments carried out with SNAP showed that in HT29-dx cells the nitration of tyrosine residues of MRP3 occurred, thus suggesting that nitration of the transporter is a possible mechanism for resistance inversion. Also, furoxan derivatives (1,2,5-oxadiazole 2-oxides), which are known to release NO under the action of thiol cofactors,8 can inhibit P-gp and MRP1 transporters in MDK (Madin−Darby canine kidney) -MDR1 and -MDRP1 cell lines, respectively, with an increase of cellular accumulation of DOXO when coadministered with the antibiotic.9 Experiments carried out with 3-phenylsulfonylfuroxan derivatives, the most potent inhibitors of the series, showed that these products were able to nitrate tyrosine residues of P-gp, which in this form is probably unable to efflux the antibiotic. Other studies report that a reduced supply of oxygen could induce MDR in solid cancers and that hypoxia-induced MDR could be reversed by low concentrations of NO mimetics.10,11
Using these bases as our starting point, we decided to synthesize new DOXO semisynthetic derivatives in which the antibiotic was joined through an ester linkage to moieties containing either 3-phenylsulfonylfuroxan or nitrooxy substructures, in view of the potential ability of these products to trigger anticancer action combined with a reduced capacity to induce resistance. As aforementioned, the furoxan system can release NO under the action of endogenous thiols. By contrast, NO release from organic nitrates occurs through enzymatic catalysis. A number of enzymes have been proposed for this conversion; in particular, the role of mitochondrial aldehyde dehydrogenase (mtALDH) and P-450 enzyme(s) was emphasized.12−14 The preliminary results obtained by studying the products 4 and 5 (NO−DOXO) (Chart 1) on HT29-dx cell populations are reported here and discussed.
Chart 1.

Compounds 4 and 5 were prepared from a mixture of 14-bromo and 14-chlorodaunorubicine hydrobromide 1 by reaction with 4-(2,3-dinitrooxypropyl)benzoic acid 2 and 3-[(3-phenylsulfonyl)furoxan-4-yloxy]propanoic acid 3 in acetone, respectively. After purification by flash chromatography, the products were isolated as hydrochlorides. The simple methyl esters 6 and 7, used for a comparison, were prepared treating 2 and 3 in refluxing methanol, in the presence of H2SO4 (see the Supporting Information).
HT29-dx cells were incubated in RPMI 1640-medium with DOXO and NO−DOXO (4 and 5) at different concentrations (0.5−25 μM) for 24 h. At the end of this period, the antibiotics were dosed on the cell lysate, using a spectrofluorimeter method with λ emission 553 nm and λ excitation 475 nm. At these wavelengths, the spectra of DOXO, 4, and 5 were superimposable (data not shown). The results, collected in Figure 1, show that NO−DOXOs accumulate into the cells in a concentration-dependent manner, unlike DOXO, whose intracellular concentration does not significantly increase, except at the highest concentration (25 μM). This effect was more accentuated for furoxan derivative 5 than for nitrooxy derivative 4 and was reversed (Figure 3A) by coincubation with 2-phenyl-4,4,5,5,-tetramethylimidazoline-1-oxyl 3-oxide (PTIO) and red blood cells, chosen as effective NO scavengers (Figure 3C). The pretreatment with phenylsulfonylfuroxan derivatives 3 or 7 increased the intracellular accumulation of DOXO (see the Supporting Information). The ester 7 was more potent than the corresponding simple acid 3, most likely because of the different permeability of the compounds. Furthermore, coincubation of 3 and 7 with DOXO increased the accumulation of the antibiotic into the cells (see the Supporting Information). Also, these experiments clearly indicate that the phenomenon is NO-dependent.
Figure 1.

Dose-dependent accumulation of DOXO and NO−DOXOs in HT29-dx cells. Cells were incubated for 24 h in the absence or presence of different concentrations (0.5, 1, 2.5, 5, and 25 μM) of DOXO (open columns), 4 (hatched columns), or 5 (solid columns), then lysed, and checked for the intracellular drug content, as reported in the Supporting Information. Data are presented as means ± SDs (n = 3) vs untreated cells (0): *p < 0.005.
Figure 3.

Effects of NO scavengers on DOXO and NO−DOXOs accumulation and toxicities. HT29-dx cells were incubated in the absence (−) or presence (CTRL) of 5 μM DOXO, 4, or 5 for 24 h; when indicated, 100 μM PTIO (PT) or 10 μL/mL of packed red blood cells was coincubated. The intracellular drug content (A), the extracellular activity of LDH (B), and the nitrite amount in the culture supernatant (C) were measured in duplicate as described in the Supporting Information (n = 3). Significance for A: vs CTRL DOXO, *p < 0.002; vs DOXO or 4 or 5, °p < 0.05. For B and C: vs CTRL DOXO, *p < 0.05; vs DOXO or 4 or 5, °p < 0.05.
Similarly NO−DOXOs increase their accumulation in a time-dependent manner: Elevation of the intracellular content of 5 μM antibiotics became significant after 1 h for 5 and after 3 h for 4 and was never significant between 1 and 24 h for DOXO (data not shown). This behavior parallels the drug efflux kinetics from HT29-dx cells. Indeed, when cell monolayers were loaded with different amounts of the three drugs (1−200 μM solutions) for 10 min and then the antibiotic intracellular content was examined spectrofluorimetrically on cell lysates collected after 10 and 20 min, the results outlined in Figure 2 were obtained. The measured Vmax values rank the order DOXO (8.38 ± 0.38 nmol of the drug transported/min/mg prot) > 4 (4.23 ± 0.18 nmol of the drug transported/min/mg prot) > 5 (2.65 ± 0.09 nmol of the drug transported/min/mg prot), showing that the efflux of NO−DOXOs decreases dramatically with respect to the simple antibiotic.
Figure 2.

Efflux kinetics of DOXO and NO−DOXOs from HT29-dx cells. HT29-dx cells were loaded in PBS buffer for 10 min at 37 °C with different amounts (1−200 μM) of DOXO (open circle), 4 (open square), or 5 (solid square) and then washed. One aliquot of each sample was checked immediately for the content of drug (time 0), whereas a second aliquot was further incubated in fresh PBS for an additional 10 min, washed, and tested for the drug content as previously described (for details, see the Supporting Information). The rate of DOXO efflux was calculated from the difference between the drug concentration at time 0 and at 10 min, respectively. Measurements (n = 3) were done in duplicate, and data are presented (means ± SDs) as the rate of efflux vs the intracellular drug concentration at time 0. The significance of 4 vs DOXO: *p < 0.01; significance of 5 vs DOXO, °p < 0.002; significance of 5 vs 4, ◊p < 0.001.
As a consequence of this behavior, the toxicity of NO−DOXOs was definitively higher than that of DOXO. Indeed, when incubated with 5 μM solutions of NO−DOXOs, the cells exhibited a significant increase in the release of lactate dehydrogenase (LDH), a sensitive index of a drug's toxicity, with respect to DOXO alone (Figure 3B). The level of LDH was higher for the furoxan derivative 5 than for the nitrooxy-substituted product 4. In both cases, the toxicity was reduced in the presence of the NO scavengers PTIO and red blood cells. By contrast, no significant increase in LDH levels was observed with the simple acids 2 and 3, used to obtain the two codrugs 4 and 5, and the related methyl esters 6 and 7. This finding indicates that the cytotoxic effect is due to the anthracycline pharmacophore rather than to the NO-releasing moieties.
To determine if NO−DOXOs are able to restore NO production in HT29-dx cells, solutions of 4 and 5 of different concentration (0.5−25 μM) were incubated with cell monolayers for 24 h, and nitrite in the culture medium was then quantified by Griess reaction. The results reported in Figure 4 show that nitrite production is concentration-dependent and that it is maximized for furoxan derivative 5. The nitrite release was time-dependent: Using a 5 μM solution of the antibiotics, a significant increase of nitrite was detected after 1 h with 5 and after 3 h with 4 but remained not significant between 1 and 24 h with DOXO (data not shown).
Figure 4.
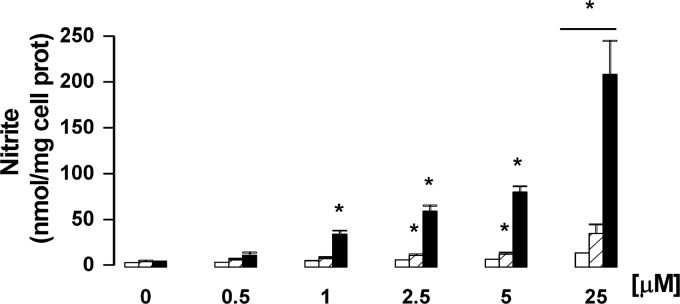
Dose-dependent production of nitrite in HT29-dx cells treated with DOXO and NO−DOXOs. Cells were incubated for 24 h in the absence or presence of different concentrations (0.5, 1, 2.5, 5, and 25 μM) of DOXO (open columns), 4 (hatched columns), or 5 (solid columns), and then, the levels of nitrite in the cell medium were evaluated by the Griess method. Data are presented as means ± SDs (n = 3) vs untreated cells (0): *p < 0.001.
It is known that drug efflux pumps P-gp and MRP3 are overexpressed in HT29-dx cells.7 To check whether the capacity of 4 and 5 to accumulate in these cells could be due to the nitration of tyrosine residues of these efflux pumps, following NO release by the products, HT29-dx cells were incubated for 24 h with the two antibiotics or with DOXO. The whole cellular lysate was immunoprecipitated with a specific antinitrotyrosine antibody, and the immunoprecipitated proteins were subjected to Western blotting, using anti-P-gp and anti-MRP3 antibodies (Figure 5). The presence of nitrotyrosine residues was detectable on MRP3 protein in HT29-dx cells incubated with 4 or 5 but not with DOXO alone. A number of reactive nitrogen species, generated under different conditions and environments, can mediate this nitration. The amount of nitro tyrosine produced by 5 appeared greater than that produced by 4. This result is in keeping with the higher levels of nitrite detected with 5. Also, the intracellular accumulation of DOXO with 5 was higher than with 4. It is known that DOXO up-regulates the inducible isoform of NO synthase (iNOS),15 thereby increasing the endogenous synthesis of NO in mammalian cells. Thus, it is possible that the nitration of MRP3 elicited by 4 and 5 is due in part to the release of NO by the products and in part to the increased production of endogenous NO via the induction of the iNOS enzyme. No nitration occurred in P-gp protein. Both P-gp and MRP3 are involved in DOXO efflux, depending on the different tissues and on the relative abundance of the transporters in tumor cells.3 Because the treatment with the NO donor compounds 3 and 7 did not modify the activity of P-gp in HT29-dx cells (see the rhodamine 123 test in the Supporting Information), we have to conclude that this transporter does not play a primary role in the effect exerted by NO on DOXO retention. This is most likely due to the lower number of tyrosine residues of the transporter with respect to MRP3 and/or to its lower expression.7,16
Figure 5.

Nitration of drug efflux pumps by DOXO and NO−DOXOs in HT29-dx cells. HT29-dx cells were incubated for 24 h with 5 μM DOXO, 4, or 5. Afterward, cells were lysed, and the whole cellular lysate was immunoprecipitated with an antinitrotyrosine polyclonal antibody. The immunoprecipitated proteins were subjected to Western blotting, using an anti-Pgp or an anti-MRP3 antibody (see the Supporting Information). An aliquot of cells lysate before immunoprecipitation was analyzed for total P-gp and MRP3. The expression of the housekeeping protein GAPDH (glyceraldehydes-3-phosphate dehydrogenase) was measured as a control of equal loading. The experiment is representative of three similar experiments.
The preliminary results disclosed in the present work show that the design of hybrid NO donor antitumor drugs could be a useful strategy to address the problem of MDR in cancer therapy.
Acknowledgments
We are indebted to Istituto Zooprofilattico Sperimentale del Piemonte, Liguria e Valle d'Aosta, for mass spectral data.
Glossary
Abbreviations
- DOXO
doxorubicin
- NO
nitric oxide
- MDR
multidrug resistance
- P-gp
P-glycoprotein
- ABC
ATP binding cassette
- SNAP
S-nitrosopenicillamine
- SNP
sodium nitroprusside
- GSNO
S-nitrosoglutathione
- ODQ
1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one
- sGC
soluble guanylate cyclase
- Rp-8-Br-cGMPS
8-bromoguanosine-3′,5′-cyclic monophosphorothioate
- 8-Br-cGMP
8-bromoguanosine cyclic monophosphate
- cGMP
guanosine-3′,5′-cyclicmonophosphate
- MDCK cells
Madin−Darby canine kidney cells
- mtALDH
mitochondrial aldehyde dehydrogenase
Supporting Information Available
Syntheses and characterization data for the compounds and procedures for biological assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Gottesman M. M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [DOI] [PubMed] [Google Scholar]
- Szakacs G.; Paterson J. K.; Ludwig J. A.; Booth-Genthe C.; Gottesman M. M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discovery 2006, 5, 219–234. [DOI] [PubMed] [Google Scholar]
- Gottesman M. M.; Fojo T.; Bates S. E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [DOI] [PubMed] [Google Scholar]
- Colabufo N. A.; Berardi F.; Cantore M.; Contino M.; Inglese C.; Niso M.; Perrone R. Perspectives of P-glycoprotein modulating agents in oncology and neurodegenerative diseases: Pharmaceutical, biological, and diagnostic potentials. J. Med. Chem. 2010, 53, 1883–1897and references therein. [DOI] [PubMed] [Google Scholar]
- www.clinicaltrials.gov (NCT00028873).
- Türk D.; Hall M. D.; Chu B. F.; Ludwig J. A.; Fales H. M.; Gottesman M. M.; Szakács G. Identification of compounds selectively killing multidrug-resistant cancer cells. Cancer Res. 2009, 69, 8293–8301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riganti C.; Miraglia E.; Viarisio D.; Costamagna C.; Pescarmona G.; Ghigo D.; Bosia A. Nitric oxide reverts the resistance to doxorubicin in human colon cancer cells by inhibiting the drug efflux. Cancer Res. 2005, 65, 516–525. [PubMed] [Google Scholar]
- Gasco A.; Schoenafinger K.. The NO-releasing heterocycles. In Nitric Oxide Donors; Wang P. G., Cai T. B., Taniguchi N., Eds.; Wiley-VCH Verlag GmbH & Co KGaA: Weinheim, 2005; pp 131−175. [Google Scholar]
- Fruttero R.; Crosetti M.; Chegaev K.; Guglielmo S.; Gasco A.; Berardi F.; Niso M.; Perrone R.; Panaro M. A.; Colabufo N. A. Phenylsulfonylfuroxans as modulators of multidrug-resistance associated protein-1 and P-glycoprotein. J. Med. Chem. 2010, 53, 5467–5475. [DOI] [PubMed] [Google Scholar]
- Matthews N. E.; Adams M. A.; Maxwell L. R.; Gofton T. E.; Graham C. H. Nitric oxide-mediated regulation of chemosensitivity in cancer cells. J. Natl. Cancer I 2001, 93, 1879–1885. [DOI] [PubMed] [Google Scholar]
- Frederiksen L. J.; Siemens D. R.; Heaton J. P.; Maxwell L. R.; Adams M. A.; Graham C. H. Hypoxia induced resistance to doxorubicin in prostate cancer cells is inhibited by low concentrations of glyceryl trinitrate. J. Urol. 2003, 170, 1003–1007. [DOI] [PubMed] [Google Scholar]
- Mayer B.; Beretta M. The enigma of nitroglycerin bioactivation and nitrate tolerance: News, views and troubles. Br. J. Pharmacol. 2008, 155, 170–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiber A.; Wenzel P.; Oelze M.; Munzel T. New insights into bioactivation of organic nitrates, nitrate tolerance and cross-tolerance. Clin. Res. Cardiol. 2008, 97, 12–20. [DOI] [PubMed] [Google Scholar]
- Grossi L. Nitrite anion: The key intermediate in alkyl nitrates degradative mechanism. J. Med. Chem. 2008, 51, 3318–3321. [DOI] [PubMed] [Google Scholar]
- Aldieri E.; Bergandi L.; Riganti C.; Costamagna C.; Bosia A.; Ghigo D. Doxorubicin induces an increase of nitric oxide synthesis in rat cardiac cells that is inhibited by iron supplementation. Toxicol. Appl. Pharmacol. 2002, 185, 85–90. [DOI] [PubMed] [Google Scholar]
- Doublier S.; Riganti C.; Voena C.; Costamagna C.; Aldieri E.; Pescarmona G.; Ghigo D.; Bosia A. RhoA silencing reverts the resistance to doxorubicin in human colon cancer cells. Mol. Cancer Res. 2008, 6, 1607–1620. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.