

Abstract

Inhibition of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase (MEK) represents a promising strategy for the discovery of a new generation of anticancer chemotherapeutics. Our synthetic efforts, beginning from the lead compound 2, were directed at improving antiproliferative activity against cancer cells as well as various drug properties. These efforts led to the discovery of N-{3-[3-cyclopropyl-5-(2-fluoro-4-iodophenylamino)-6,8-dimethyl-2,4,7-trioxo-3,4,6,7-tetrahydro-2H-pyrido[4,3-d]pyrimidin-1-yl]phenyl}acetamide dimethylsulfoxide solvate (GSK1120212, JTP-74057 DMSO solvate; 1), a selective and highly potent MEK inhibitor with improved drug properties. We further confirmed that the antiproliferative activity correlates with cellular MEK inhibition and observed significant antitumor activity with daily oral dosing of 1 in a tumor xenograft model. These qualities led to the selection of 1 for clinical development.
Keywords: GSK1120212, JTP-74057, antiproliferative, MEK inhibitor
Activation of the p42/44 MAPK signaling pathway comprising mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) kinase (MEK)−ERK has been implicated in the pathogenesis and progression of various human malignant tumors.1,2 The MEK−ERK pathway is often activated by mutation of upstream factors, BRAF or Ras, or by the signals of constitutively activated cell-surface receptors.1−6 Therefore, inhibition of the p42/44 MapK pathway is an attractive therapeutic strategy for multiple cancers. Indeed, several small molecules that target the pathway, including MEK and Raf inhibitors, are being tested in human clinical studies.1,2,6
During a high-throughput screening for compounds that can induce expression of the cyclin-dependent kinase (CDK) 4/6 inhibitor p15INK4b, we identified 2 (Figure 1).7 Subsequent experiments confirmed that 2 has an antiproliferative activity against human cancer cell lines ACHN (renal adenocarcinoma) and HT-29 (colorectal adenocarcinoma) with IC50 values of 4800 and 990 nM, respectively. We conducted a medicinal chemistry campaign, chemically modifying 2 to optimize for these antiproliferative effects. Our synthetic efforts led to the discovery of orally bioavailable GSK1120212 (JTP-74057 DMSO solvate) 1 (Figure 1), demonstrating selective inhibition of proliferation in various BRAF mutant cancer cell lines. This compound was confirmed through molecular target analyses7,8 to be a highly potent and selective inhibitor of MEK1/2. We describe herein the structure−activity relationship (SAR) studies on 2 guided by ACHN and HT-29 cancer cell lines growth inhibitory activity and the detailed characteristics of 1.9
Figure 1.

Structures (1−3) and cellular activities (2 and 3). aNumbers in parentheses represent numbers of determinations.
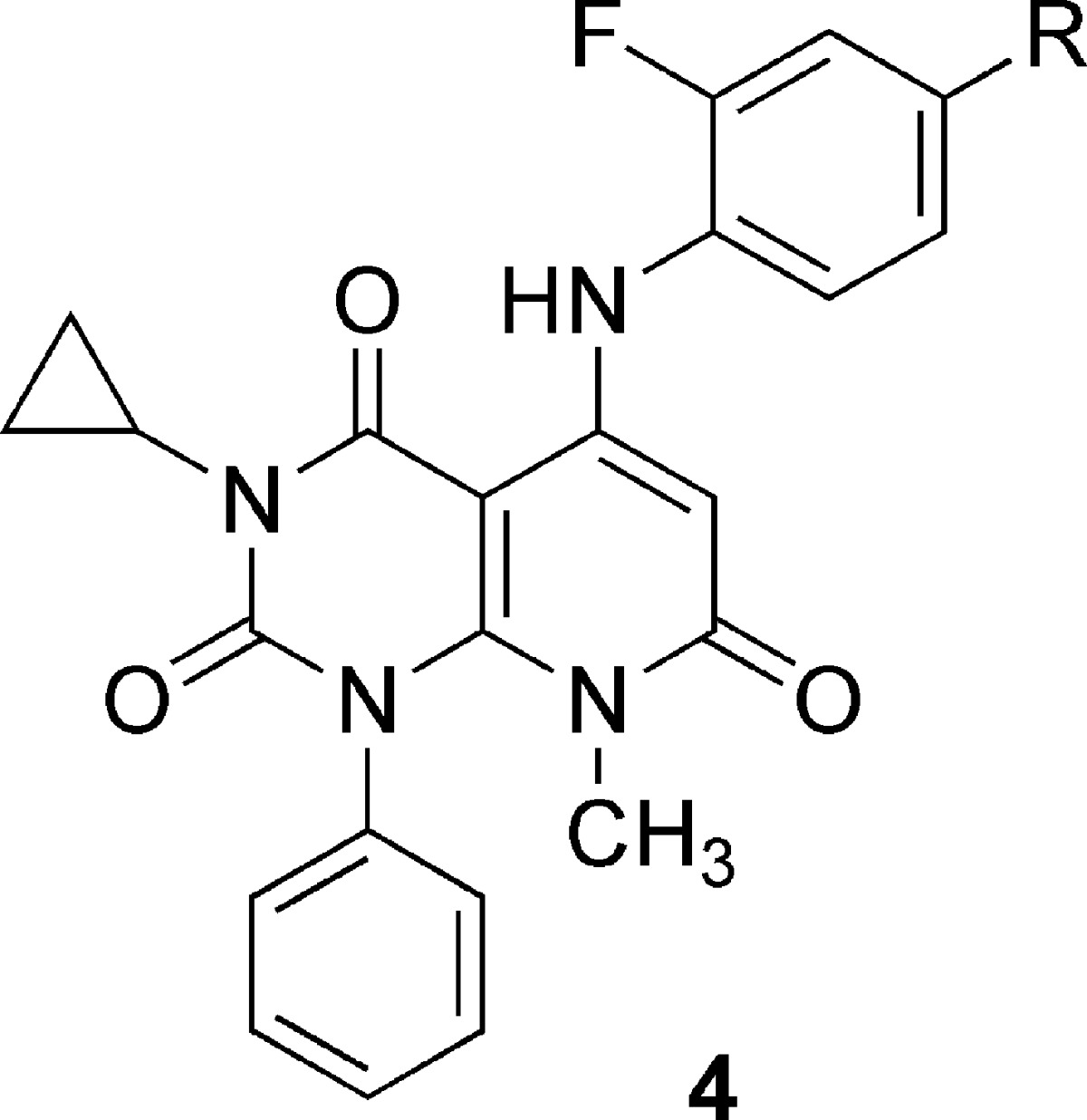
Compound 2 has three aromatic rings on the pyridopyrimidine core, making it an unattractive starting point for further optimization due to the high hydrophobicity (ClogP 6.3). With the aim to reduce hydrophobicity of 2, we replaced each aromatic ring with small alkyl groups by turns. Replacement of the benzene ring at the 3-position (upper left) of the pyridopyrimidine core with a cyclopropyl ring and a chlorine atom on the aniline ring with bromine resulted in 3 (Figure 1), which exhibited reduced hydrophobicity (ClogP 5.0) and improved potency by 4-fold. Replacement of benzene rings at position 1 (lower left) or 5 (upper right) with lower alkyl groups resulted in a loss of potency (data not shown). Encouragingly, 3 showed the first sign of antitumor effect in an in vivo model, the nude mouse HT-29 xenograft model (data not shown). We next introduced a small substituent (e.g., methyl, halogen, methoxy, etc.) at the 2- or 3-position of the aniline ring of 3. Introduction of fluorine atom at the 2-position of the aniline ring (4l, Table 1) improved the potency by ca. 4-fold as compared to 3 with a small increase in ClogP (5.2).
Table 1. Substitution at the 4-Position of Aniline SARa.
| IC50 (nM) |
|||
|---|---|---|---|
| compd | R | ACHN | HT-29 |
| 4a | H | 10000 (1) | 1300 (1) |
| 4b | Me | 1300 (2) | 110 (2) |
| 4c | Et | 185 (3) | 27 (3) |
| 4d | Pr | 2100 (2) | 230 (2) |
| 4e | cPr | 290 (2) | 39 (2) |
| 4f | vinyl | 104 (3) | 15 (3) |
| 4g | C≡CH (ethynyl) | 42 (2) | 5.9 (2) |
| 4h | C≡C−Me | >3000 (1) | >1000 (1) |
| 4i | CN | >10000 (1) | 1600 (1) |
| 4j | F | >3000 (1) | >1000 (1) |
| 4k | Cl | 4600 (2) | 102 (2) |
| 4l | Br | 325 (4) | 15 (4) |
| 4m | I | 13 (2) | 1.5 (2) |
Numbers in parentheses represent numbers of determinations.
To further increase the potency, we proceeded to explore the substitution at the 4-position on the aniline ring (Table 1). To investigate the size limit of the substituent at the 4-position, we first replaced the bromine atom of 4l with alkyl groups (4b−e). As the size increased from hydrogen (4a) to ethyl (4c), the compounds became more potent, while removal of the bromine atom of 4l resulted in loss of activity (4a). Propyl (4d) showed less potency, but cyclopropyl (4e) restored potency to a similar level of 4c. Introduction of the unsaturated alkyls vinyl (4f) and ethynyl (4g) further increased the potency, although one-carbon extension deprived 4g of activity (4h). These results suggested the existence of a sterically constrained region of the target protein of these compounds in the vicinity of the para-substituents. The compound with cyano (4i) showed weak potency in spite of having similar shape and size as the ethynyl group of potent compound 4g. We next introduced halogen atoms (4j−m) and found that iodine, synthetic intermediate for 4g etc., significantly increased the potency to IC50 values of 13 and 1.5 nM for ACHN and HT-29, respectively (4m). Thus, we succeeded in improving potency by 500-fold from the lead 2 to 4m.
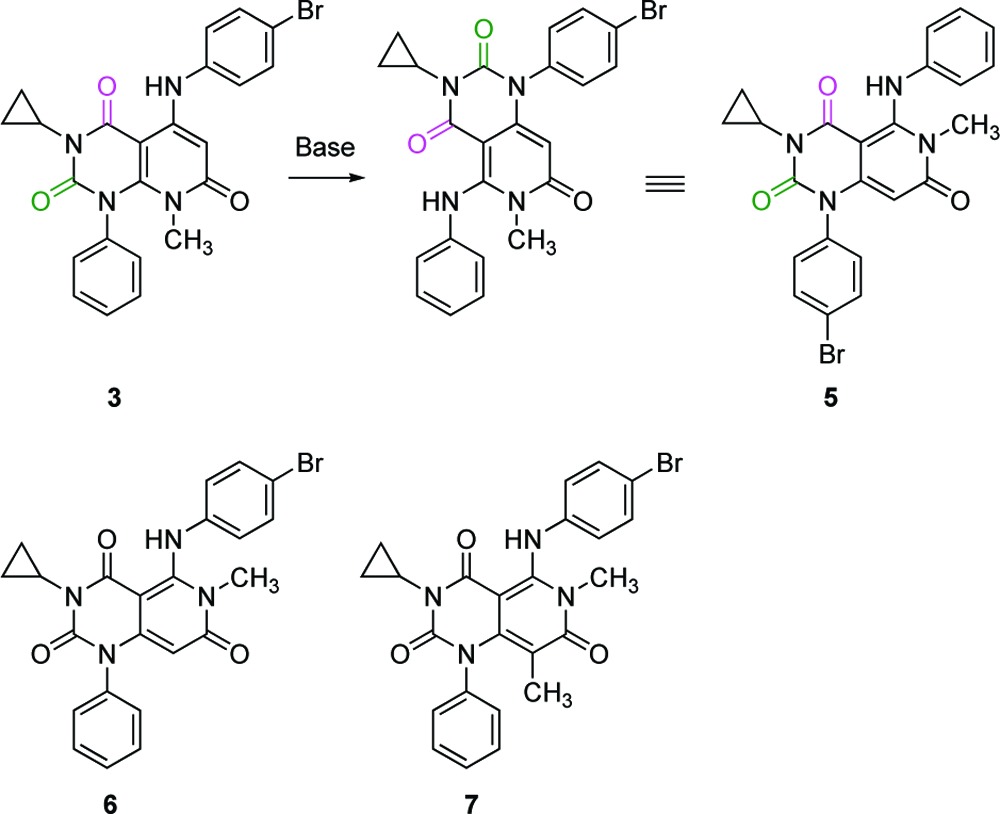
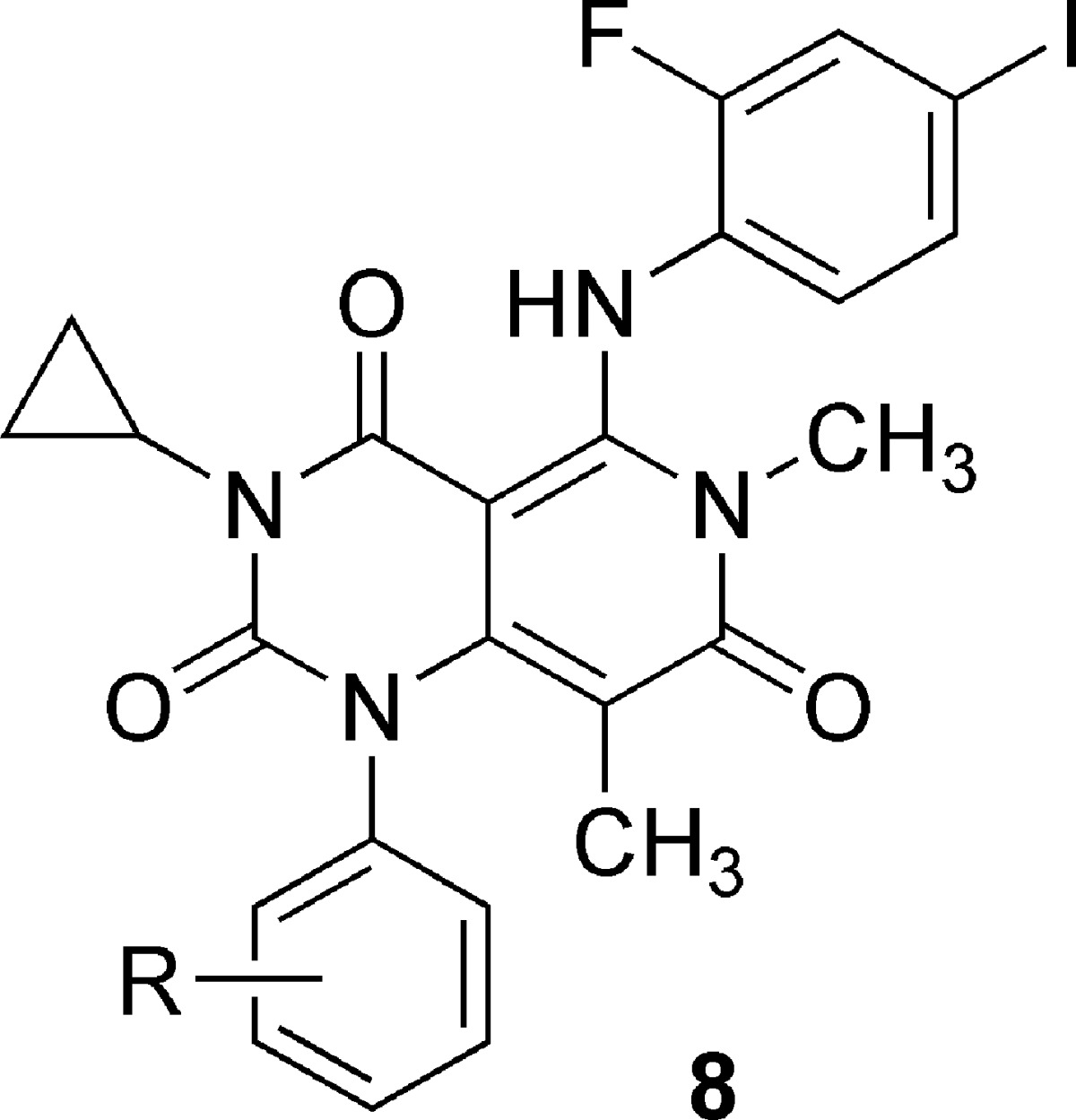
Unfortunately, the compounds detailed above, possessing a pyrido[2,3-d]pyrimidine core including 4m, were proven to be unstable under weak basic conditions and not appropriate for further development as drug candidates. For example, treatment of 3 with potassium carbonate in methanol/chloroform at ambient temperature provided 5 having a pyrido[4,3-d]pyrimidine core in 97% yield. We assumed that 5 was formed through the opening of the pyrimidinedione ring by the attack of nucleophile (in this case methanol) at the C-2 carbonyl and subsequent ring closure to another pyrimidinedione involving the neighboring nitrogen atom of the other aniline (upper right). Compound 5 was totally inactive (Table 2). However, we hypothesized that placement of appropriate substituents in the same alignment as 4m to the postarrangement stable scaffold might afford chemically stable and highly active compounds, because the spatial arrangement of two benzene and cyclopropane rings and three carbonyls on two heterocyclic cores of 3 and 5 was almost identical. Although the inhibitory activity was not restored by just a transposition of bromine atom from lower left phenyl (5) to the aniline ring at the 5-position (6), incorporation of the methyl to 6 at the 8-position of the heterocyclic core to yield 7 raised the potency up to the level of prerearranged 3. We reasoned that this was due to a potential effect on the orientation of the neighboring phenyl ring at the 1-position of the heterocyclic core. Finally, introduction of 2-F,4-I aniline moiety in the same substitution fashion as 4m to give 8a provided a substantial boost in potency as we anticipated (Table 3).
Table 2. Heteroaromatic Core Transformation SARa.
| IC50 (nM) |
||
|---|---|---|
| compd | ACHN | HT-29 |
| 3 | 1270 (2) | 100 (2) |
| 5 | >3000 (1) | >1000 (1) |
| 6 | >3000 (1) | >1000 (1) |
| 7 | 830 (2) | 135 (2) |
Numbers in parentheses represent numbers of determinations.
Table 3. Polar Group Substitution SARa.
| IC50 (nM) |
|||
|---|---|---|---|
| compd | R | ACHN | HT-29 |
| 8a | H | 25 (2) | 1.7 (2) |
| 8b | 3-NHCOMe | 9.8 (5) | 0.57 (10) |
| 8c | 4-NHCOMe | 195 (2) | 14 (2) |
| 8d | 3,5-diNHCOMe | >1000 (1) | >100 (1) |
| 8e | 3-NHSO2Me | 6.4 (5) | 0.52 (5) |
| 8f | 4-NHSO2Me | 22 (5) | 1.4 (5) |
Numbers in parentheses represent numbers of determinations.
Although 8a exhibited IC50 values of 25 and 1.7 nM for ACHN and HT-29, respectively, this compound displayed very low aqueous solubility due to the high ClogP of 6.0. We investigated polar group substitution of the phenyl ring at the 1-position of the heterocyclic core as a means to optimize physicochemical properties while further improving potency. An acetamide and a methanesulfonamide analogues were found to be the most active among a variety of polar groups explored. Representative examples are shown in Table 3. Compounds 8b and 8e substituted at the meta-position improved potency by 3-fold and were coupled with the reduced hydrophobicity (ClogP 8b, 5.0; 8e, 4.8). Disubstituted analogue 8d exhibited no activity up to 100 nM. Compounds 8c and 8f each had a para-substituent and showed moderately less or similar potency as compared to 8a. Consistent with their reduced hydrophobicity, 8b and 8e showed improved solubility and permeability as compared to 8a (Table 4) as well as better bioavailability than 4l. Compound 8b (JTP-74057) represented the best combination of desired features, with IC50 values of 9.8 and 0.57 nM for ACHN and HT-29, respectively, and was subsequently determined to be a candidate for further development based on the results of in vivo xenograft model and toxicity studies.
Table 4. PK Related Parametersa.
| compd | solubility (PBS) | Caco2 permeability | AUCb | % Fb |
|---|---|---|---|---|
| 4l | <0.4 | <0.1 | 7.7 | 2.1 |
| 4m | <0.3 | <0.1 | ||
| 8a | <0.2 | 14.0 | ||
| 8b | 5.3 | 26.0 | 88.3 | 30 |
| 8e | 3.3 | 17.1 | 59.2 | 59 |
Units: solubility, μM; Caco2 permeability, cm/s 10−6; and AUC, μM h.
AUC and % F were determined after dosing orally to mice at 30 mg/kg in 0.5% methyl cellulose suspension.
Initial batches of 8b having a mp of 180 °C showed acceptable oral bioavailability as shown in Table 4. However, through crystalline polymorphism studies, the most stable form of mp 300 °C appeared and was found to show poor oral exposure. Compound 8b was weakly basic compound and not suitable for stable salt formation. In hope of restoring oral bioavailability, we prepared various solvates. Fortunately, we found that two solvates with class 3 solvent acetic acid and DMSO showed similar oral bioavailability to 8b with an mp of 180 °C. DMSO solvate 1 showed desirable solid state properties and was selected for development.
Because the molecular target of this compound series was still unknown at that time, we carried out a compound-immobilized affinity chromatography in which the analogues of 8b were immobilized to resin. As a result, we found that 8b binds to MEK1/2 and inhibits its kinase activity with high specificity.7,8,10,11
To confirm that cellular antiproliferative activity is due to cellular inhibition of MEK1/2 kinase activity, the DMSO solvate 1 was tested in both a cellular phospho-ERK1/2 assay and cell proliferation assays. In BRAF mutant SK-MEL-28 cells and KRAS mutant HCT116 cells, 1 caused dose-dependent inhibition of ERK1/2 phosphorylation as well as dose-dependent growth inhibition. We then considered the question of the relationship between the extent of ERK1/2 phosphorylation (p-ERK1/2) inhibition and the extent of growth inhibition in similar time frames. To address that relationship, we compared the respective curves of phosphoinhibition and growth inhibition after a 72 h drug treatment (Figure 2). In both SK-MEL-28 and HCT116 cells, 1 inhibits 50% p-ERK1/2 at nearly equivalent concentrations (0.8 and 1.8 nM, respectively). However, as the slopes of the curves reflect, in SK-MEL-28 cells, 1 inhibits 90% p-ERK1/2 at a lower concentration (3.4 nM) than in HCT116 (33.3 nM). Furthermore, in both cell lines, 50% growth inhibition was only achieved at concentrations 1 that produced near complete ERK1/2 inhibition (85 and 90%, respectively). These data suggest that near complete inhibition of ERK1/2 phosphorylation is required to achieve significant cell growth inhibition and that cells inherently differ in the slope of their MEK inhibition dose−response curve.
Figure 2.

In both BRAFV600E SK-MEL-28 cells and KRASG13D HCT116 cells, compound 1 (GSK1120212) potently inhibits both phosphorylation of ERK1/2 (filled circles) and cell growth (open circles) over 72 h. Differences in growth assay IC50 values between cell lines appear to correlate with the concentrations required for ca. 90% inhibition of ERK1/2 phosphorylation.
In addition, 1 was then evaluated in vivo in an A549 (KRAS mutant cell line) xenograft model, orally dosing daily for 21 days (qd × 21). In this study (Figure 3), near complete tumor growth inhibition was observed at 5.0 and 2.5 mg/kg [92 and 87% tumor growth inhibition (TGI), respectively] and to a lesser degree at 0.5 and 0.1 mg/kg (62 and 58% TGI). [Notably, although 5 mg/kg was the maximally tolerated dose (MTD) in this study, 3 mg/kg is the typically observed MTD.] Dose-dependent antitumor activity with 1 treatment has been similarly reported for several other KRAS and BRAF mutant tumor models.10
Figure 3.
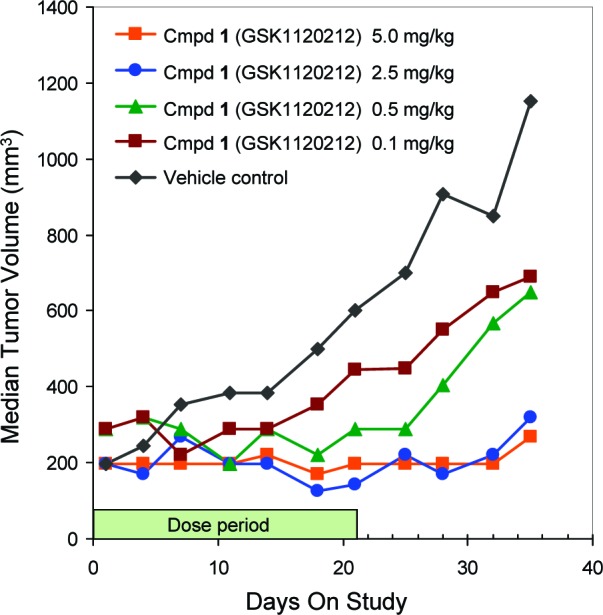
Compound 1 causes significant antitumor activity in KRASG12S A549 tumor xenograft model. Daily oral administration for 21 days resulted in significant and dose-dependent tumor growth inhibition.
Although SAR studies from the lead compound to JTP-74057 were guided by cancer cell lines growth inhibitory activity rather than MEK inhibitory activity, we realized after confirmation of the enzymatic target that the resultant compounds shared SAR features to those of known MEK inhibitors.12,13 In particular, the best substitution fashion of the aniline ring, 2-F,4-I, of 1 is also employed in the second-generation MEK inhibitor PD032590112 (Pfizer). The methyl group of 1, introduced at the 8-position of the heterocyclic core to boost the potency, is found in the corresponding position of pyridone type MEK inhibitors.14−16 A marked difference of 1 from the previously identified MEK inhibitors is its unique pyrido[4,3-d]pyrimidine core structure. This unique feature likely contributes to 1's good pharmacokinetic profile, with sustained plasma drug concentrations and narrow “peak/trough” ratio,10 resulting in significant anticancer activity with once-daily dosing.
In conclusion, starting from an initial lead 2, SAR studies to improve antiproliferative activity against cancer cells culminated in the discovery of 1, GSK1120212 (JTP-74057 DMSO Solvate), a highly potent, selective, and orally active MEK inhibitor. Compound 1 displays remarkable potency in cellular assays for ERK1/2 phosphorylation and growth inhibition and excellent in vivo activity. Inhibition of the MEK−ERK pathway offers a very promising therapeutic strategy for cancers with activating mutant Ras and Raf, and 1 has been advanced into phase III trials for advanced or metastatic BRAF mutant melanoma as well as phase I and II studies in subjects with solid tumors or leukemia.
Acknowledgments
We thank our Analytical Research and Development Laboratories for collecting analytical data and Dr. Jun-ichi Haruta for helpful discussions.
Glossary
Abbreviations
- MEK
mitogen-activated protein kinase/extracellular signal-regulated kinase kinase
- ERK
mitogen-activated protein kinase/extracellular signal-regulated kinase
Supporting Information Available
Biological assays and experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Sebolt-Leopold J. S.; Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat. Rev. Cancer 2004, 4, 937–947. [DOI] [PubMed] [Google Scholar]
- McCubrey J. A.; Milella M.; Tafuri A.; Martelli A. M.; Lunghi P.; Bonati A.; Cervello M.; Lee J. T.; Steelman L. S. Targeting the Raf/MEK/ERK pathway with small-molecule inhibitors. Curr. Opin. Invest. Drugs 2008, 9, 614–630. [PubMed] [Google Scholar]
- Davies H.; Bignell G. R.; Cox C.; Stephens P.; Edkins S.; Clegg S.; Teague J.; Woffendin H.; Garnett M. J.; Bottomley W.; Davis N.; Dicks E.; Ewing R.; Floyd Y.; Gray K.; Hall S.; Hawes R.; Hughes J.; Kosmidou V.; Menzies A.; Mould C.; Parker A.; Stevens C.; Watt S.; Hooper S.; Wilson R.; Jayatilake H.; Gusterson B. A.; Cooper C.; Shipley J.; Hargrave D.; Pritchard-Jones K.; Maitland N.; Chenevix-Trench G.; Riggins G. J.; Bigner D. D.; Palmieri G.; Cossu A.; Flanagan A.; Nicholson A.; Ho J. W.; Leung S. Y.; Yuen S. T.; Weber B. L.; Seigler H. F.; Darrow T. L.; Paterson H.; Marais R.; Marshall C. J.; Wooster R.; Stratton M. R.; Futreal P. A. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [DOI] [PubMed] [Google Scholar]
- Adjei A. A. Blocking oncogenic Ras signaling for cancer therapy. J. Natl. Cancer Inst. 2001, 93, 1062–1074. [DOI] [PubMed] [Google Scholar]
- Porter A. C.; Vaillancourt R. R. Tyrosine kinase receptor-activated signal transduction pathways which lead to oncogenesis. Oncogene 1998, 17, 1343–1352. [DOI] [PubMed] [Google Scholar]
- Frémin C.; Meloche S. From basic research to clinical development of MEK1/2 inhibitors for cancer therapy. J. Hematol. Oncol. 2010, 3, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi T.; Yoshida T.; Kurachi R.; Kakegawa J.; Hori Y.; Nanayama T.; Hayakawa K.; Abe H.; Takagi K.; Matsuzaki Y.; Koyama M.; Yogosawa S.; Sowa Y.; Yamori T.; Tajima N.; Sakai T. Identification of JTP-70902, a p15INK4b-inductive compound, as a novel MEK1/2 inhibitor. Cancer Sci. 2007, 98, 1809–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T.;et al. Unpublished results.
- Sakai T.; Kawasaki H.; Abe H.; Hayakawa K.; Iida T.; Kikuchi S.; Yamaguchi T.; Nanayama T.; Kurachi H.; Tamaru M.; Hori Y.; Takahashi M.; Yoshida T.. 5-Amino-2,4,7-trioxo-3,4,7,8-tetrahydro-2H-pyrido-[2,3-d]-pyrimidine derivatives and related compounds for the treatment of cancer. WO-2005121142, 2005.
- Gilmartin A. G.; Bleam M. R.; Groy A.; Moss K. G.; Minthorn E. A.; Kulkarni S. G.; Rominger C. M.; Erskine S; Fisher K. E.; Yang J.; Zappacosta F.; Annan R.; Sutton D.; Laquerre S. G.. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin. Cancer Res., published online January 18, 2011; DOI: 10.1158/1078-0432. [DOI] [PubMed] [Google Scholar]
- The specificity of GSK1120212 for MEK1/2 was confirmed against a panel of >180 kinases including B-Raf, C-RAf, and MEK5, the closest kinase homologue. See ref (10) andYamaguchi T.; et al. Unpublished results.
- Barrett S. D.; Bridges A. J.; Dudley D. T.; Saltiel A. R.; Fergus J. H.; Flamme C. M.; Delaney A. M.; Kaufman M.; LePage S.; Leopold W. R.; Przybranowski S. A.; Sebolt-Leopold J.; Van Becelaere K; Doherty A. M.; Kennedy R. M.; Marston D.; Howard W. A. Jr.; Smith Y.; Warmus J. S.; Tecle H. The discovery of the benzhydroxamate MEK inhibitors CI-1040 and PD 0325901. Bioorg. Med. Chem. Lett. 2008, 18, 6501–6504. [DOI] [PubMed] [Google Scholar]
- Wallace E.; Yang H. W.; Lyssikatos J. P.. Bicyclic inhibitors of MEK and methods of use thereof. WO-05051300, 2004.
- Spicer J. A.; Rewcastle G. W.; Kaufman M. D.; Black S. L.; Plummer M. S.; Denny W. A.; Quin J. III; Shahripour A. B.; Barrett S. D.; Whitehead C. E.; Milbank J. B. J.; Ohren J. F.; Gowan R. C.; Omer C.; Camp H. S.; Esmaeil N; Moore K.; Sebolt-Leopold J. S.; Pryzbranowski S.; Merriman R. L.; Ortwine D. F.; Warmus J. S.; Flamme C. M.; Pavlovsky A. G.; Tecle H. 4-Anilino-5-carboxamido-2-pyridone Derivatives as Noncompetitive Inhibitors of Mitogen-Activated Protein Kinase Kinase. J. Med. Chem. 2007, 50, 5090–5102. [DOI] [PubMed] [Google Scholar]
- Wallace E. M.; Lyssikatos J.; Blake J. F.; Seo J.; Yang H. W.; Yeh T. C.; Perrier M.; Jarski H.; Marsh V.; Poch G.; Livingston M. G.; Otten J.; Hingorani G.; Woessner R.; Lee P.; Winkler J.; Koch K. Potent and Selective Mitogen-Activated Protein Kinase Kinase (MEK) 1,2 Inhibitors. 1. 4-(4-Bromo-2-fluorophenylamino)-1-methylpyridin-2(1H)-ones. J. Med. Chem. 2006, 49, 441–444. [DOI] [PubMed] [Google Scholar]
- In ref (14), it was pointed out that N-Me pyridones are active in the kinase and cellular assays, but N-H pyridones are not potent. They speculated that the drop of activity of N-H pyridones may be attributed to the loss of the key hydrogen bond to Ser212 due to partial tautomerization of the pyridone system. However, in our case, introduction of a methyl group in the corresponding position, that is, 6 to 7, does not change the tautomerization state of the pyrido[4,3-d]pyrimidine core, so we speculated that methyl group affected the orientation of neighboring phenyl ring.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.