

Abstract
The process of immobilizing enzymes onto solid supports for bioreactions has some compelling advantages compared to their solution-based counterpart including the facile separation of enzyme from products, elimination of enzyme autodigestion, and increased enzyme stability and activity. We report the immobilization of λ-exonuclease onto poly(methylmethacrylate) (PMMA) micropillars populated within a microfluidic device for the on-chip digestion of double-stranded DNA. Enzyme immobilization was successfully accomplished using 3-(3-dimethylaminopropyl) carbodiimide/N-hydroxysuccinimide (EDC/NHS) coupling to carboxylic acid functionalized PMMA micropillars. Our results suggest that the efficiency for the catalysis of dsDNA digestion using λ-exonuclease, including its processivity and reaction rate, were higher when the enzyme was attached to a solid support compared to the free solution digestion. We obtained a clipping rate of 1.0 × 103 nucleotides s–1 for the digestion of λ-DNA (48.5 kbp) by λ-exonuclease. The kinetic behavior of the solid-phase reactor could be described by a fractal Michaelis–Menten model with a catalytic efficiency nearly 17% better than the homogeneous solution-phase reaction. The results from this work will have important ramifications in new single-molecule DNA sequencing strategies that employ free mononucleotide identification.
Recently, solid-phase bioreactors have found interesting applications in the areas of single-molecule enzymology,1 biochemical manufacturing,2 and nanotechnology.3,4 A subgroup of solid-phase bioreactors called immobilized microfluidic enzymatic reactors (IMERs)5,6 comprises systems in which an enzyme is immobilized within the channels of a microfluidic device. There are several advantages associated with these systems as opposed to their homogeneous (liquid-based) counterparts. These include enhanced stability and activity of the tethered enzyme relative to the enzyme in free solution,7−9 reduced interference from catalytic enzymes during the analysis phase of the assay,10 and reusability of the enzyme.11−13 In the case of proteolytic enzymes such as trypsin, immobilization of the enzyme can prevent autodigestion as well.14 The reported success in the attachment of enzymes onto solid supports stems from the availability of several enzyme/solid surface attachment chemistries.15,16 Based on the plethora of available attachment chemistries, solid supports such as silicon, glass, or polymers, can be selected to accommodate the pendant functional groups available on most proteins and the fabrication strategies used to produce the fluidic devices associated with IMERs.17,18
Of the numerous chemical strategies for protein attachment, many rely upon reactions between functional groups within the protein (amine and/or carboxylic acids) and complementary groups on the solid surface.17 In general, noncovalent19 and covalent20 attachment chemistries have been used to immobilize proteins to solid surfaces with the latter reported to provide more robust linkages, hence, less susceptibility to detachment or denaturation.3 If the interactions between the protein and support are not carefully designed, there is a tendency to produce reactors possessing randomly oriented proteins with some orientations providing inactive forms.21 Attachment chemistries involving the use of affinity tags such as poly-His and glutathione S-transferase have been shown to eliminate issues with random attachment of enzymes to solid surfaces; nevertheless, they form chemical bonds that can become unstable over time or after multiple usages of the reactor.3 A recent study suggested that limiting the surface functional group density of a substrate can induce single site-attachment minimizing the generation of multisite attachment potentially deactivating the immobilized biomolecule.22
In the past decade, polymer substrates have become beneficial for the design of biological reactors due to their exceptional biocompatibility, widespread surface functionality, attachment chemistries that are relatively stable over a wide range of pH values, and the ease of surface activation for the generation of functional scaffolds for protein attachment.23−26 In many cases, polymer substrates can exhibit glass-like properties, such as high optical transparency and low autofluorescence, and provide production of low-cost fluidic devices with good fidelity, appropriate for in vitro diagnostics.27−29 Some polymeric materials which possess the aforementioned characteristics are poly(methylmethacrylate) (PMMA)27 and cyclic olefin copolymer (COC).30 In particular, PMMA has been a substrate of choice in the design of fluidic devices for biological assays due to its favorable biocompatibility,27 excellent optical properties, and simplicity in the surface modification techniques that can be employed.24 Previously, we have shown that IMERs can be generated using PMMA substrates for the proteolytic digestion of proteins.31,32 PMMA IMERs with immobilized trypsin have shown enhanced enzyme stability, high reaction rates, and the absence of trypsin autodigestion, thereby simplifying protein identification using mass spectrometry.31,32
Exonucleases, which cleave double-stranded DNAs (dsDNA) or single-stranded DNA (ssDNA) along the phosphate backbone to generate mononucleotides, are involved in biological processes such as replication, repair, and recombination.33 Lambda-Exonuclease (λ-Exo), isolated from lambda bacteriophage, is a toroidally shaped processive enzyme composed of three identical subunits with a tapered pore active site, 30 Å diameter on one face for entry of dsDNA and 15 Å diameter on the opposite face for the exit of ssDNA.34−36 λ-Exo, which digests only dsDNAs with phosphorylated 5′ ends, has been suggested to possess a processivity of ∼3 000 nucleotides in free-solution and generate an intact ssDNA byproduct with an electrostatic ratchet digestion mechanism.37 Though its clipping rate is highly variable, single-molecule measurements have revealed an average value of ∼1 000 nt s–1.38 The digestion properties of λ-Exo offer several unique applications.39 For example, λ-Exo has been suggested to be useful in single-molecule DNA sequencing strategies,40 one format of which involves the exonuclease and an α-hemolysin nanopore. Previous simulation and experimental reports have suggested the use of immobilized λ-Exo for the systematic clipping of DNA into mononucleotides with each unit identified via a molecular-dependent flight time through nanochannels.41,42 The unique capabilities of λ-Exo and its immobilization onto solid supports can serve as a useful tool in the design of biosensors directed toward the sequence analysis of DNAs.10
A recent study demonstrated the digestion of dsDNA by λ-Exo with the dsDNA electrostatically anchored onto the substrate surface and the enzyme introduced in free solution and allowed to randomly bind to the free end(s) of the anchored dsDNA.43−45 Single-molecule fluorescence studies, with a fluorescently stained dsDNA target revealed that λ-Exo digestion occurred in three modes; (i) incomplete digestion at only one end of the dsDNA molecule; (ii) full simultaneous digestion at both ends; and (iii) incomplete digestion at both ends.43 While this single-molecule enzyme study provided valuable information concerning the catalytic action of λ-Exo, it did not address the scenario in which the enzyme was immobilized and the dsDNA target was present in free solution.40 A report by Perkins et al., which showed the successful immobilization of a single λ-Exo apoenzyme/dsDNA complex onto a quartz substrate, revealed that the activity of λ-Exo remained comparable to the free solution digestion.46
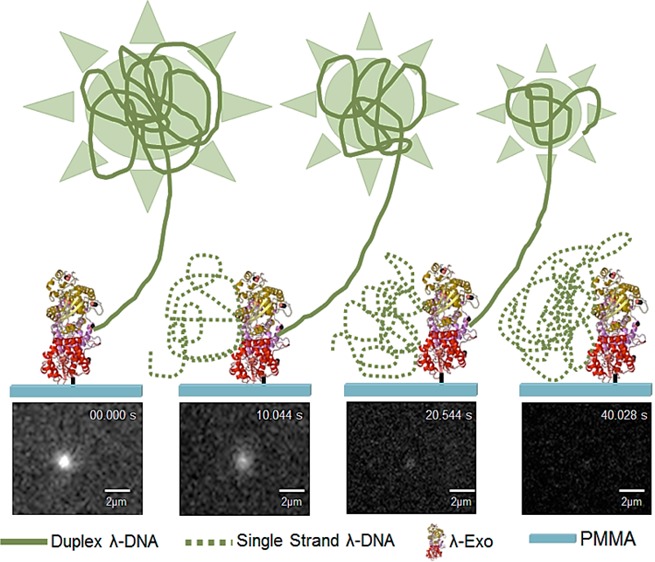
In this work, we report the first IMER involving λ-Exo as the immobilized enzyme for the digestion of dsDNA. λ-Exo was immobilized onto a PMMA device consisting of an array of micropillars populating a bioreactor. This device geometry allowed for an increased enzyme load and reduction in the diffusional kinetic barriers associated with open-channel IMERs.47 The immobilization was accomplished using 3-(3-dimethylaminopropyl) carbodiimide/N-hydroxysuccinimide (EDC/NHS) coupling chemistry for the conjugation of λ-Exo to UV-generated carboxylic acids on the PMMA surface. Atomic force microscopy (AFM) revealed the absence of nonspecific attachment (physisorption) of the enzyme to the polymer surface indicating that the enzyme was only attached covalently to the surface. Capillary electrophoresis using laser-induced fluorescence detection (CE-LIF) of the digestion products provided information on the lengths of the fragments remaining after digestion. Fluorescence microscopy studies of YOYO-1 stained dsDNA allowed for real-time observation of the digestion from which the enzyme clipping rate and apparent processivity were deduced.
Experimental Section
IMERs Fabrication
The IMERs used in this study consisted of a 1.4 mm × 24 mm polymer microchannel populated with 3 600 microposts, each 50 μm tall and 100 μm in diameter.48 Details on the device fabrication are provided in the Supporting Information. Parts A and B of Figure S1 in the Supporting Information show the CAD drawing and SEM image of the device, respectively.
Enzyme Immobilization onto PMMA IMERs
λ-Exo was anchored onto the IMERs’ surfaces using EDC-NHS coupling chemistry previously outlined by our group for the immobilization of amine-containing biological entities onto photoactivated PMMA substrates.49−52 A discussion of the immobilization chemistry is found in the Supporting Information with the reaction scheme shown in Figure S1C.
Digestion Studies of dsDNA
Duplexed λ-DNA (48 502 bp), purchased from New England Biolabs (Ipswich, MA), was incubated in the enzyme-modified IMERs for various reaction times. The desired reaction times were achieved by hydrodynamic pumping (PHD2000 syringe pump, Harvard Apparatus, Holliston, MA) a λ-DNA solution through the IMERs at an appropriate flow rate. An experimental control, which involved the introduction of a solution containing λ-DNA into the IMER bed in the absence of immobilized enzyme, was performed. The control revealed that there was neither a loss nor breakage of the dsDNA from nonspecific adsorption onto the reactor wall or shearing, respectively. On-chip enzymatic reactions were temperature controlled at 37 °C via a custom-built thermocouple heating stage. The effluent was collected at the device outlet for downstream analyses with CE and bulk fluorescence measurements.
Fluorescence Measurements of IMER-Digested dsDNA
PicoGreen intercalating dye (Life Technologies, Grand Island, NY) was used to determine the amount of intact dsDNA remaining after passage through the IMERs. PicoGreen shows high specificity for binding to dsDNA with a 1 000-fold fluorescence enhancement after intercalation to dsDNA. Because the dye displays minimal amounts of fluorescence upon binding to ssDNA (<10% that of dsDNA) and does not bind to mononucleotides with an associated fluorescence increase,53 it is suitable for determining specifically the dsDNA content from a λ-Exo reaction, which should consist of ssDNA, dsDNA, and mononucleotides. The DNA staining dye was added postdigestion to avoid perturbation in enzymatic activity of λ-Exo that may result from nuclear staining.54 The dye-labeled samples were excited at 480 nm and fluorescence spectra (490–700 nm) were collected and analyzed using a Fluorolog-3 spectrofluorimeter (Horiba JobinYvon, Edison, NJ) and DataMax Software 2.20. Kinetic data was acquired using a PicoGreen-stained digested dsDNA sample at varying input concentrations (20–5 μg/mL) using a 60 s reaction time with 4.96 pmol of surface-bound enzyme.
CE-LIF
Digestion products and the HIND III sizing ladder were analyzed using a home-built CE instrument with LIF detection. A schematic of this system can be found in Figure S2 in the Supporting Information.
Enzyme Quantification
The amount of λ-Exo in solution was determined using a spectrophotometric assay (Pierce 660 nm protein assay kit, Thermo Fisher Scientific; Rockford, IL). Details of enzyme quantification can be found in the Supporting Information.
AFM Characterization of PMMA/λ-Exo Surfaces
To deduce the surface coverage and possible orientation of the enzyme, cleaned PMMA sheets (1.7 cm × 1.7 cm squares, 3 mm thick) were activated with UV light and incubated with a λ-Exo solution overnight at 4 °C in the absence and presence of EDC/NHS coupling reagents. Samples were rinsed with reaction buffer and ddH2O and gently dried with compressed air prior to AFM analysis. Surface characterization was performed using an Asylum Research MFP3D AFM at a 1.00 Hz scanning rate in ac (tapping) mode. At this scanning frequency, we speculate that there would be negligible damage of the immobilized enzyme from the tapping force exerted by the tip.
Real-Time Digestion Analysis Using Fluorescence Microscopy
The microscope used in this study was a Zeiss Axiovert 200 M inverted microscope (75 W Xe lamp, Zeiss, Germany) fitted with a 100×/1.3 NA oil-immersion microscope objective and an Andor iXon3 EMCCD camera (20 fps acquisition rate). A custom mount was machined to hold the assembled IMERs onto the microscope stage. All images were acquired using MetaMorph Advanced 7.7.6.0 Software (Molecular Devices LLC, Sunnyvale, CA) and analyzed using ImageJ (National Institutes of Health, Bethesda, MD).
PMMA IMERs were modified as previously described. λ-Exo reaction buffer (glycine-KOH in ultrapure water at pH 9.4, 0.1% (v/v) Triton X-100, reagents purchased from Sigma Aldrich) was prepared without the cofactor (Mg2+). Following enzyme attachment, λ-DNA stained with YOYO-1 in a 1:50 dye-to-base pair ratio was introduced into the reactor in a Mg2+ free buffer to allow for the generation of the necessary enzyme/DNA complexes. Next, the reaction buffer (1×) containing 25 mM MgCl2 was introduced into the IMERs to initiate the enzymatic reaction after which the system was heated to 37 °C with real-time monitoring of the digestion process. Reagent introduction into the IMER was achieved using a PHD2000 syringe pump (Harvard Apparatus, Holliston, MA). The pump was connected to the inlet and outlet reservoirs of the IMERs through peak tubing sealed via epoxy with the inlet tube connected to a syringe using a leur-lock connector.
Results and Discussion
Enzyme Attachment and Characterization
To determine if λ-Exo was covalently attached to the activated PMMA surface of the IMERs reactor bed, we performed spectrophotometric analysis (660 nm) using a protein quantification kit. In this analysis, an aliquot of the reaction solution containing the enzyme, before and after running through the IMERs, was evaluated. The differences in the pre- and postfilling absorbance values were used as an indicator of the amount of enzyme remaining on the reactor surfaces. A calibration plot (R2 = 0.992) using the UV absorbance intensities of a protein calibration standard was used to determine the amount (in picomoles) of enzyme attached to the polymer surface for three different input amounts (75, 90, and 100 pmol). The amount of enzyme immobilized range from 3.25 to 6.40 pmol, yielding a reaction efficiency of 4.3–6.4%. On the basis of the available surface area of the IMERs bed (1.17 cm2), the surface concentration (pmol/cm2) was determined to range from 2.78 to 5.47. The fact that the surface concentration increased over the concentration range studied indicated that the surface was below monolayer coverage (see Table S1A in the Supporting Information for results).
Successful attachment of λ-Exo onto PMMA was further confirmed by AFM analysis. From the AFM scan depicted in Figure 1A for the activated-PMMA/λ-Exo reaction performed in the absence of the EDC/NHS coupling reagents, there was no indication of the presence of surface features consistent with the size of the λ-Exo enzyme. This confirmed that physisorption of enzyme onto the activated polymer surface did not occur under these reaction conditions. AFM images acquired from the PMMA surface in which the EDC/NHS coupling reagents were used revealed the presence of surface features consistent in height with λ-Exo (Figure 1B). Substrates containing covalently attached λ-Exo had an average RMS roughness of 1.58 ± 0.18 nm as compared to 0.34 ± 0.01 nm for surfaces without enzyme. Further AFM scans of the EDC/NHS/λ-Exo functionalized PMMA surface over a 15 μm × 15 μm area (Figure 1C) revealed surface features that possessed an average height of 14.3 ± 2.3 nm (see Figure 1D). This value is similar to the reported dimensions of λ-Exo measured from X-ray crystallography at angles α = β = γ = 90° (15.6 nm × 15.6 nm × 13.1 nm).36
Figure 1.

Tilted view of a 3 μm × 3 μm AFM scan of a PMMA surface following UV activation and incubation with 7 μg/mL λ-Exo enzyme without (A) and with (B) EDC/NHS coupling reagents. (C) A 15 × 15 μm phase image of a PMMA surface incubated with 7 μg/mL λ-Exo enzyme with EDC/NHS coupling. Surface AFM analysis revealed an RMS roughness of 1.58 ± 0.18 nm. (D) Histogram of the height of features on the activated PMMA surface and subsequently functionalized with λ-Exo determined by taking an AFM line scan across each immobilized enzyme and measuring the maximum height of the feature.
Although it is difficult to directly visualize the orientation of the immobilized enzyme on PMMA, the closeness of the average measured height of each feature to the protein crystallographic size indicates that the enzyme is primarily oriented with its access pore normal to the polymer surface. Though qualitative, this data indicated that the UV dose and enzyme concentration used for the immobilization reaction did not lead to surface cross-linking. Cross-linking may result in the enzyme laying parallel to the surface making its pore inaccessible to dsDNA based on surface steric considerations. This data also confirmed that the conjugation of the complete homotrimer was achieved with little if any dissociation into its monomer units.55
After UV-activation of the PMMA surface, the carboxylic acid group density was measured using a Toluidine Blue assay. For a UV dose of 16.0 mW/cm2 for 15 min, a carboxyl surface density of 32 pmol/cm2 was obtained, a value 10-fold higher than the λ-Exo surface concentration stated above.49,56 The Toluidine Blue assay, while effective for approximate surface carboxylate quantification, has the propensity to label carboxylic acid groups below the substrate surface where enzyme attachment is not possible due to inaccessibility issues.49
Surface Enzyme Activity
Figure 2 shows the fluorescence spectra of a free solution λ-Exo digestion of λ-DNA and one carried out in the IMERs for the same effective reaction time (60 s). The control for this experiment consisted of a 50 μg/mL λ-DNA stock solution exposed to an enzyme-free reactor for 60 s, which was collected at the outlet of the reactor and measured to determine if any loss of dsDNA resulted from transport through the reactor. To determine the extent of DNA digestion in the IMER, PicoGreen was added to the digestion products from the IMER and the free solution reaction. The amount of dsDNA remaining after digestion was monitored using fluorescence microscopy. As can be seen from Figure 2, the amount of fluorescence observed from the digestion products of the free solution was higher than that from the IMERs. This indicated that more dsDNA was remaining for the free solution digestion compared to the IMERs digestion. For the reactions carried out here, peak area analysis of Figure 2 revealed that 91.7% of the dsDNA was digested for the IMERs compared to 83.3% for the free solution digestion.
Figure 2.
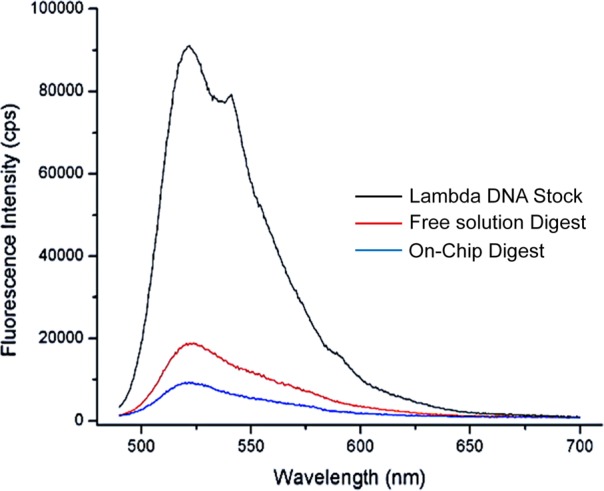
Plot of fluorescence intensity for a λ-DNA stock solution, free enzyme digestion, and the effluent from an IMERs digestion. The emission spectra were taken from 490 to 700 nm with 480 nm excitation. The spectrum labeled in black depicts the intensity of the λ-DNA stock. The blue line represents the spectrum of the IMERS digestion and the red line was that for the free solution digestion. For the IMERs digestion, the amount of immobilized enzyme was 4.96 pmol. For the λ-DNA stock, the IMERs was free of immobilized enzyme. In all cases, the solutions were incubated with PicoGreen following the reaction.
To evaluate the effects of enzyme surface concentration on the activity of the immobilized enzyme, experiments were conducted in which the reaction time and dsDNA substrate concentrations were kept constant and the enzyme surface concentration used for the digestion varied (Table S1B in the Supporting Information). Our data revealed that ∼96% digestion of λ-DNA was achieved when loading the IMERs with 3.25 pmol of enzyme (Table S1B in the Supporting Information) with a slight decrease in digestion efficiency at higher enzyme loads (∼85% at a load of 6.20 pmol). However, over the range of λ-Exo surface loads investigated, no statistical difference in the percent λ-DNA digestion was observed at the 95% confidence level.
Next, experiments were performed to carefully determine the effect of changing the reaction time of the dsDNA with the immobilized enzyme on the digestion efficiency. The IMERs were exposed to λ-DNA for 60, 300, and 1200 s, which was controlled by changing the linear velocity of the input λ-DNA through the reactor. The reactor generated digestion efficiencies >90% for all reaction times investigated (Table S1C in the Supporting Information).
Analysis of λ-Exo Reaction Products Using CE-LIF
As shown in the electropherogram obtained for the digestion products of the IMERs, the Hind III sizing ladder and intact λ-DNA (Figure S3 in the Supporting Information), there was the absence of peaks corresponding to the intact λ-DNA following IMERs digestion suggesting that most of the λ-DNA was digested. This is consistent with the data shown in Tables S1B,C in the Supporting Information and Figure 2. The digestion reaction will proceed until (1) the end of the dsDNA molecule is reached, (2) the enzyme dissociates into its monomers, and/or (3) the DNA is expelled from the enzyme.36 Furthermore, it is possible that a dsDNA molecule after threading through the pore of λ-Exo could be partially digested, disengaged, and re-engaged with another enzyme molecule within the IMER and undergoing further digestion from its complementary phosphorylated strand. Because the CE results indicated that the dominant dsDNA fragment size remaining was ∼7 kbp, this indicated an apparent processivity of ∼41 kbp if the re-engagement of the dsDNA molecule, which the CE-LIF results cannot determine, is ignored.
Reactor Reusability
We also tested whether the enzyme could be used for subsequent rounds of digestion by running different batches of λ-DNA through the reactor with different resident times (see Figure S5 in the Supporting Information). For the initial reaction, the digestion efficiency was found to be 95% for a 60 s reaction (see Table S1C in the Supporting Information). The reactor was then washed with buffer and a second round of digestion was undertaken by infusing λ-DNA through the IMERs. For the second round (60 s reaction time), the digestion efficiency dropped to 80% and for the third round, 53%.
Real-Time Digestion of λ-DNA
The digestion of a single dsDNA molecule with an immobilized enzyme was studied in real-time using fluorescence microscopy. λ-Exo was immobilized onto PMMA using EDC/NHS coupling chemistry and a solution of YOYO-1 stained dsDNA in a 1:50 dye to base-pair ratio was introduced into the enzyme reaction buffer (glycine-KOH in ultrapure water at pH 9.4, 0.1% (v/v) Triton X-100) without Mg2+. Previous work by Kang et al. revealed that stained dsDNA with a 1:50 dye-to-base pair ratio has comparable digestion rates to unstained dsDNA in the presence of λ-Exo.43
Real time monitoring of the enzyme-threaded DNA’s fluorescence was conducted. Some λ-DNA molecules within the microscopic region were observed to be immobile at one end due to complexation with the λ-Exo enzyme and freely moving at the opposite end due to shear forces. Uncomplexed DNAs remained in the bulk flow and eventually disappeared from the field-of-view. Following complexation, the reaction buffer containing the necessary Mg2+ cofactor for λ-Exo was added to initiate clipping and the reaction was monitored in real time as depicted in Figure 3 under nonflow conditions. To ascertain that the reduction in fluorescence intensity was a result of digestion and not photobleaching or photonicking, control experiments were performed by exposing an enzyme/DNA complex to the excitation light in the absence of Mg2+. Under these conditions, minimal amounts of fluorescence were lost during the time course of the experiment (60 s).
Figure 3.

(A–D) Fluorescence still images for the real-time digestion of dsDNA using λ-Exo covalently immobilized to a PMMA substrate configured in the IMER device. (E–H) The corresponding fluorescence intensity line plots taken from the still images shown in parts A–D. In these cases, the line plot was secured from a horizontal line that crossed the section in the still image containing the stained DNA molecule. (I) Graphical depiction of the relative fluorescence intensity of a single dsDNA that was digested by an immobilized λ-Exo molecule as a function of reaction time, where possible pausing events were seen in each inset. Immobilization of λ-Exo was accomplished using EDC/NHS onto a PMMA substrate. The λ-DNA was labeled in a 1:50 dye/bp ratio with YOYO-1. The fluorescence intensity was measured in the presence (black) or absence (red) of the enzyme cofactor, Mg2+. The dotted line for the intensity profile in the presence of Mg2+ indicates the time at which the cofactor was infused into the IMER.
When the reaction was fortified with Mg2+ ions, there was an observed decrease in the bulk fluorescence of the λ-DNA/λ-Exo complex (Figure 3). There were two regions in the Mg2+ fortified enzymatic reaction where the fluorescence intensity remained relatively constant for a short period of time indicating that the digestion paused. According to previous work, pauses are likely sequence-dependent; λ-Exo has the propensity to pause in regions with GGCGATTCT sequences, which includes the GGCGA 5-bp motif.46 This study also suggested that sequences associated with pausing could also be contained within a GGCGATTCT domain.46 Upon examination of the sequence of λ-DNA, it was determined that two regions within its sequence contained the first 7 of the 9 bases within the GGCGATTCT motif at 37 701 bp and 43 372 bp positions. This is consistent with the pauses we observed in the fluorescence intensity profile shown in Figure 3. The fluorescence intensity was monitored until the signal strength became indistinguishable from the background.
Fluorescence measurements were used to determine the size of the smallest detectable dsDNA fragment stained using a 50:1 base-pair to dye ratio. From the calibration plot, the smallest detectable fragment was 4.6 kbp (see Figure S4 in the Supporting Information). The average digestion rate was determined based on the total number of base-pairs for λ-DNA (48 502 bp) minus the size of the smallest detectable fragment (4.6 kbp) and the time required for the fluorescence to reach the baseline. This calculation yielded a digestion rate of 1.0 × 103 ± 100 nt/s (n = 4), a value similar to that reported by Kang et al. for electrostatically immobilized dsDNA.43
We also estimated the degree of processivity from a single digestion event (see Figure 3) and the shortest fragment we could observe using fluorescence (4.6 kbp, Figure S4 in the Supporting Information). As seen in Figure 3, a single λ-DNA molecule engaged with the immobilized λ-exonuclease resulted in a complete loss of fluorescence indicating that the remaining fragment was <4.6 kbp in length; the apparent processivity based on this analysis would be >40 kbp. This number is in close agreement to that seen by the CE-LIF data (see Figure S3 in the Supporting Information). Our value was approximately 10-fold higher than previous reports for free solution digestions using λ-Exo.38,43
The higher apparent processivity of the solid-phase reactor relative to the free-solution case could be attributed to increased stability of the enzyme when anchored to the support. Previous reports have shown that enzyme attachment may prevent the dissociation of λ-Exo into its monomer units during a digestion event that terminates the enzymatic reaction and, thus, limits its processivity.55 In addition, the processivity observed could be associated with a dependence on dsDNA substrate length;34 as the dsDNA length increases from 0.5 to 23 kbp, it is less likely the enzyme will dissociate from the DNA.34 We note that the processivity reported here was labeled as apparent because of the indirect evidence used to secure this value. As discussed above, the CE-LIF data did not account for re-engagement of a partially digested λ-DNA molecule. In addition, the single-molecule fluorescence observations could not account for digestion at both ends of a single dsDNA molecule.
Kinetic Description of Immobilized λ-Exo
Attempts to correlate the kinetic behavior observed for IMER digestions to the classical Michaelis–Menten (MM) model or the Lilly–Hornby model for packed solid-phase reactors have been unsuccessful,57,58 due to limitations associated with the MM model that assumes free diffusion and thermodynamically driven collision of enzyme/substrate. This is not the case for IMERs in which molecular mobility of the enzyme is restricted due to immobilization. Also, the model for continuous-flow enzymatic reactors, when the Lilly–Hornby model was applied, was determined to be insufficient due to the strong reaction dependence on flow rate of substrate through the reactor.58 Alternatively, the IMER reactions can be described by fractal-like MM kinetics.59−61 Furthermore, enzymatic reactions involving polymerized substrates like DNA can exhibit characteristics of fractal/MM kinetic behavior.62
To explain the kinetic behavior of our system, the fractal MM model proposed by Xu was used, which modifies the classical MM approach by incorporating a fractal contribution offering a more detailed explanation of MM like behavior.62 From this formalism, a fractal dimension, f, in the rate coefficient was used to account for the fractal behavior observed using enzyme concentration [E]a,b, and time ta,b, where a and b denote two different concentrations and times as seen from eq 1:
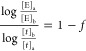 |
1 |
For a solubilized enzyme acting on a polymer substrate processively, the reaction may be considered as 1-dimensional with f = 0.5. For an immobilized enzyme acting on a soluble substrate, the reaction can be considered 2-dimensional with f ∼ 0.3. The system depicted in our work involves both cases and was determined to have a theoretical f value of ∼0.7 based on eq 1 and values obtained from experimental data. We then plotted our experimental data at various λ-DNA concentrations as a Lineweaver–Burke (double reciprocal) plot using the fractal formalism by Xu et al. shown in eq 2:
| 2 |
where ν is the initial rate of the reaction and its corresponding change (difference in concentration divided by reaction time), [E] and [S] are the enzyme and substrate concentrations, respectively, where [E] was determined from the 660 nm colorimetric assay for protein quantification as previously mentioned, t is reaction time, k2′ is the enzyme turnover rate, and Km′ is the Michaelis constant; the primes indicate modified Michaelis’ constants based on the fractal behavior of our system (see eq 2). As can be seen in Figure 4, the double reciprocal plot for immobilized λ-Exo digestion of dsDNA was linear and yielded k2′ = 5.24 s–1. This turnover rate was found by taking the reciprocal of the intercept from the double reciprocal plot depicted in Figure 4 and incorporating the enzyme concentration, fractal number, and reaction time as noted in eq 2. Taking the reciprocal of the intercept and multiplying it by the slope of the line from Figure 4 yielded Km′, which was found to be 4.8 × 10–6 mM. According to Berg et al.,63 enzymes that have upper limit catalytic efficiencies, k2′/Km′, on the order of 108 to 109 M–1 s–1 are said to have attained kinetic perfection, which means that the reaction they catalyze occurs as quickly as the reactants diffuse to the enzyme. From Figure 4, the catalytic efficiency for our system was determined to be 1.1 × 109 M–1 s–1. According to Berg et al., this suggests that the catalysis is restricted only by the rate at which the enzyme encounters substrate in the system.63 The efficiency of our solid-phase reactor as denoted by k2′/Km′ when compared to a homogeneous digestion (0.9 × 109 M–1 s–1)34 indicated a 17% increase in catalytic efficiency for duplex disassembly into individual mononucleotides for the IMERs.
Figure 4.

Double reciprocal plot depicting fractal-like Michaelis–Menten kinetics of λ-Exo based on eq 2. Experiment parameters were [S] = 1.5, 1.8, 2.1, 5, 3, or 6.6 (× 10–6) mM; [E] = 4.96 × 10–4 mM; f = 0.7; Km = 4.8 × 10–6 mM; and k2 = 5.24 s–1. These values were determined by extrapolation of 1/ν = (Km/Vmax)/[S] + 1/Vmax, where the initial rate was calculated by the difference in concentration as a measure of the fluorescence of the remaining dsDNA divided by the reaction time (60 s).
Conclusion
In this study, we demonstrated for the first time, to the best of our knowledge, the attachment of a processive exonuclease (λ-Exo) to a PMMA solid phase reactor configured in a microfluidic device (IMERs). The covalent attachment to a photoactivated PMMA support was accomplished using EDC/NHS coupling chemistry, which utilized carboxylic acid groups that were generated by UV-activation of the PMMA surface. The data presented in this work suggested that λ-Exo, when immobilized onto a solid support with controlled carboxylic acid surface density, adopted primarily a single-point attachment configuration with the pore of the enzyme accessible to dsDNA. The IMERs demonstrated increased efficiency as determined by the k2′/Km′ value when compared to a homogeneous digestion of dsDNA.34 In addition, our IMERs exhibited an increased apparent processivity compared to a homogeneous reaction and displayed fractal-like enzyme kinetics due to the heterogeneous nature of the IMER and the processive digestion of dsDNAs. The findings secured in this study will provide important information on strategies for immobilizing exonuclease enzymes to solid supports for potential applications in single-molecule DNA sequencing.40,41
Acknowledgments
This research was supported, in part, by the National Institutes of Health (Grant R21 HG006278). N.J.O.-C. would like to thank the National GEM Consortium Fellowship and the National Institutes of Health (NIH) F31 training grant for financial support as well as partial support from the National Science Foundation (Eastern Asian Pacific Summer Institute, EAPSI) and Prof. Cho at Ulsan National Institute of Science & Technology for hosting N.J.O.-C. during the EAPSI fellowship. The authors would like to extend thanks to Dr. Feng Xu of Novozyme Inc. for his helpful discussion regarding the fractal kinetic results. The authors would also like to thank Dr. Amar S. Kumbhar at the Chapel Hill Analytical and Nanofabrication Laboratory (CHANL) for help with the AFM images and Mr. Matthew Verber of the Electronics facility at the University of North Carolina at Chapel Hill for writing the LabView program for our CE-LIF system. Finally, the authors would like to thank Prof. Francis Barany for his insight and thoughts regarding the biology of exonuclease enzymes.
Supporting Information Available
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Löffelholz C.; Husemann U.; Greller G.; Meusel W.; Kauling J.; Ay P.; Kraume M.; Eibl R.; Eibl D. Chem. Ing. Tech. 2013, 85, 40–56. [Google Scholar]
- Hambor J. E. BioProc. Int. 2012, 10, 11. [Google Scholar]
- Liu W.; Wang L.; Jiang R. Top. Catal. 2012, 55, 1146–1156. [Google Scholar]
- Ansari S. A.; Husain Q. Biotechnol. Adv. 2012, 30, 512–523. [DOI] [PubMed] [Google Scholar]
- Le Nel A.; Minc N.; Smadja C.; Slovakova M.; Bilkova Z.; Peyrin J. M.; Viovy J. L.; Taverna M. Lab Chip 2008, 8, 294–301. [DOI] [PubMed] [Google Scholar]
- Urban P. L.; Goodall D. M.; Bruce N. C. Biotechnol. Adv. 2006, 24, 42–57. [DOI] [PubMed] [Google Scholar]
- Laurell T.; Drott J.; Rosengren L.; Lindström K. Sens. Actuators, B: Chem 1996, 31, 161–166. [Google Scholar]
- Létant S. E.; Hart B. R.; Kane S. R.; Hadi M. Z.; Shields S. J.; Reynolds J. G. Adv. Mater. 2004, 16, 689–693. [Google Scholar]
- Manjon A.; Obon J. M.; Casanova P.; Fernandez V. M.; Ilborra J. L. Biotechnol. Lett. 2002, 24, 1227–1232. [Google Scholar]
- Matosevic S.; Szita N.; Baganz F. J. Chem. Technol. Biotechnol. 2011, 86, 325–334. [Google Scholar]
- Seong G. H.; Zhan W.; Crooks R. M. Anal. Chem. 2002, 74, 3372–3377. [DOI] [PubMed] [Google Scholar]
- Davidson Y. Y.; Soper S. A.; Margolis S.; Sander L. C. J. Sep. Sci. 2001, 24, 10–16. [Google Scholar]
- Mao H.; Yang T.; Cremer P. S. Anal. Chem. 2002, 74, 379–385. [DOI] [PubMed] [Google Scholar]
- Calleri E.; Temporini C.; Perani E.; Stella C.; Rudaz S.; Lubda D.; Mellerio G.; Veuthey J. L.; Caccialanza G.; Massolini G. J. Chromatogr., A 2004, 1045, 99–109. [DOI] [PubMed] [Google Scholar]
- Wong L. S.; Khan F.; Micklefield J. Chem. Rev. 2009, 109, 4025–4053. [DOI] [PubMed] [Google Scholar]
- Hanefeld U.; Cao L.; Magner E. Chem. Soc. Rev. 2013, 42, 6211–6212. [DOI] [PubMed] [Google Scholar]
- Ducker R. E.; Montague M. T.; Leggett G. J. Biointerphases 2008, 3, 59–65. [DOI] [PubMed] [Google Scholar]
- Křenková J.; Foret F. Electrophoresis 2004, 25, 3550–3563. [DOI] [PubMed] [Google Scholar]
- Petrov P.; Pavlova S.; Tsvetanov C. B.; Topalova Y.; Dimkov R. J. Appl. Polym. Sci. 2011, 122, 1742–1748. [Google Scholar]
- Zimmermann J. L.; Nicolaus T.; Neuert G.; Blank K. Nat. Protoc. 2010, 5, 975–985. [DOI] [PubMed] [Google Scholar]
- Calleri E.; Ambrosini S.; Temporini C.; Massolini G. J. Pharm. Biomed. Anal. 2012, 69, 64–76. [DOI] [PubMed] [Google Scholar]
- Ye P.; Wan R.-B.; Wang X.-P. J. Mol. Catal. B: Enzym. 2009, 61, 296–302. [Google Scholar]
- Goddard J. M.; Hotchkiss J. H. Prog. Polym. Sci. 2007, 32, 698–725. [Google Scholar]
- Dominick W. D.; Berhane B. T.; Mecomber J. S.; Limbach P. A. Anal. Bioanal. Chem. 2003, 376, 349–354. [DOI] [PubMed] [Google Scholar]
- Qu H.; Wang H.; Huang Y.; Zhong W.; Lu H.; Kong J.; Yang P.; Liu B. Anal. Chem. 2004, 76, 6426–6433. [DOI] [PubMed] [Google Scholar]
- Rodrigues R. C.; Ortiz C.; Berenguer-Murcia A.; Torres R.; Fernandez-Lafuente R. Chem. Soc. Rev. 2013, 42, 6290–6307. [DOI] [PubMed] [Google Scholar]
- Tennico Y. H.; Koesdjojo M. T.; Kondo S.; Mandrell D. T.; Remcho V. T. Sens. Actuators, B: Chem. 2010, 143, 799–804. [Google Scholar]
- Soper S. A.; Ford S. M.; Qi S.; McCarley R. L.; Kelly K.; Murphy M. C. Anal. Chem. 2000, 72, 642a–651a. [DOI] [PubMed] [Google Scholar]
- Chantiwas R.; Park S.; Soper S. A.; Kim B. C.; Takayama S.; Sunkara V.; Hwang H.; Cho Y. K. Chem. Soc. Rev. 2011, 40, 3677–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Midwoud P. M.; Janse A.; Merema M. T.; Groothuis G. M.; Verpoorte E. Anal. Chem. 2012, 84, 3938–3944. [DOI] [PubMed] [Google Scholar]
- Lee J.; Musyimi H. K.; Soper S. A.; Murray K. K. J. Am. Soc. Mass Spectrom. 2008, 19, 964–972. [DOI] [PubMed] [Google Scholar]
- Lee J.; Soper S. A.; Murray K. K. Analyst 2009, 134, 2426–2433. [DOI] [PubMed] [Google Scholar]
- Cassuto E.; Lash T.; Sriprakash K. S.; Radding C. M. Proc. Natl. Acad. Sci. U.S.A. 1971, 68, 1639–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsis P. G.; Kwagh J. G. Nucleic Acids Res. 1999, 27, 3057–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little J. W. J. Biol. Chem. 1967, 242, 679–686. [PubMed] [Google Scholar]
- Kovall R.; Matthews B. W. Science 1997, 277, 1824–1827. [DOI] [PubMed] [Google Scholar]
- Zhang J.; McCabe K. A.; Bell C. E. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 11872–11877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura S.; Komatsu J.; Hirano K.; Yasuda H.; Takashima K.; Katsura S.; Mizuno A. Nucleic Acids Res. 2001, 29, E79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H.; Li B.; Zhong R.; Qin J.; Zhu Y.; Lin B. Electrophoresis 2008, 29, 4956–4963. [DOI] [PubMed] [Google Scholar]
- Clarke J.; Wu H. C.; Jayasinghe L.; Patel A.; Reid S.; Bayley H. Nat. Nanotechnol. 2009, 4, 265–270. [DOI] [PubMed] [Google Scholar]
- Novak B. R.; Moldovan D.; Nikitopoulos D. E.; Soper S. A. J. Phys. Chem. B 2013, 117, 3271–3279. [DOI] [PubMed] [Google Scholar]
- Ayub M.; Hardwick S. W.; Luisi B. F.; Bayley H. Nano Lett. 2013, 13, 6144–6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S.; Lee S.; Yeung E. S. Analyst 2010, 135, 1759–1764. [DOI] [PubMed] [Google Scholar]
- Lee G.; Yoo J.; Leslie B. J.; Ha T. Nat. Chem. Biol. 2011, 7, 367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.; Kang S. H.; Yeung E. S. Talanta 2011, 85, 2135–2141. [DOI] [PubMed] [Google Scholar]
- Perkins T. T.; Dalal R. V.; Mitsis P. G.; Block S. M. Science 2003, 301, 1914–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B.; Basuray S.. Microfluidics: Technologies and Applications; Springer: Heidelberg, Germany, 2011. [Google Scholar]
- Njoroge S. K.; Witek M. A.; Battle K. N.; Immethun V. E.; Hupert M. L.; Soper S. A. Electrophoresis 2011, 32, 3221–3232. [DOI] [PubMed] [Google Scholar]
- Jackson J. M.; Witek M. A.; Hupert M. L.; Brady C.; Pullagurla S.; Kamande J.; Aufforth R. D.; Tignanelli C. J.; Torphy R. J.; Yeh J. J.; Soper S. A. Lab Chip 2014, 14, 106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarley R. L.; Vaidya B.; Wei S.; Smith A. F.; Patel A. B.; Feng J.; Murphy M. C.; Soper S. A. J. Am. Chem. Soc. 2005, 127, 842–843. [DOI] [PubMed] [Google Scholar]
- Subramanian B.; Kim N.; Lee W.; Spivak D. A.; Nikitopoulos D. E.; McCarley R. L.; Soper S. A. Langmuir 2011, 27, 7949–7957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S.; Vaidya B.; Patel A. B.; Soper S. A.; McCarley R. L. J. Phys. Chem. B 2005, 109, 16988–16996. [DOI] [PubMed] [Google Scholar]
- Singer V. L.; Jones L. J.; Yue S. T.; Haugland R. P. Anal. Biochem. 1997, 249, 228–238. [DOI] [PubMed] [Google Scholar]
- Subramanian K.; Rutvisuttinunt W.; Scott W.; Myers R. S. Nucleic Acids Res. 2003, 31, 1585–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aissaoui N.; Landoulsi J.; Bergaoui L.; Boujday S.; Lambert J.-F. Enzyme Microb. Technol. 2013, 52, 336–343. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Bachman M.; Sims C. E.; Li G. P.; Allbritton N. L. Langmuir 2006, 22, 2719–2725. [DOI] [PubMed] [Google Scholar]
- Hornby W. E.; Lilly M. D. Biochem. J. 1968, 107, 669–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong G. H.; Heo J.; Crooks R. M. Anal. Chem. 2003, 75, 3161–3167. [DOI] [PubMed] [Google Scholar]
- Savageau M. A. J. Theor. Biol. 1995, 176, 115–124. [DOI] [PubMed] [Google Scholar]
- Schnell S.; Turner T. E. Prog. Biophys. Mol. Biol. 2004, 85, 235–260. [DOI] [PubMed] [Google Scholar]
- Kopelman R. Science 1988, 241, 1620–1626. [DOI] [PubMed] [Google Scholar]
- Xu F.; Ding H. Appl. Catal., A 2007, 317, 70–81. [Google Scholar]
- Berg J. M.; Tymoczko J. L. Stryer L.. Biochemistry, 7th ed.; W.H. Freeman: New York, 2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.