

Abstract
Due to the high cost of failed runs and suboptimal data yields, quantification and determination of fragment size range are crucial steps in the library preparation process for massively parallel sequencing (or “Next Generation Sequencing”). Current library quality control methods commonly involve quantification using real-time quantitative PCR and size determination using gel or capillary electrophoresis. These methods are laborious and subject to a number of significant limitations that can be responsible for unreliable library calibration. Herein we propose and test an alternative method for the quality control of sequencing libraries using Droplet Digital PCR (ddPCR). By exploiting a correlation that we have discovered between droplet fluorescence and amplicon size, we achieve the joint quantification and size determination of target DNA with a single ddPCR assay. We demonstrate the accuracy and precision of applying this method to the preparation of sequencing libraries.
Keywords: droplet digital PCR, absolute quantification, next-generation sequencing, massively-parallel sequencing, library preparation, quality control, size determination
Introduction
Massively-parallel next-generation sequencing (NGS) technology is rapidly revolutionizing the fields of genomics, molecular diagnostics, and personalized medicine through the increasingly efficient and economical generation of unprecedented volumes of data (1-7). A common characteristic of the various commercially available NGS technologies is the need to load a precise number of viable DNA library molecules onto the instrument to optimize the yield of data from an individual sequencing experiment (8-11). Performing a sequencing run with either too many or too few library molecules results in compromised data yields or completely failed sequencing runs that waste sample, user time, instrument time, and expensive reagents. Similarly, if library molecules are not the appropriate length to fully utilize the capabilities of the sequencing platform fewer bases can be sequenced in an NGS run and the throughput is wasted. Thus, accurate quantification and size determination of library DNA is essential for achieving optimal data yield and maximizing a laboratory’s efficiency and sequencing throughput.
Protocols for the preparation of NGS libraries include quality control steps to validate the size and concentration of amplifiable library molecules (i.e. molecules properly ligated to NGS adapter sequences) before committing to a sequencing run. Manufacturers typically recommend quantification with quantitative real-time PCR (qPCR) and size determination with gel or capillary electrophoresis. It is also possible to enumerate library DNA using UV spectrophotometry, the Quant-iT PicoGreen assay, or the Agilent BioAnalyzer, but these methods are not ideal because they quantify amplifiable and non-amplifiable molecules equally (12-14). These methods are also only capable of measuring mass per volume which must be converted to copy number using an estimated average size of library molecules which can introduce further error (15). Although qPCR is widely considered the best option for library quantification, there are considerable drawbacks to the method including amplification biases due to template size and GC-content and the need for a standard curve to estimate the absolute quantity of DNA (16). Creating a standard curve for each sample to be analyzed is a difficult and uncertain process that leads to inaccuracies in measurements of absolute target quantity (15,17). When intercalating dyes are used for quantification, the concentration reading can include non-amplifiable DNA as dyes measure dsDNA indiscriminately. Because of these potential inaccuracies, some NGS platform manufacturers recommend performing titration runs on their instrument to determine the proper loading amount. The high cost of reagents and the length of NGS runs make this an expensive and time-consuming step.
We have developed a new assay capable of concurrently measuring the absolute concentration and the length of unknown amplifiable DNA templates, making it well suited for quality control of NGS libraries. The assay, which we have termed “QuantiSize”, is based on the previously validated Droplet Digital PCR (ddPCR) absolute quantification system (18,19) and adds the ability to calculate the size of target DNA by exploiting a linear correlation that we have discovered between the fluorescence amplitude of ddPCR droplets and the size of amplicons within them.
Method summary
QuantiSize allows for the determination of absolute quantity and size distribution of target DNA molecules in a single experiment. This assay exploits a correlation, reported herein, between the length of an amplified DNA molecule and the fluorescence amplitude produced in droplet digital PCR (ddPCR), to allow the user to calculate the size of unknown DNA. As ddPCR simultaneously measures the concentration of target DNA, the user can accurately determine the target population size and quantity in a single step As a quantification method, ddPCR has demonstrated greater precision and sensitivity than real-time PCR (18). We demonstrate that QuantiSize is capable of accurately measuring the size and concentration of target DNA simultaneously while avoiding the limitations of other quantification systems and we highlight the utility of this assay for the preparation of NGS libraries.
Materials and methods
Purification of DNA size standards
An exACTGene 50 bp DNA Ladder (Fisher, Waltham, MA) and 1 kb Plus DNA Ladder (Fisher) were run on a 1.0% UltraPure Low-Melting Point Agarose (Invitrogen, Carlsbad, CA) electrophoresis gel and the 25, 50, 100, 200, 300, 400, 500, 600, 700, 800 and 1000 bp ladder bands were manually excised. The DNA in these gel slices was purified using the QIAcube automated gel extraction protocol with the QIAquick Gel Extraction Kit (QIAGEN, Hilden, Germany). The size and purity of all DNA fragments were verified by gel electrophoresis.
Ligation of DNA size standards to adapter sequences
The DNA fragments were ligated to the following sequences containing the Nextera version 1 adapters (Epicentre Biotechnologies, Madison, WI): Adapter 1:
5′-CAAGCAGAAGACGGCATACGAGATTCGCCTTACGGTCTGCCTTGCCAGC CCGCTCAGAGATGTGTATAAGAGACAG[index 1]CCC3′
Adapter 2: 5′-GGG[index 2]CTGTCTCTTATACACATCTCTGATGGCGCGAGGGAGGCGCG ATCTAGTGTAGATCTCGGTGGTCGCCGTATCATT-3′
All size standard fragments were ligated to adapters with the following 7-bp indices:
Index 1: 5′-TACCTCT-3′
Index 2: 5′-ACACATT-3′
Ligations were carried out in 20 μL reactions containing 1 μL T4 DNA Ligase (New England BioLabs, Ipswich, MA) and 2 μL 10× T4 DNA Ligase Buffer. The ligation reactions were incubated at room temperature for 2 hours. The ligated DNA was purified with a phenol/chloroform/isoamyl alcohol extraction. All samples were sent to the Fred Hutchinson Cancer Research Center ABI capillary sequencing facility to verify that the correct insert had been ligated to the adapters in each sample.
Library preparation for Illumina MiSeq
Samples of the plasmid pET-23a were sheared to an average size of 150 bp using the Covaris S220 Ultrasonicator (Covaris, Woburn, MA). The sheared DNA was run on a 1.0% UltraPure Low-Melting Point Agarose gel and a gel slice corresponding to ~100-200 bp was manually excised and purified using the QIAcube gel extraction protocol. The sheared DNA was blunted and phosphorylated using the Quick Blunting Kit (New England BioLabs) and purified with a phenol/chloroform/isoamyl alcohol extraction.
Eight samples of sheared DNA were ligated to adapter sequences with unique 7-bp indices in separate 20 μL reactions containing 1 μL T4 DNA Ligase (New England BioLabs) and 2 μL 10× T4 DNA Ligase Buffer (New England BioLabs). The ligation reactions were incubated at room temperature for 2 hours. All ligations were purified with a phenol/chloroform/isoamyl alcohol extraction.
All ligated libraries were amplified with 20 cycles of standard PCR using the following primer sequences:
Primer 1: 5′-AATGATACGGCGACCACCGA-3′
Primer 2: 5′-CAAGCAGAAGACGGCATACGA-3′
The amplified libraries were quantified using the ddPCR system (Bio-Rad, Hercules, CA) and the Quant-iT PicoGreen assay (Invitrogen). In the ddPCR experiment, the libraries were run in parallel with adapter-ligated size standards to allow for the estimation of library size distribution. The measured concentrations of the eight differently indexed libraries were used to dilute and combine the libraries in a molar ratio of 100:50:10:1 with two libraries at each concentration. The combination of libraries was denatured and diluted in preparation for loading onto the MiSeq flow cell (Illumina, San Diego, CA) as per the Illumina protocol.
Genomic DNA was purified from the human colon cancer cell line HCT 116 using the QIAcube automated purification protocol with the DNeasy Kit (QIAGEN). The Nextera XT DNA Sample Preparation Kit (Illumina) was used to generate a MiSeq compatible library from the HCT 116 DNA. The optional bead-based normalization step in the Nextera XT protocol was omitted and the library was instead normalized by quantification with ddPCR and the volume was adjusted to 2 nM as required by the MiSeq loading protocol. The 2 nM library was denatured, further diluted as per the manufacturer’s guidelines, and the standard phi X control library (Illumina) was spiked into the denatured HCT 116 library at 5% by volume. The library and phi X mixture was finally loaded into a MiSeq 300-Cycle v2 Reagent Kit (Illumina).
TaqMan probe and primer design
The following primers (Invitrogen) and TaqMan probe (Applied Biosystems, Foster City, CA) were designed to hybridize to the Nextera adapter sequences:
Primer 1: 5′-GCGACCACCGAGATCTACAC-3′
Primer 2: 5′-AGCAGAAGACGGCATACGAG-3′
Probe: 5′-FAM-CTGT+CT+CT+TA+TA+CA+CATC-IBFQ-3′
The ‘+’ indicates that the previous base is a Locked Nucleic Acid (LNA) base.
Droplet digital PCR
All quantified DNA libraries and size standards were prepared for droplet PCR in 25 μL reactions containing 2× ddPCR Master Mix (Bio-Rad), 250 nM TaqMan probe, 900 nM each of the appropriate flanking primers, and ~10,000 copies of target DNA. Emulsified 1 nL reaction droplets were made by adding 20 μL of each reaction mixture to the sample wells of a droplet generator DG8 cartridge (Bio-Rad) and 70 μL ddPCR Droplet Generation Oil (Bio-Rad) to the oil wells of the cartridge for use in the QX100 Droplet Generator (Bio-Rad). Forty microliters of the generated droplet emulsions were transferred to Twin.tec semi-skirted 96-well PCR plates (Eppendorf, Hamburg, Germany), which were then heat sealed with pierceable foil sheets. To amplify the target DNA, the droplet emulsions were thermally cycled using the following protocol: initial denaturation at 95 °C for 10 min, followed by 40 cycles of 94 °C for 30 sec and 60°C for 1 min. The fluorescence of each thermally cycled droplet was measured using the QX100 Droplet Reader. All measurements were performed in triplicate.
Data analysis
The equation of the line fitting the correlation between amplicon size and fluorescence amplitude for the size standards was generated using Excel (Microsoft, Redmond, WA) and applied to the measured fluorescence amplitude of each sequencing library to calculate amplicon size. The .fastq data files produced by the MiSeq were imported to Sequencher (Gene Codes, Ann Arbor, MI) and aligned to the pET-23a plasmid sequence to generate a sequence alignment/map file (SAM). A perl script was used to count the length of each read pair by retrieving the number corresponding to the “TLEN” field of the SAM file. Only library molecules for which both paired-end reads passed the quality filter were included in the analysis.
Results and discussion
The ability of the QuantiSize assay to combine quantification and size determination in a single ddPCR experiment is derived from a correlation that exists between the fluorescence amplitude of droplets and the size of amplicon within them. With standard ddPCR reagent concentrations, DNA amplification is eventually limited by the availability of dNTPs and inhibited by the presence of pyrophosphate (19,20); thus long DNA templates, which consume more dNTPs and generate more pyrophosphate, will have produced fewer products than short templates at the endpoint of a standard reaction. Because the final number of products generated within a droplet determines its level of fluorescence, the measured fluorescence amplitude of droplets containing short templates will be greater than that of droplets containing long templates. The QuantiSize assay exploits this fact to generate an equation relating fluorescence amplitude to amplicon size by using measurements of known size standards. The equation describing the relationship between fluorescence amplitude and amplicon size can be used to calculate the size of any unknown ddPCR template that shares common primer and probe binding sites with the size standards. Creating size standards that have primer and probe binding sites in common with DNA samples can be accomplished in a number of ways including cloning sample DNA into a vector and appending adapter sequences to both the sample DNA and size standards (21).
We created a set of size standards applicable to Illumina NGS libraries containing inserts ranging from 25 to 1000 base pairs flanked by adapter sequences compatible with the Illumina MiSeq platform. A pair of primers and a fluorescent TaqMan probe were designed to hybridize to the adapter sequences such that the length of each amplicon is 160 base pairs plus the length of the insert. As the primers and probe are specific to the MiSeq adapter sequences, only the adapter-ligated molecules that will be amplifiable on the MiSeq flow cell will be quantified.
A ddPCR experiment was performed with the aforementioned size standards in separate wells of a 96-well plate. Droplets containing the target (positive) increased in fluorescence following amplification of the target whereas droplets lacking the target (negative) remained at the background level of fluorescence (Fig. 1A). The distribution of droplet amplitudes is consistent across most amplicon lengths, but the 760 and 860 bp amplicons show a broader distribution of amplitudes (Fig. 1B). An inverse, linear correlation between amplicon size and mean fluorescence amplitude was observed (R2=0.99436) (Fig. 1C). The equation describing this correlation allows for the calculation of amplicon size given a measured fluorescence amplitude. The slope of this equation provides a measure of the difference in mean fluorescence amplitude that is expected with a given difference in amplicon size. Maximizing the magnitude of this slope maximizes the resolution of size standards, which is advantageous for the purpose of determining the length of unknown amplicons more accurately. The size standards used for QuantiSize are highly analogous to the standards used in gel and capillary electrophoresis. The size of unknown DNA can be determined by visually comparing the fluorescence amplitude of the size references to that of the unknown DNA or by entering the fluorescence amplitude value into the equation describing the relationship between average fluorescence amplitude and amplicon size for the size standards.
Figure 1. ddPCR amplification of 10 size standards designed for use with the QuantiSize assay.

All size standards were amplified in parallel with standard reagent and thermal cycling conditions. (A) Scatter plot of fluorescence amplitude of individual droplets for each size standard. Droplets whose fluorescence amplitude is above a specified threshold (“positives”) are shown in black and droplets with fluorescence amplitude below the threshold (“negatives”) are shown in grey. (B) Box-and-whisker plots showing distribution of fluorescence amplitudes of positive droplets. Horizontal bars mark the mean fluorescence amplitude, boxes mark the interquartile range, and whiskers mark the 95% confidence interval. (C) Plot of mean fluorescence amplitude ± SEM versus amplicon size showing a linear correlation (R2=0.9943).
The droplet reader software counts positive and negative droplets by using a threshold of fluorescence between the well-defined populations of high and low fluorescence amplitude droplets. For one particular TaqMan probe tested, the fluorescence amplitude of droplets containing amplicons larger than 660 bp was too low to reliably discriminate between positive and negative droplets when templates are amplified with a one-minute elongation time. When this is the case, the average fluorescence amplitude for these amplicons cannot be calculated. Increasing the elongation time to two minutes increases the fluorescence amplitude of all droplets containing amplifiable template (Fig. 2). This enables the acquisition of accurate concentration and fluorescence amplitude data for longer templates, but the slope of the relationship between amplicon size and fluorescence amplitude is decreased (from m = −11.66 to m = −9.12), which decreases the ability to resolve small differences in amplicon size (Fig. 2). Decreasing the elongation time to 30 seconds increases the resolution of the relationship between amplicon size and fluorescence amplitude, but prevents targets longer than 460 bp from amplifying to the point that they fluoresce detectably above the background fluorescence (Fig. 2). This is likely due to the fact that longer products require more time for complete polymerization of nascent strands to occur. Thus, there is a tradeoff between the resolution and range of QuantiSize, though the assay can be easily adjusted to fit particular experimental needs.
Figure 2. Effect of ddPCR elongation time on the relationship between fluorescence amplitude ± SEM and amplicon size.

Three ddPCR experiments were carried out with the same size standards using 0.5, 1, and 2 minute elongation times during droplet thermal cycling. With a 0.5 minute elongation time (blue), the slope of the regression line relating fluorescence amplitude to amplicon size was -13.760 (R2=0.9905). With a 1 minute elongation time (red), the slope was -11.460 (R2=0.9906). With a 2 minute elongation time (green), the slope was -9.123 (R2=0.9975). As the magnitude of the slope of the relationship between fluorescence amplitude and amplicon size increases, so does the ability to accurately resolve small differences in amplicon size. Larger templates require longer elongation times for positive droplets to fluoresce discernibly above the background level of droplet fluorescence.
To validate the use of the QuantiSize assay for the sizing and quantification steps in library preparation for NGS, we compared the quantity and size distribution of library DNA predicted by the QuantiSize assay to the quantity and size distribution of reads generated by the Illumina MiSeq platform. Eight uniquely indexed test libraries were generated by ligating DNA sheared to an average size of 150 bp onto MiSeq compatible adapter sequences similar to those used to create the aforementioned size standards. The libraries were run in individual wells of a ddPCR experiment alongside the set of size standards. Using the concentrations measured by ddPCR, the eight uniquely indexed libraries were diluted and combined in a molar ratio of 100:50:10:1 with two libraries at each concentration. The observed number of MiSeq reads containing each index was compared to the expected number of copies of each uniquely indexed library loaded onto the MiSeq. The observed ratio of the number of reads containing each index very closely matched the expected ratio of 100:50:10:1 that was measured by ddPCR and the correlation between the expected and actual number of library molecules gave an R2 value of 0.9693 (Fig. 3A).
Figure 3. Cluster density and number of sequencing reads ± SEM across multiple sequencing runs performed using QuantiSize.
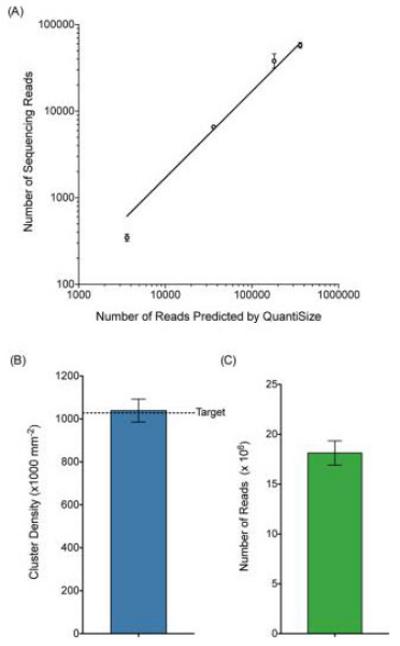
(A) Eight uniquely indexed libraries were loaded onto the MiSeq with two libraries at each concentration. The libraries were loaded in a concentration ratio of 100:50:10:1 based on ddPCR measurements. Due to the binding kinetics of library molecules on the MiSeq flow cell, the number of reads generated by the MiSeq is expected to be a fraction of the number of library molecules loaded. The relative numbers of MiSeq reads for each library closely correspond to the relative numbers of molecules loaded according to ddPCR measurements (R2=0.9693). (B) Mean cluster density (± SEM) resulting from three separate sequencing runs using Nextera-prepared samples normalized based on QuantiSize measurements. The target cluster density (represented by a horizontal dashed line) was 1.028×106 clusters/mm2 including a 5% phi X control and the observed mean cluster density from the three runs was 1.039 ± 0.053×106 cluster/mm2. (C) Mean number of reads (± SEM) resulting from three separate sequencing runs. The observed mean number of reads was 1.813 ± 0.070×107.
The Nextera XT DNA Sample Preparation Kit was used to prepare a sequencing library from genomic DNA extracted from the human colon cancer cell line HCT 116. In lieu of the optional bead normalization step in the Nextera XT protocol, the concentration and size distribution of the library were measured with QuantiSize and the library concentration was adjusted to the proper concentration based on this measurement. The process of quantifying, normalizing, denaturing, and loading the library onto the MiSeq was repeated three times to demonstrate the precision of QuantiSize for predicting cluster density. The target cluster density for each MiSeq run was 1.0×106 clusters/mm2 (1.028×106 clusters/mm2 including the phi X control recommended by Illumina). The mean cluster density ± SEM obtained was 1.039 ± 0.053×106 clusters/mm2 (Fig. 3B). The mean number of reads ± SEM obtained was 1.813 ± 0.070×107 (Fig. 3C). There are several potential sources of error in the MiSeq sample loading process including the pipetting of small volumes, variability in the efficiency of the denaturation reaction, variability of flow cell surface area, and user error. These factors may account for some of the observed variance in cluster density. However, even with the potential error caused by these factors, the target cluster density was achieved with high precision using the QuantiSize method.
The equation relating amplicon size and fluorescence amplitude can be applied either to the average (mean or median) fluorescence amplitude of a sample or to the fluorescence amplitude of individual droplets. Applying the equation to individual droplets allows for a more detailed analysis of the distribution of product sizes present in a sample. We applied the equation generated by the adapter-ligated size standards to the fluorescence amplitude of individual droplets containing library DNA to calculate the expected amplicon size within each droplet and compared the distribution of sizes to the distribution of read sizes measured by the MiSeq (Fig. 4). The frequency distribution shows a high degree of overlap and a common center point for the estimation made using the QuantiSize assay and the observations from the MiSeq. As depicted in Figure 2A, there is an inherent variance in droplet amplitude that occurs even within a completely homogeneous sample of amplicon lengths. This variance likely accounts for the wider distribution of product sizes estimated by ddPCR than were observed in the MiSeq data. Size determination with QuantiSize provides a detailed calculation of the distribution of fragment sizes present in a sample, whereas gel or capillary electrophoresis provide only a range of sizes.
Figure 4. Comparison of library molecule size distribution.

The QuantiSize assay was performed on a DNA library prepared for the MiSeq in order to predict the distribution of library molecule sizes. The DNA library was amplified in parallel with a set of size standards using the same primers and TaqMan probe, allowing us to estimate the expected amplicon size within each individual droplet. The resulting size distribution is shown in blue. The actual size distribution was determined through paired-end sequencing on the Illumina MiSeq system (shown in red). Both histograms show the relative frequency of measured molecule sizes in 10 base pair bins. The size distribution measured by QuantiSize is naturally wider than the distribution measured by the MiSeq due to the inherent variance in droplet amplitude that occurs even with amplicons of the same length. The DNA library was amplified in parallel with a set of size standards using the same primers and TaqMan probe, allowing us to estimate the expected amplicon size within each individual droplet.
The QuantiSize assay demonstrates accuracy, reliability, and flexibility through the strength of the correlation between fluorescence amplitude and amplicon size in ddPCR experiments, and the ease with which the assay can be adjusted to fit specific experimental needs. Applying the QuantiSize assay to NGS library preparation avoids the limitations of other independent quantification and size determination methods and has the potential to increase the average yield of usable data generated from sequencing runs, thereby increasing the efficiency and throughput. The ability to determine the absolute quantity and the detailed size distribution of target DNA with a single experiment will be useful for a broad range of applications that require the quantification and sizing of target DNA.
Acknowledgements
We would like to thank Nolan Ericson and Mariola Kulawiec for helpful discussion and critical reading of our manuscript. This work was supported by an Ellison Medical Foundation New Scholar award (AG-NS-0577-09), an Outstanding New Environmental Scientist Award (ONES) (R01) from the National Institute of Environmental Health Sciences (R01ES019319), and a grant (W81XWH-10-1-0563) from the Congressionally Directed Medical Research Programs/U.S. Department of Defense.
Footnotes
Competing interests
The authors declare that they have no competing interests.
REFERENCES
- 1.Didelot X, Bowden R, Wilson DJ, Peto TE, Crook DW. Transforming clinical microbiology with bacterial genome sequencing. Nature reviews. Genetics. 2012;13:601–612. doi: 10.1038/nrg3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Biesecker LG, Burke W, Kohane I, Plon SE, Zimmern R. Next-generation sequencing in the clinic: are we ready? Nature reviews. Genetics. 2012;13:818–824. doi: 10.1038/nrg3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martin JA, Wang Z. Next-generation transcriptome assembly. Nature reviews. Genetics. 2011;12:671–682. doi: 10.1038/nrg3068. [DOI] [PubMed] [Google Scholar]
- 4.Voelkerding KV, Dames SA, Durtschi JD. Next-generation sequencing: from basic research to diagnostics. Clinical chemistry. 2009;55:641–658. doi: 10.1373/clinchem.2008.112789. [DOI] [PubMed] [Google Scholar]
- 5.Su Z, Ning B, Fang H, Hong H, Perkins R, Tong W, Shi L. Next-generation sequencing and its applications in molecular diagnostics. Expert review of molecular diagnostics. 2011;11:333–343. doi: 10.1586/erm.11.3. [DOI] [PubMed] [Google Scholar]
- 6.Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nature reviews. Genetics. 2010;11:685–696. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- 7.Zhang J, Chiodini R, Badr A, Zhang G. The impact of next-generation sequencing on genomics. Journal of genetics and genomics = Yi chuan xue bao. 2011;38:95–109. doi: 10.1016/j.jgg.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu L, Li Y, Li S, Hu N, He Y, Pong R, Lin D, Lu L, Law M. Comparison of next-generation sequencing systems. Journal of biomedicine & biotechnology. 2012;2012:251364. doi: 10.1155/2012/251364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, Pallen MJ. Performance comparison of benchtop high-throughput sequencing platforms. Nature biotechnology. 2012;30:434–439. doi: 10.1038/nbt.2198. [DOI] [PubMed] [Google Scholar]
- 10.Glenn TC. Field guide to next-generation DNA sequencers. Molecular ecology resources. 2011;11:759–769. doi: 10.1111/j.1755-0998.2011.03024.x. [DOI] [PubMed] [Google Scholar]
- 11.Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, Bertoni A, Swerdlow HP, Gu Y. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC genomics. 2012;13:341. doi: 10.1186/1471-2164-13-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linnarsson S. Recent advances in DNA sequencing methods - general principles of sample preparation. Experimental cell research. 2010;316:1339–1343. doi: 10.1016/j.yexcr.2010.02.036. [DOI] [PubMed] [Google Scholar]
- 13.Meyer M, Briggs AW, Maricic T, Hober B, Hoffner B, Krause J, Weihmann A, Paabo S, Hofreiter M. From micrograms to picograms: quantitative PCR reduces the material demands of high-throughput sequencing. Nucleic acids research. 2008;36:e5. doi: 10.1093/nar/gkm1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buehler B, Hogrefe HH, Scott G, Ravi H, Pabon-Pena C, O’Brien S, R Formosa, S Happe. Rapid quantification of DNA libraries for next-generation sequencing. Methods. 2010;50:S15–18. doi: 10.1016/j.ymeth.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 15.White RA, 3rd, Blainey PC, Fan HC, Quake SR. Digital PCR provides sensitive and absolute calibration for high throughput sequencing. BMC genomics. 2009;10:116. doi: 10.1186/1471-2164-10-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valasek MA, Repa JJ. The power of real-time PCR. Advances in physiology education. 2005;29:151–159. doi: 10.1152/advan.00019.2005. [DOI] [PubMed] [Google Scholar]
- 17.Yun JJ, Heisler LE, Hwang, Wilkins O, Lau SK, Hyrcza M, Jayabalasingham B, Jin J, et al. Genomic DNA functions as a universal external standard in quantitative real-time PCR. Nucleic acids research. 2006;34:e85. doi: 10.1093/nar/gkl400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Analytical chemistry. 2011;83:8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hori M, Fukano H, Suzuki Y. Uniform amplification of multiple DNAs by emulsion PCR. Biochemical and biophysical research communications. 2007;352:323–328. doi: 10.1016/j.bbrc.2006.11.037. [DOI] [PubMed] [Google Scholar]
- 20.Xiao M, Phong A, Lum KL, Greene RA, Buzby PR, Kwok PY. Role of excess inorganic pyrophosphate in primer-extension genotyping assays. Genome research. 2004;14:1749–1755. doi: 10.1101/gr.2833204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Zhang D, Li W, Chen J, Peng Y, Cao W. A novel real-time quantitative PCR method using attached universal template probe. Nucleic acids research. 2003;31:e123. doi: 10.1093/nar/gng123. [DOI] [PMC free article] [PubMed] [Google Scholar]