

Abstract
Background
Hepatospecific deletion of PTEN results in constitutive activation of Akt and increased lipogenesis. In mice, the addition of a high fat diet (HFD) downregulates lipogenesis. The aim of this study was to determine the effects of a HFD on hepatocellular damage induced by deletion of PTEN.
Methods
12 Week old male flox/flox hepatospecific PTEN mice (PTENf/f) or Alb-Cre controls were fed a HFD composed of 45% fat-derived calories (from corn oil) or a normal chow. Animals were then analyzed for hepatocellular damage, oxidative stress and expression of enzymes involved in fatty acid metabolism.
Results
In the Alb-Cre animals, the addition of a HFD resulted in a significant increase in liver triglycerides and altered REDOX capacity as evidenced by increased GPX activity, decreased GST activity and decreased hepatic concentrations of GSSG. In addition, SCD2, ACLY and FASN were all downregulated by the addition of HFD. Furthermore, expression of PPARα and PPARα-dependent proteins Cyp4a and ACSL1 were upregulated. In the PTENf/f mice, HFD resulted in significant increased in ALT, serum triglycerides and decreased REDOX capacity. Although expression of fatty acid synthetic enzymes was elevated in the chow fed PTENf/f group, the addition of HFD resulted in SCD2, ACLY and FASN downregulation. Compared to the Alb-Cre HFD group, expression of PGC1α, PPARα and its downstream targets ACSL and Cyp4a were upregulated in PTENf/f mice.
Conclusions
These data suggest that during conditions of constitutive Akt activation and increased steatosis, the addition of a HFD enhances hepatocellular damage due to increased CD36 expression and altered REDOX status. In addition, this work indicates HFD-induced hepatocellular damage occurs in part, independently of Akt signaling.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is a leading cause of liver disease in the United States today. A common phenotype of NAFLD is an environment characterized by pronounced hepatic lipid accumulation and enhanced oxidative stress. In a subset of NAFLD patients, symptoms progress to nonalcoholic steatohepatitis (NASH) and in this inflammatory environment a further subset will progress to fibrosis and ultimately cirrhosis [1], [2], [3], [4].
Many animal models of NAFLD utilize a long term feeding of a diet high in polyunsaturated fatty acids to induce hepatocellular steatosis. Frequently, additional hepatic insults such as cholesterol or oxidized low density lipoproteins are added as well [5], [6], [7]. In NASH, steatosis is frequently regarded as the first “hit” and is hypothesized to be the prerequisite for progression to steatohepatitis. A second, not yet definitively identified, “hit” is required for the progression to steatohepatitis. This second hit has been proposed to include cellular processes such as mitochondrial injury, oxidative stress, innate immunity or proinflammatory cytokines [8].
The phosphatase and tensin homolog deleted on chromosome 10 (PTEN)/Akt pathway is well documented in its ability to directly regulate de novo lipogenesis (DNL) in the liver [9]. PTEN is a dual specificity phosphatase possessing both lipid and protein phosphatase activity and is a member of the protein tyrosine phosphatase (PTP) family of phosphatases [10], [11]. PTEN negatively regulates Akt activation through its ability to dephosphorylate the 3-position phosphate from PtdIns (3,4,5) P3 to produce PtdIns (4,5) P2. Inactivation of PTEN leads to sustained Akt activation in both cellular and animal models. Hepatospecific deletion of PTEN (PTENf/f) is an established model to examine the effects of a NASH-like condition [1]. In the liver, PTENf/f results in insulin hypersensitivity, hepatomegaly, triglycerides, and constitutive activation of DNL. As these mice age, a progression into steatohepatitis and ultimately hepatocellular carcinoma occurs in mice fed normal chow diets [12], [13]. PTEN expression in other organs and tissues is normal but there is an overall reduction in overall body fat [1], [3], [12], [13], [14].
In the present study, the effects of short term feeding of a HFD was used as a second hit and examined in a background of enhanced steatosis that occurs in PTENf/f mice. We demonstrate that addition of a HFD significantly exacerbates hepatocellular damage and oxidative stress in PTENf/f mice. Furthermore, HFD suppresses expression of de novo synthetic enzymes downstream of Akt and upstream of SREBP1. This study also provides additional insight into the mechanism of HFD-induced oxidative stress and delineates the relative contribution of the PTEN/Akt pathway in HFD-induced hepatocellular damage.
Materials and Methods
Animal model
PTENf/f mice and Alb-Cre mice on a C57Bl6/J background were bred as previously described [12]. Mice, 12 weeks of age in groups of six were fed a liquid HFD (45% fat derived calories from corn oil) (Bio-Serv, Frenchtown, NJ) or standard chow for six weeks. Upon completion of the study, animals were anesthetized via intraperitoneal injection with sodium pentobarbital and euthanized by exsanguination. Blood was collected from the inferior vena cava and plasma was separated through centrifugation @4°C and assayed for alanine aminotransferase (ALT) activity (Sekisui Diagnostics, P.E.I., Canada). Excised livers were weighed, sections of the liver caudate and median lobes collected, fixed in 10% neutral buffered formalin and embedded in paraffin for histological and immunohistochemical examination and prepared for hematoxylin and eosin staining. The remaining portion was subjected to differential centrifugation and subcellular fractionation as previously described [15]. All procedures involving animals were approved by the Institutional Animal Care and Use Committee of the University of Colorado and were performed in accordance with published National Institutes of Health guidelines.
Western blotting
Proteins from either whole liver extracts or subcellular fractions were subjected to standard SDS-PAGE and transferred to PVDF (GE Healthcare, Picataway, NJ). Membranes were blocked for 60 minutes with a tris-buffered saline solution containing 1% Tween-20 (TBST) and 5% non-fat dry milk and probed overnight with primary antibodies directed according to Table S1. A horseradish peroxidase conjugated secondary (Jackson ImmunoResearch Inc. West Grove, PA) was then applied and membranes developed using ECL-Plus Reagent (GE Healthcare). Chemiluminescence was visualized using either film or a Storm 860 scanner from Molecular Dynamics (Sunnyvale, CA).
Biochemical analysis
Liver triglycerides were measured in a 2∶1 chloroform:methanol extract of liver homogenate using a kit from Diagnostic Research Inc. Serum adiponectin was determined using serum diluted 1∶500 in assay buffer and an ELISA kit according to the manufacturer's protocol (Millipore, Billerica, MA). GSH, GSSG, GST activity, Gpx, TrxR activity and biotin hydrazide detection of carbonylated proteins were detected as previously described [16], [17], [18]. Protein concentrations were determined using a modified Lowry Protein Assay from Bio-Rad (Hercules, CA).
Gene expression
Liver (50 mg) was taken from Cre and PTEN chow fed, and HFD-treated animals, and homogenized in 0.6 ml of RLT plus buffer (Qiagen) using a Precellys homogenizer (Bertin Technologies, Rockville, MD). Total RNA was isolated according to the manufacturer's instructions accompanying the RNeasy Plus RNA isolation kit (Qiagen). RNA samples were quantified by UV spectrometry, and total RNA was reversed transcribed using iScript cDNA synthesis (Bio-Rad Laboratories, Hercules, CA) as per manufacturer's instructions. Subsequent real-time PCR analysis was carried out using SYBR green and an ABI 7500 sequencing detection system (Applied Biosystems, Foster City, CA). Results were quantified using deltaCT method relative to GAPDH. Gene specific primers for each gene tested are listed in Table S2.
Statistical analysis
Relative densitometry of Western blots was quantified using ImageJ (http://rsb.info.nih.gov/ij/). Statistical analysis of data was performed using 1-way Analysis of Variance, 2-way Analysis of Variance or a student's t-test and Prism 4 for Windows (GraphPad Software, San Diego, CA). All data are expressed as mean +/− S.E. and p values <0.05 were considered significant.
Results
HFD results in an increase in hepatocellular damage and hepatic triglycerides in PTENf/f mice
To validate the penetrance of the Alb-Cre promoter, lysates were prepared from Chow and HFD fed Alb-Cre and PTENf/f livers and Western blotted for PTEN, pSer473Akt and total Akt. As shown in Figure S1A-C, PTENf/f results in a greater than 95% deletion of hepatic PTEN and a 3-fold increase in Akt phosphorylation.
The data presented in Table 1 describes the effect of hepatospecific PTEN deletion on HFD-induced hepatotoxicity. In the Alb-Cre animals, a HFD resulted in a mildly elevated serum ALT (1.15-fold). As expected, in the PTENf/f chow fed animals, serum ALT increased 6.7-fold when compared to Alb-Cre chow-fed animals. The addition of a HFD significantly elevated this marker of hepatocellular damage by 11.93, 10.3 and 1.81-fold when compared to chow-fed Alb-Cre, high fat fed Alb-Cre and chow-fed PTENf/f animals respectively. Overall, the addition of a HFD resulted in a significant increase in body weight in both genotypes. Comparing PTENf/f and Alb-Cre liver weights, PTENf/f resulted in significant increase in overall liver weight. Surprisingly, compared to chow-fed animals, HFD feeding resulted in decreased liver weight in both the Alb-Cre and the PTENf/f groups. This decrease in liver weight corresponded to decreased liver to body weight ratios in both HFD groups. In the chow-fed groups, PTENf/f resulted in a significant 6-fold increase in hepatic triglycerides compared to respective Alb-Cre controls. In both models, liver triglycerides were increased in the HFD-fed groups; (1.55-fold Alb-Cre, 1.66-fold PTENf/f-).
Table 1. Biochemical and global analysis of serum and liver homogenates of chow-fed and HFD-fed Alb-Cre and PTENf/f mice.
| Alb-Cre | PTENf/f | Two-Way ANOVA P value | |||||||
| Parameter╪ | Chow | HFD | Chow | HFD | Genotype | HFD | Interaction | ||
| ALT (U/L) | 14.51±3.14a | 16.70±3.23b | 95.50±15.17a,c | 173.05±25.65b,c | <0.0001 | 0.0516 | 0.0646 | ||
| Liver weight (grams) | 1.35±0.09a | 0.95±0.05a,b | 3.47±0.40a | 2.60±0.21b | <0.0001 | 0.0324 | 0.4044 | ||
| Change in body weight (grams) | 2.97±0.42a | 7.02±0.43a,b | 2.34±0.35c | 3.83±0.39b,c | 0.0002 | <0.0001 | 0.0059 | ||
| Liver/Body Weight | 4.57±0.08a | 3.20±0.05a,b | 12.26±0.89a | 10.53±0.48b | <0.0001 | 0.0438 | 0.8728 | ||
| Liver Triglycerides (µg/mg tissue) | 0.002±0.0002a | 0.0031±0.0007b | 0.01±0.001a,c | 0.02±0.001b,c | <0.0001 | 0.0004 | 0.0035 |
Serum ALT, liver weight, change in body weight, liver to body weight and liver triglycerides were determined as described in methods. Data are means± SEM as analyzed by two-way ANOVA with a Bonferroni post hoc analysis (Alb-Cre group compared to PTENf/f group). Means with a common superscript letter are significantly different (N = 6 mice/group (p<0.05)).
Data are presented as mean± SEM. Statistical significance was determined by Two-Way ANOVA followed by Bonferroni post hoc analysis.
Letter with similar superscripts (a, b, c) denote significant difference of P<0.05.
HFD results in an increase in hepatic steatosis but not fibrosis in PTENf/f mice
Given the finding that the addition of a HFD resulted in a significant increase in hepatocellular damage as determined by ALT, the effects of HFD in the PTENf/f background was examined in H&E stained liver sections. From Figure 1B, using hematoxylin and eosin staining, the addition of a HFD resulted in a mild increase in steatosis in Alb-Cre liver sections. In the chow-fed PTENf/f group, a dramatic increase in steatosis occurred primarily in hepatic zones 2 and 3. In addition, significant enlargement of the bile ducts (BD) was evident. The addition of a HFD resulted in a significant increase in overall hepatic steatosis in all three zones. Liver sections were then graded for steatosis and inflammatory foci [19]. In the Alb-Cre animals, no steatosis or inflammatory foci were evident in the chow-fed group. Addition of HFD resulted in a mildly increased in steatosis with no inflammatory foci (score 0–0.25). In the PTENf/f group, chow diet resulted in a significant increase in steatosis (score 2.25±0.38) with 0.775±0.35 lobular inflammatory foci/200X field. The HFD significantly increased both overall steatosis score (2.75±0.32) and inflammatory foci (1.3±0.24) in PTENf/f mice. As shown in Figure 2, no significant change in fibrosis was evident following HFD in either genotype but PTENf/f exhibited significantly more picrosirius red staining than the Alb-Cre animals.
Figure 1. High fat diet results in increased lipid accumulation in PTENf/f mice.
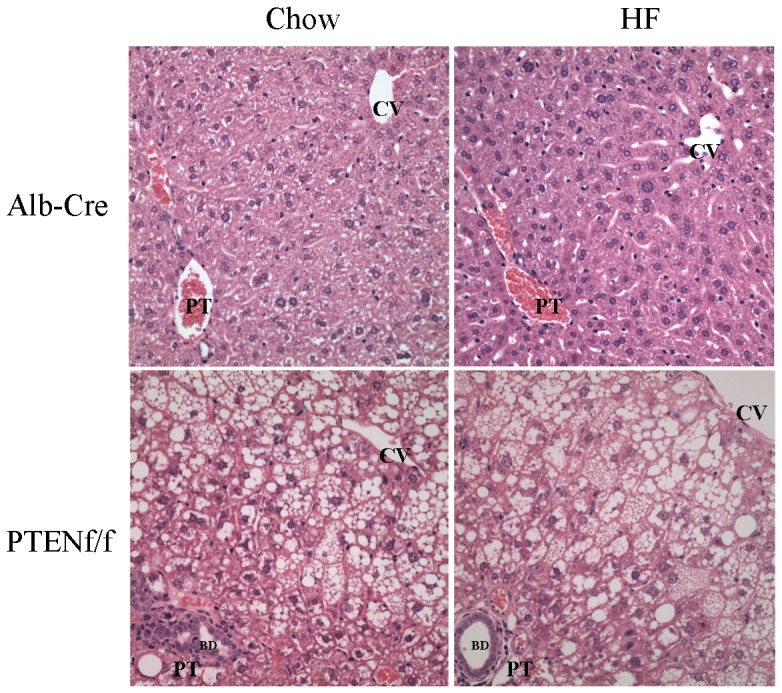
Hematoxylin and eosin staining of tissue sections from chow/HFD-fed Alb-Cre and PTENf/f mice. (CV, central vein, PT, portal triad, BD, bile ducts). Original Magnification 400X.
Figure 2. HFD does not alter fibrosis in PTENf/f mice.

Picrosirius Red staining of tissue Hematoxylin and eosin staining of tissue sections from chow/HFD-fed Alb-Cre and PTENf/f mice. (A) White light. (B) Polarized light. (CV, central vein, PT, portal triad). Original Magnification 400X.
Effects of a HFD and hepatospecific PTEN deletion on oxidative stress and overall glutathione REDOX capacity
Reactive aldehydes are a validated measure of overall cellular oxidative stress in the liver [20]. In humans, lipid aldehydes are increased in NASH patients [21], [22], [23]. To determine the effects of a HFD on overall protein lipid peroxidation, whole cell extracts were treated with biotin hydrazide and examined by Western blotting. As shown in Table 2, HFD feeding did not result in an increase in overall protein carbonylation in the Alb-Cre groups. Compared to Alb-Cre controls, hepatospecific deletion of PTEN resulted in a significant decrease in protein carbonylation in the chow fed group. The addition of a HFD had very little effect in the Alb-Cre group but significantly increased overall protein carbonylation in PTENf/f animals. In hepatocytes, reactive aldehydes are removed via conjugation to glutathione [24]. Therefore, using whole hepatic tissue, the relative amounts of GSH, GSSG and ratio of GSH:GSSG was determined in all groups. From Table 2, in this system, HFD consumption did not significantly affect GSH in the Alb-Cre group. Compared to Alb-Cre controls, PTENf/f resulted in a significant 1.56-fold increase in GSH. Addition of HFD resulted in a decrease in GSH in PTENf/f group. Oxidation of glutathione occurs under conditions of oxidative stress. Comparing both models, deletion of PTEN resulted in a significant increase in GSSG. The addition of a HFD significantly reduced GSSG in the Alb-Cre group whereas GSSG was significantly increased in the PTENf/f group. A phenotype of increased cellular oxidative stress reflects significant decrease in the ratio of GSH:GSSG signifying decreased antioxidant capacity [25], [26]. Surprisingly, following HFD, GSH:GSSG was increased by 35% in the Alb-Cre mice suggesting a decrease in overall cellular oxidative stress. In the PTENf/f mice, HFD feeding resulted in a marked 40% reduction in GSH:GSSG indicating an increase in cellular oxidative stress. Compared to both groups of Alb-Cre mice, cellular redox ratios significantly decreased in the PTENf/f mice. 2-way ANOVA revealed an interaction between HFD and PTENf/f. Combined, these data indicate that hepatospecific deletion of PTEN results in increased hepatocellular oxidative stress that is further exacerbated by the addition of HFD.
Table 2. Oxidative stress measurements in hepatic tissue isolated from chow/HFD-fed Alb-Cre and PTENf/f mice.
| Alb-Cre | PTENf/f | Two-Way ANOVA P value | |||||||
| Parameter╪ | Chow | HFD | Chow | HFD | Genotype | HFD | Interaction | ||
| GSH (µmol/g tissue) | 3.39±0.26a | 3.37±0.24 | 5.29±0.44a | 4.01±0.64 | 0.0142 | 0.1797 | 0.1923 | ||
| GSSG (µmol/g tissue) | 0.27±0.01a | 0.17±0.01a,b | 0.52±0.02a | 0.61±0.03b | <0.0001 | 0.1633 | 0.0011 | ||
| GSH:GSSG | 12.64±0.76a | 19.43±1.30a,b | 10.23±0.76c | 6.51±0.68b,c | <0.0001 | 0.1675 | 0.0002 | ||
| GST activity (U/mg protein) | 19.93±2.28a | 13.89±1.09a | 19.52±1.29c | 14.59±0.94c | 0.9181 | 0.0007 | 0.6944 | ||
| GPX activity (U/mg protein) | 17.98±0.83a | 26.67±1.30a,b | 15.46±1.25 | 16.85±0.75b | <0.0001 | 0.0005 | 0.005 | ||
| TrxR activity (Percent Alb-Cre Chow) | 100±12.23 | 108±4.23a | 134.00±11.18 | 152.46±7.742a | 0.0013 | 0.175 | 0.6356 | ||
| GSTμ expression | 100±10.16a | 104.62±4.77 | 169.640±21.15a,b | 104.70±10.16b | 0.0277 | 0.0494 | 0.0279 | ||
| GSTπ expression | 100±13.76 | 103.83±4.23a | 65.44±7.98 | 49.43±4.92a | 0.001 | 0.5 | 0.283 | ||
| GSTA4 expression | 100±12.57 | 101.07±7.21a | 79.96±4.05 | 58.26±4.98a | 0.0042 | 0.2303 | 0.1881 | ||
| Carbonylation (Percent Chow Alb-Cre) | 100.00±7.28a | 103.90±7.70b | 53.59±4.86a,c | 85.16±8.33b,c | 0.0007 | 0.0291 | 0.0774 |
Data are means± SEM as analyzed by two-way ANOVA with a Bonferroni post hoc analysis (Alb-Cre group compared to PTENf/f group). Means with a common superscript letter are significantly different (N = 6 mice/group (p<0.05)).
Data are presented as mean± SEM. Statistical significance was determined by Two-Way ANOVA followed by Bonferroni post hoc analysis. Letter with similar superscripts (a, b, c) denote significant difference of P<0.05.
Analysis of the effects of PTENf/f and HFD on GST, GPx and TrxR activity
The data presented in Table 2 suggests that hepatospecific deletion of PTEN results in an increase in oxidative stress and altered glutathione metabolism. In human patients with livers staged as NASH, GST activity was increased [27]. In other studies however, GST activity was reduced during the progression of NAFLD to NASH [28]. To further elucidate these processes in our models, overall GST and GPx activity was assessed. Comparing each genotype, chow feeding did not significantly affect GST activity. In both groups, the addition of a HFD resulted in a significant decrease in GST activity. Examining GPx activity, compared to Alb-Cre chow-fed mice, PTENf/f significantly suppressed GPx activity. The addition of a HFD resulted in a significant 1.5-fold increase in GPx activity in the Alb-Cre group. Surprisingly, PTENf/f prevented HFD-induced increases in GPx activity compared to Alb-Cre control mice. An alternative indicator of oxidative stress is protein carbonylation [29]. Examining overall levels of carbonylation, PTENf/f significantly decreased protein carbonylation compared to chow fed Alb-Cre animals. Although the addition of HFD did not have a significant effect in Alb-Cre animals, in the PTENf/f group, carbonylation was increased 1.6-fold. The thioredoxin/thioredoxin reductase system assists in the antioxidant response by reducing oxidized cysteine residues [25]. PTEN has also been demonstrated to directly interact with thioredoxin 1 altering activity [30], [31]. Therefore, the effect of PTENf/f and a HFD on thioredoxin reductase activity was examined. From Table 2, a HFD had no effect in the Alb-Cre animals. In PTENf/f, thioredoxin reductase activity was significantly increased when compared to respective Alb-Cre Chow fed as well as HFD fed animals.
Based on our activity data, we sought to determine the specific isoforms of GST that are contributing to the observed changes in activity. From the Western blot and quantifications in Figure 3 and Table 2, a HFD did not significantly affect expression of GSTμ, π or A4 in the Alb-Cre mice. Comparing genotypes, PTENf/f resulted in a significant increase in overall GSTμ expression but surprisingly, expression of GSTπ and A4 was significantly decreased. Furthermore, the addition of a HFD resulted in only GSTμ expression. Using 2-way ANOVA, a significant interaction was observed for only GSTμ. All three isoforms tested exhibited genotype specific effects although not in the same direction. Overall, the data presented in Table 2 and Figure 3 PTENf/f induces cellular oxidative stress and may exert a selective effect on glutathione metabolism.
Figure 3. Effects of HFD on expression of GST's in Alb-Cre and PTENf/f.

Western blotting analysis of GSTA4, μ and μ using whole cell extracts isolated from chow/HFD-fed Alb-Cre and PTENf/f mice.
Analysis of the effects of HFD and PTENf/f on serum adiponectin
As shown in Figure 1 and Table 1, the addition of a HFD resulted in a significant increase in both liver triglycerides and steatosis in the PTENf/f model. In NASH, serum adiponectin inversely correlate to body fat mass and negatively correlates with disease progression [32]. The relative levels of serum adiponectin have not been evaluated in PTENf/f mice with a HFD. As shown in Table 3, as expected given that PTENf/f mice have previously been reported to possess reduced visceral fat mass, in the chow-fed group, PTENf/f resulted in a 70% decrease in serum adiponectin [12]. Surprisingly, the addition of a HFD had no effect in the Alb-Cre groups but significantly increased serum adiponectin 2-fold in the PTENf/f group. This increase however, did not restore serum adiponectin to Alb-Cre chow fed levels. This suggests that although not directly measured, the increase in adiponectin in HFD PTENf/f correlates to an increase in body fat mass that would correlate with the decreased liver to body weight ratios found in HFD PTENf/f mice.
Table 3. Statistic analysis of the Western blots presented in Figure 2.
| Alb-Cre | PTENf/f | P value | |||||||
| Parameters (actin normalized)╪ | Chow | HFD | Chow | HFD | vs genotype | vs HFD | Interaction | ||
| β-oxidation/fatty acid transport | |||||||||
| pAMPK | 100±9.60 | 107.80±4.13 | 104.77±1.48 | 110.32±3.05 | 0.5638 | 0.2731 | 0.1276 | ||
| total AMPK | 100±13.64 | 109.04±1.79 | 116.39±5.21 | 105.97±3.86 | 0.4073 | 0.9309 | 0.237 | ||
| pACC | 100±9.30a | 89.33±2.43b | 252.19±12.07a | 206.83±16.87b | <0.0001 | 0.0399 | 0.1677 | ||
| total ACC | 100±8.64a | 108.90±9.84b | 199.21±3.18a | 178.21±4.76b | <0.0001 | 0.4218 | 0.0698 | ||
| CPT1α | 100±5.95a | 92.67±5.69 | 76.0±1.84a,b | 135.06±18.01b | 0.3824 | 0.0316 | 0.103 | ||
| PPARα | 100±30.00a | 128.98±18.34b | 192.81±11.85a | 174.10±38.65b | 0.033 | 0.8524 | 0.3994 | ||
| Cyp4a | 100±0.72a | 142.29±9.63a,b | 123.23±11.41a,c | 187.86±6.67b,c | 0.003 | 0.0002 | 0.208 | ||
| ACOX1 | 100±6.32 | 86.56±11.56 | 102.83±4.34a | 83.76±1.60a | 0.9984 | 0.0483 | 0.697 | ||
| LFABP | 100±4.74 | 107.88±0.72 | 106.94±6.28 | 80.71±4.02 | 0.5295 | 0.0489 | 0.2966 | ||
| ACSL | 100±8.76a | 144.85±5.33a | 168.98±17.84a | 147.84±2.32 | 0.0084 | 0.2852 | 0.0129 | ||
| serum adiponectin (µg/ml) | 27.80±2.64a | 28.79±1.77c | 8.65±1.4a,b | 15.63±1.73b,c | <0.0001 | 0.0477 | 0.1682 | ||
| lipogenesis | |||||||||
| FASN | 100±9.61a | 82.67±4.26b | 148.84±12.41a | 137.01±7.13b | 0.0004 | 0.1392 | 0.7647 | ||
| ACLY | 100±16.41a | 28.32±10.06a,b | 192.26±6.85a | 159.32±15.82b | <0.0001 | 0.0037 | 0.1722 | ||
| SCD-2 | 100±11.26a | 67.64±2.3a,b | 179.40±17.25a,c | 97.25±10.92b,c | 0.0017 | 0.0012 | 0.0676 | ||
| SCD-1 | 100±7.85a | 58.06±7.29a,b | 161.40±6.39a | 145.72±9.76b | <0.0001 | 0.0066 | 0.1359 | ||
| PPARγ | 100±2.20a | 81.18±8.13b | 150.19±8.59a,c | 113.60±7.23b,c | 0.0002 | 0.0042 | 0.2409 | ||
| PGC1α | 100±2.85 | 96.70±19.16 | 121.99±27.40 | 145.79±3.62 | 0.0683 | 0.5604 | 0.4452 | ||
| nuclear transcription factors | |||||||||
| PPARγ | 100±27.84a | 228.28±67.94b | 2363.87±419.19a | 2889.97±321.36b | <0.0001 | 0.2547 | 0.477 | ||
| PPARα | 100±34.34a | 147.82±43.40b | 501.93±79.21a | 830.64±108.96b | <0.0001 | 0.0323 | 0.0899 | ||
| SREBP 1 | 100±3.16a | 81.34±4.68b | 118.17±5.22a | 86.07±2.30b | 0.0213 | 0.0002 | 0.1325 | ||
| SREBP 2 | 100±5.77a | 130.91±7.97a | 57.55±18.94 | 108.68±12.90 | 0.032 | 0.011 | 0.4409 | ||
| Nrf-2 | 100±10.61 | 75.5±3.37 | 71.2±7.28 | 76.4±8.62 | 0.1168 | 0.2563 | 0.0981 |
Data are means± SEM as analyzed by two-way ANOVA with a Bonferroni post hoc analysis (Alb-Cre group compared to PTENf/f group). All proteins were normalized to either actin (whole cell extracts) or lamin B1 (nuclear extracts). Means with a common superscript letter are significantly different (N = 3 mice/group (p<0.05)).
Data are presented as mean± SEM. Statistical significance was determined by Two-Way ANOVA followed by Bonferroni post hoc analysis.
Letter with similar superscripts (a, b, c) denote significant difference of P<0.05.
Analysis of effects of HF-diet and PTENf/f on expression of key enzymes involved in hepatic fatty acid metabolism
In the liver, adiponectin binds the adiponectin receptors resulting in an increase in 5′ AMP activated phosphorylation (AMPK) on Thr172 [33]. Activated pThr172AMPK subsequently phosphorylates acetyl CoA Carboxylase (ACC) inhibiting ACC activity and decreasing malonyl CoA, an inhibitor of β-oxidation [34], [35]. AMPK signaling has not been examined under PTENf/f conditions. Whole cell extracts were prepared from hepatic tissue isolated from chow-fed/HFD-fed Alb-Cre and PTENf/f mice and probed for total and phosphorylated forms of AMPKα and ACC. From Figure 4, neither the addition of a HFD nor deletion of PTEN had an effect on AMPKα expression or phosphorylation indicating that there was not a compensatory β-oxidative response to increased lipid accumulation. Surprisingly, pSer79ACC was significantly increased corresponding to an increase in overall ACC expression in PTENf/f mice [13], [36]. Combined these data suggest that PTENf/f does not induce changes in AMPK but significantly effects its immediate downstream target ACC.
Figure 4. Effects of HFD on regulation of β-oxidation in Alb-Cre and PTENf/f.

Western blotting analysis of key enzymes involved in β-oxidation using whole cell extracts isolated from chow/HFD-fed Alb-Cre and PTENf/f mice.
In murine models of diet induced obesity, PPARα expression has been show to increase [37]. Elevated pSer79ACC in both PTEN groups led us to further investigate by examining expression of PPARα and expression of the PPARα-dependent enzymes carnitine palmitoyltransferase (CPT1α), acyl CoA oxidase 1 (ACOX1), long chain Acyl CoA synthetase (ACSL1), cytochrome P450-4a (Cyp4a) and liver fatty acid binding protein (LFABP). From Figure 4, PPARα expression was mildly elevated by a HFD in the Alb-Cre group. Hepatospecific deletion of PTEN resulted in a significant increase in PPARα expression. Comparing chow and HFD feeding, expression of CPT-1α was not significantly altered in Alb-Cre animals. In the PTENf/f group, compared to chow-fed Alb-Cre animals, expression CPT1α was decreased. The addition of HFD resulted in increased CPT1α expression in PTENf/f animals. Examining expression of other PPARα-dependent proteins, changes in ACSL1 expression were similar to PPARα whereas Cyp4A was increased by high fat in both models. Surprisingly, both ACOX1 and LFABP were suppressed by HFD and did not exhibit an increase in expression in PTENf/f mice. Combined, these data indicate a selective effect concerning HFD and PTENf/f on cellular β-oxidative processes.
It has previously been reported that hepatospecific deletion of PTEN results in increased expression of fatty acid synthetic enzymes [12], [14], [36]. The effects of a HFD on expression of fatty acid synthetic enzymes in PTENf/f mice have not been examined. From Figure 5 and Table 3, stearoyl CoA desaturase (SCD-1/2), fatty acid synthase (FASN), and ATP citrate lyase (ACLY) were all significantly decreased following HFD in the Alb-Cre groups. As expected, SCD-1, SCD-2, FASN and ACLY were significantly increased in the PTENf/f group. With the exception of SCD-1, the addition of a HFD resulted in a significant decrease in expression. This expression however, was elevated when compared to Alb-Cre HFD animals. Compared to chow-fed PTENf/f animals, HFD did not significantly decrease SCD-1 expression. PPARγ is directly regulated downstream of PTEN and regulates a variety of cellular processes including lipid metabolism, glucose metabolism, inflammatory responses and angiogenesis [13]. The addition of HFD resulted in a significant decrease in PPARγ expression in both Alb-Cre and PTENf/f mice. Comparing genotypes, PTENf/f resulted in a significant increase in PPARγ compared to Alb-Cre animals. PPARγ Coactivator 1α (PGC1α) also promotes de novo lipogenesis. The addition of HFD had no effect on PGC1α expression in Alb-Cre or PTENf/f mice. Comparing the two genotypes, PGC1α was elevated in PTENf/f animals.
Figure 5. Effects of HFD on regulation of hepatic lipogenesis in Alb-Cre and PTENf/f.

Western blotting analysis of de novo lipogenic enzymes using whole cell extracts isolated from chow/HFD-fed Alb-Cre and PTENf/f mice.
Although substantial changes in expression of both β-oxidative as well as de novo lipogenic proteins is shown in Figures 4 and 5 the effects of HFD in combination with PTENf/f on nuclear localization of relevant transcription factors has not been described. Using Western blotting of nuclear extracts, nuclear localization of PPARγ, SREBP1, SREBP2, Nrf2 and β-catenin was evaluated (Figure 6, Table 3). Nuclear translocation of SREBP1 is a critical factor in the regulation of fatty acid synthetic proteins [38]. From Figure 6 and Table 3, nuclear localization of SREBP1 is decreased by HFD feeding in both models. PTEN is a direct regulator of PPARγ. As expected, PPARγ expression is dramatically upregulated by PTENf/f as well as HFD. From Table 2, there is evidence of increased oxidative stress in our model. Following nuclear localization, Nrf2 stimulates expression of a variety of oxidative stress related proteins [28], [39]. Following HFD, nNrf2 is decreased in Alb-Cre animals. Hepatospecific PTEN deletion resulted in a decrease in nNrf2 but HFD had no significant effect. In cells, nuclear localization of β-catenin is an indicator of increased cellular proliferation. Examining the Western blot, nuclear β-catenin is significantly increased in the PTEN animals irrespective of diet compared to Cre controls.
Figure 6. Effects of HFD on nuclear localization of metabolic transcription factors in Alb-Cre and PTENf/f mice.

Western blotting analysis of metabolic transcription factors using nuclear fractions isolated from chow/HFD-fed Alb-Cre and PTENf/f mice.
Effects of HFD and PTEN deletion on expression of selected hepatic genes
As shown in Table 1, feeding a HFD resulted in a significant increase in hepatocellular damage in PTENf/f. To further characterize the effects of PTENf/f and HFD, mRNA analysis of TNFα, CD14, IL-6 and IL-10 was performed using mRNA isolated from fresh frozen tissue sections. As shown in Table 4, in the Alb-Cre groups, although TNFα and CD14 trended upward, HFD did not result in a significant increase in either proinflammatory (TNFα, CD14, IL-6) or anti-inflammatory cytokines (IL-10). Compared to the Alb-Cre groups, PTENf/f resulted in a significant increase in both TNFα and CD14 but there was no significant change in either rIL-6 or IL-10. This suggests that in the PTEN model inflammation is increased independently of consumption of 6 weeks of HFD.
Table 4. mRNA expression analysis of selected cytokines and fatty acid metabolic proteins from tissue isolated from chow/HFD-fed Alb-Cre PTENf/f mice.
| Table 4 mRNA analysis of selected genes | Alb-Cre | PTENf/f | P value | ||||||
| inflammation | Chow | HFD | Chow | HFD | vs genotype | vs HFD | Interaction | ||
| TNFα | 0.02±0.14a | 0.11±0.16c | 0.41±0.14a | 0.48±0.12c | 0.023 | 0.605 | 0.974 | ||
| CD14 | 0.09±0.09a | 0.17±0.11b | 0.41±0.09a | 0.36±0.08b | 0.018 | 0.862 | 0.521 | ||
| IL-6 | 0.07±0.06 | 0.08±0.05 | 0.12±0.05 | 0.05±0.05 | 0.964 | 0.616 | 0.541 | ||
| IL-10 | 0.02±0.01 | 0.03±0.01 | 0.03±0.01 | 0.01±0.01 | 0.794 | 0.955 | 0.118 | ||
| lipid transport and metabolism | |||||||||
| PPARα | 0.02±0.02a,b | 0.07±0.02b | 0.04±0.02a,c | 0.08±0.12c | 0.419 | 0.034 | 0.664 | ||
| PPARγ | 0.51±0.64a | 0.76±0.74b | 5.48±0.58a | 6.41±0.58b | <0.001 | 0.374 | 0.609 | ||
| CD36 | 0.01±0.02a | 0.02±0.02b | 0.11±0.01a,c | 0.18±0.01b,c | <0.001 | 0.055 | 0.241 | ||
| FATP2 | 0.30±0.11a | 1.1±0.13a,c | 0.40±0.11b | 0.60±0.11b,c | 0.188 | <0.001 | 0.027 |
Data are means± SEM as analyzed by two-way ANOVA with a Holm-Sidak post hoc analysis (Alb-Cre group compared to PTENf/f group).
Values are mean (SEM); statistical analysis performed by two-way ANOVA followed by Holm-Sidak analysis.
Letter with similar superscripts (a, b, c) denote significant difference of P<0.05.
From Table 3, Western blotting analysis indicated a significant increase in both PPARα and PPARγ following PTEN deletion, to validate these data, mRNA analysis was performed. As shown in Table 4, mRNA expression of both PPARα and PPARγ are significantly increased in PTENf/f mice. This increase was further enhanced by the addition of a HFD. The lipid transporter CD36 is a direct target of PPARγ and is upregulated in mice fed diets rich in fatty acids [19]. Fatty acid transport protein 2 (FATP2) assists in hepatic free fatty acid uptake [40]. In our study, Western blotting for CD36 was not successful, therefore mRNA expression of lipid transporters CD36 and FATP2 was performed. From Table 4, PTENf/f resulted in a significant increase in CD36 but not FATP2. The addition of a HFD significantly increased FATP2 in both models but CD36 only in the PTENf/f model. Analysis of CD36 and FATP2 by two-way ANOVA indicated a significant interaction between HFD and PTENf/f with respect to FATP2 but not CD36. In summary, a HFD exerts differential effects on lipid transport proteins when combined with hepatospecific PTEN deletion.
Discussion
The accumulation of fat is the first step in the progression of NASH. In this study, we utilized an initial hit of steatosis due to hepatospecific deletion of PTEN and followed it by the addition of a second hit in the form of a HFD over a short time course. Not surprisingly, in our Alb-Cre animals, the addition short term HFD only induced a mild accumulation of hepatic triglycerides and only demonstrated a trend in increased hepatocellular damage. This is an expected result, a longer duration of feeding is necessary to produce hepatocellular damage in normal mice [41], . In PTENf/f mice, a HFD significantly increased triglycerides, body weight and ALT. Comparing both genotypes, PTENf/f resulted in a dramatic increase in overall liver weight, liver:body weight ratios, hepatocellular damage (as evidenced by ALT) and hepatic triglycerides. As expected, HFD resulted in a significant increase in overall body weight. In this study, hepatic triglycerides, body weight, and ALT were significantly increased in the HFD PTENf/f group. This indicates that the effects of a HFD on these parameters are independent of the PTEN pathway and that there is a combinatorial effect in the mutant group.
Not surprisingly, adiponectin levels were decreased in the chow fed PTENf/f group. Previous measurement of fat mass in chow fed PTENf/f animals indicated a significant decrease in body fat [12]. What is surprising is the increase in serum adiponectin in the HFD PTENf/f group. A recent study using Leprdb/db mice has identified adiponectin as an independent predictor of NASH [44]. With HFD feeding, increased hepatic steatosis, triglycerides as well as adiponectin (albeit to levels still well below the Alb-Cre mice) occurred in the PTENf/f model. Yet the increase in adiponectin did not correlate with phosphorylation of AMPK and expression of PGC1α in the PTENf/f groups. Furthermore, both phosphorylation and total expression of ACC levels was significantly elevated regardless of diet and AMPK phosphorylation. The mechanism of this is not known at this time.
In the Alb-Cre model, HFD promoted alterations in cellular REDOX homeostasis as evidenced by decreased GST activity, increased GPx activity and decreased GSSG concentrations. This is in agreement with other studies where HFD was fed for longer periods of time [45]. When we examined individual isoforms of GST (μ, π and A4) we did not detect significant differences in expression following HFD in the Alb-Cre groups. These proteins did exhibit genotype specific effects in the PTENf/f groups. Overall, expression of the GST isoforms tested did not correlate well with our activity data suggesting that other GST isoforms may be in part responsible for the observed differences in GST activity. When combined with a preexisting propensity for lipid accumulation (PTENf/f), the overall REDOX capacity and GPx activity were suppressed yet GST activity exhibited the same effect regardless of genotype. Concurrently, TrxR activity and the concentrations of both reduced and oxidized glutathione significantly increased in the PTENf/f group. With the exception of GST activity, this is in agreement with a previous study that demonstrated increased levels of oxidative stress in the PTENf/f animals [46], [47]. Given the further suppression of overall REDOX capacity in the HFD PTENf/f model, it is not surprising that there was a significant increase in hepatic levels of carbonylation [48], [49]. What is surprising is that comparing the 2 genotypes, carbonylation was significantly suppressed in both PTENf/f groups despite increased oxidative stress. Furthermore, deletion of PTEN results in increased mitochondrial respiration [48]. Based on these data, we propose that increased mitochondrial respiration and mitochondrial oxidative stress results in increased resistance to carbonylation. The mechanism of resistance however remains to be elucidated.
Although we demonstrate an interaction between PTEN/HFD with respect to triglycerides and body weight, this interaction is not reflected in parameters concerning fatty acid synthesis. In our Alb-Cre model, expression of key fatty acid synthetic enzymes was suppressed by a HFD. In the PTENf/f model, rates of fatty acid synthesis and expression of fatty acid synthetic enzymes are significantly upregulated [12], [13]. This upregulation has been proposed to be directly downstream of Akt2 [9], [50]. Yet in these animals, albeit a modest effect, HFD still suppresses expression of FASN and ACLY indicating that HFD may alter FASN/ACLY expression downstream of Akt or that suppression of these enzymes by HFD is independent of Akt. Mammalian target of rapamycin 2 (mTORC2) phosphorylates Akt on Ser473 resulting in full Akt activation in the liver [51]. Using liver specific mTORC2 deletion, the addition of constitutively active Akt2 was not able to restore activation of hepatic lipogenesis [52]. Thus, in our study, downregulation of hepatic lipogenesis by a HFD may be due to alterations in mTORC2 signaling independent of Akt2 activation or downstream of Akt2.
In the PTENf/f mice, significant upregulation of SREBP1 has been demonstrated [14]. However, in our Alb-Cre and PTENf/f mice, HFD resulted in decreased nuclear accumulation of SREBP1. In the livers of SREBP1 overexpressing mice, SCD2 is upregulated [53]. Although both SCD isoforms are suppressed by the addition of a HFD in the Alb-Cre model, in the PTENf/f groups only SCD-2 is significantly suppressed following HFD feeding. SCD-2 is thought to have an important role in the synthesis of monounsaturated fatty acids during early skin and liver development [54]. Although trending downward, expression of SCD1 is not significantly changed suggesting possible alternative mechanisms of regulation that are either downstream of Akt, independent of SREBP1 or via alternative transcription factors in the PTENf/f mice.
The addition of a HFD induces PPARα and PPARα-dependent fatty acid β-oxidation [37]. In PPARα KO mice downstream genes such as ACSL and Cyp4a are significantly decreased following a HFD [37]. We find that expression of ACSL as well as Cyp4a correlate with an increase in PPARα expression in both the Alb-Cre HFD group as well as in both PTENf/f groups. Yet alternative PPARα targets such as ACOX1 and are only induced by the addition of HFD and not by PTENf/f. Furthermore, LFABP is not induced in the Alb-Cre group following HFD and is suppressed in the PTENf/f animals. This suggests that PTENf/f exerts a selective effect on PPARα-dependent proteins.
In conclusion, this study clearly indicates that even short term feeding of a HFD and PTEN deletion have an additive effect on hepatocellular damage and steatosis. Thus, patients with NAFLD should avoid diets rich in PUFAs even in the short term. Although HFD decreases nuclear localization of SREBP1, this decrease is not sufficient to restore levels of most fatty acid synthetic enzymes to normal levels. Furthermore, our data clearly demonstrates that the effects of HFD on de novo lipogenesis occur downstream of Akt or via independent mechanisms such as changes in lipid transport. Given the additive effect of PTENf/f and HFD on hepatocellular damage, these data also provide support that both dietary lipids and lipids derived from de novo lipogenesis contribute to hepatocellular steatosis in PTENf/f mice. It also suggests that in PTENf/f mice, increased β-oxidative processes are not sufficient to reduce hepatic damage.
Supporting Information
Effects of PTENf/f and HFD on PTEN signaling. (A) Western blotting analysis of PTEN, pSer473Akt and total Akt using whole cell extracts isolated from chow/HFD-fed Alb-Cre and PTENf/f mice. (B) Quantification of PTEN expression. (C) Quantification of Akt phosphorylation. Data are means± SEM as analyzed by two-way ANOVA with a Bonferroni post hoc analysis (Alb-Cre group compared to PTENf/f group). Means with a common superscript letter are significantly different (N = 3 mice/group (***p<0.001)).
(TIF)
Antibodies used for Western blotting in this study.
(XLSX)
Primers used for this study.
(XLSX)
Acknowledgments
The authors wish to thank E. Erin Smith, HTL(ASCP)CMQIHC, April Otero, HT(ASCP)CMQIHC and Kathy Lux, HT(ASCP) of the University of Colorado Denver Cancer Center Research Histology Core for their contributions to this manuscript.
Funding Statement
Studies were supported by the National Institutes of Health/National Institutes of Alcoholism and Alcohol Abuse under grant numbers 5F32AA018613-03 CTS, 5R37AA009300-18 DRP, 5R01DK074487-06 DRP. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Watanabe S, Horie Y, Suzuki A (2005) Hepatocyte-specific Pten-deficient mice as a novel model for nonalcoholic steatohepatitis and hepatocellular carcinoma. Hepatol Res 33: 161–166. [DOI] [PubMed] [Google Scholar]
- 2. Hashizume H, Sato K, Takagi H, Hirokawa T, Kojima A, et al. (2007) Primary liver cancers with nonalcoholic steatohepatitis. Eur J Gastroenterol Hepatol 19: 827–834. [DOI] [PubMed] [Google Scholar]
- 3. Watanabe S, Horie Y, Kataoka E, Sato W, Dohmen T, et al. (2007) Non-alcoholic steatohepatitis and hepatocellular carcinoma: lessons from hepatocyte-specific phosphatase and tensin homolog (PTEN)-deficient mice. J Gastroenterol Hepatol 22 Suppl 1S96–S100. [DOI] [PubMed] [Google Scholar]
- 4. Richardson MM, Jonsson JR, Powell EE, Brunt EM, Neuschwander-Tetri BA, et al. (2007) Progressive fibrosis in nonalcoholic steatohepatitis: association with altered regeneration and a ductular reaction. Gastroenterology 133: 80–90. [DOI] [PubMed] [Google Scholar]
- 5. Ioannou GN, Haigh WG, Thorning D, Savard C (2013) Hepatic cholesterol crystals and crown-like structures distinguish NASH from simple steatosis. J Lipid Res 54: 1326–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yimin, Furumaki H, Matsuoka S, Sakurai T, Kohanawa M, et al. (2011) A novel murine model for non-alcoholic steatohepatitis developed by combination of a high-fat diet and oxidized low-density lipoprotein. Lab Invest 92: 265–281. [DOI] [PubMed] [Google Scholar]
- 7. Wouters K, van Gorp PJ, Bieghs V, Gijbels MJ, Duimel H, et al. (2008) Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology 48: 474–486. [DOI] [PubMed] [Google Scholar]
- 8. Basaranoglu M, Basaranoglu G, Senturk H (2013) From fatty liver to fibrosis: a tale of “second hit”. World J Gastroenterol 19: 1158–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leavens KF, Easton RM, Shulman GI, Previs SF, Birnbaum MJ (2009) Akt2 is required for hepatic lipid accumulation in models of insulin resistance. Cell Metab 10: 405–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ross SH, Lindsay Y, Safrany ST, Lorenzo O, Villa F, et al.. (2007) Differential redox regulation within the PTP superfamily. Cell Signal. [DOI] [PubMed]
- 11. Maehama T, Dixon JE (1999) PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol 9: 125–128. [DOI] [PubMed] [Google Scholar]
- 12. Stiles B, Wang Y, Stahl A, Bassilian S, Lee WP, et al. (2004) Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected]. Proc Natl Acad Sci U S A 101: 2082–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, et al. (2004) Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest 113: 1774–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sato W, Horie Y, Kataoka E, Ohshima S, Dohmen T, et al. (2006) Hepatic gene expression in hepatocyte-specific Pten deficient mice showing steatohepatitis without ethanol challenge. Hepatol Res 34: 256–265. [DOI] [PubMed] [Google Scholar]
- 15. Galligan JJ, Fritz KS, Tipney H, Smathers RL, Roede JR, et al. (2011) Profiling impaired hepatic endoplasmic reticulum glycosylation as a consequence of ethanol ingestion. J Proteome Res 10: 1837–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shearn CT, Smathers RL, Stewart BJ, Fritz KS, Galligan JJ, et al. (2011) Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) inhibition by 4-hydroxynonenal leads to increased Akt activation in hepatocytes. Mol Pharmacol 79: 941–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shearn CT, Smathers RL, Backos DS, Reigan P, Orlicky DJ, et al. (2013) Increased carbonylation of the lipid phosphatase PTEN contributes to Akt2 activation in a murine model of early alcohol-induced steatosis. Free Radic Biol Med 65C: 680–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shearn CT, Smathers RL, Jiang H, Orlicky DJ, Maclean KN, et al. (2013) Increased dietary fat contributes to dysregulation of the LKB1/AMPK pathway and increased damage in a mouse model of early-stage ethanol-mediated steatosis. J Nutr Biochem 24: 1436–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ronis MJ, Baumgardner JN, Marecki JC, Hennings L, Wu X, et al. (2012) Dietary fat source alters hepatic gene expression profile and determines the type of liver pathology in rats overfed via total enteral nutrition. Physiol Genomics 44: 1073–1089. [DOI] [PubMed] [Google Scholar]
- 20. Smathers RL, Galligan JJ, Stewart BJ, Petersen DR (2011) Overview of lipid peroxidation products and hepatic protein modification in alcoholic liver disease. Chem Biol Interact 192: 107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weltman MD, Farrell GC, Hall P, Ingelman-Sundberg M, Liddle C (1998) Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology 27: 128–133. [DOI] [PubMed] [Google Scholar]
- 22. Seki S, Kitada T, Yamada T, Sakaguchi H, Nakatani K, et al. (2002) In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J Hepatol 37: 56–62. [DOI] [PubMed] [Google Scholar]
- 23. Chalasani N, Deeg MA, Crabb DW (2004) Systemic levels of lipid peroxidation and its metabolic and dietary correlates in patients with nonalcoholic steatohepatitis. Am J Gastroenterol 99: 1497–1502. [DOI] [PubMed] [Google Scholar]
- 24. Hartley DP, Ruth JA, Petersen DR (1995) The hepatocellular metabolism of 4-hydroxynonenal by alcohol dehydrogenase, aldehyde dehydrogenase, and glutathione S-transferase. Arch Biochem Biophys 316: 197–205. [DOI] [PubMed] [Google Scholar]
- 25. Jones DP (2008) Radical-free biology of oxidative stress. Am J Physiol Cell Physiol 295: C849–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Go YM, Pohl J, Jones DP (2009) Quantification of redox conditions in the nucleus. Methods Mol Biol 464: 303–317. [DOI] [PubMed] [Google Scholar]
- 27. Nobili V, Pastore A, Gaeta LM, Tozzi G, Comparcola D, et al. (2005) Glutathione metabolism and antioxidant enzymes in patients affected by nonalcoholic steatohepatitis. Clin Chim Acta 355: 105–111. [DOI] [PubMed] [Google Scholar]
- 28. Hardwick RN, Fisher CD, Canet MJ, Lake AD, Cherrington NJ (2010) Diversity in antioxidant response enzymes in progressive stages of human nonalcoholic fatty liver disease. Drug Metab Dispos 38: 2293–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sampey BP, Korourian S, Ronis MJ, Badger TM, Petersen DR (2003) Immunohistochemical characterization of hepatic malondialdehyde and 4-hydroxynonenal modified proteins during early stages of ethanol-induced liver injury. Alcohol Clin Exp Res 27: 1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee SR, Yang KS, Kwon J, Lee C, Jeong W, et al. (2002) Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem 277: 20336–20342. [DOI] [PubMed] [Google Scholar]
- 31. Meuillet EJ, Mahadevan D, Berggren M, Coon A, Powis G (2004) Thioredoxin-1 binds to the C2 domain of PTEN inhibiting PTEN's lipid phosphatase activity and membrane binding: a mechanism for the functional loss of PTEN's tumor suppressor activity. Arch Biochem Biophys 429: 123–133. [DOI] [PubMed] [Google Scholar]
- 32. Chandran M, Phillips SA, Ciaraldi T, Henry RR (2003) Adiponectin: more than just another fat cell hormone? Diabetes Care 26: 2442–2450. [DOI] [PubMed] [Google Scholar]
- 33. Sid B, Verrax J, Calderon PB (2013) Role of AMPK activation in oxidative cell damage: Implications for alcohol-induced liver disease. Biochem Pharmacol 86: 200–209. [DOI] [PubMed] [Google Scholar]
- 34.Shearn CT, Smathers RL, Jiang H, Orlicky DJ, Maclean KN, et al.. (2013) Increased dietary fat contributes to dysregulation of the LKB1/AMPK pathway and increased damage in a mouse model of early-stage ethanol-mediated steatosis. J Nutr Biochem. [DOI] [PMC free article] [PubMed]
- 35. You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW (2004) The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology 127: 1798–1808. [DOI] [PubMed] [Google Scholar]
- 36. Muir K, Hazim A, He Y, Peyressatre M, Kim DY, et al. (2013) Proteomic and Lipidomic Signatures of Lipid Metabolism in NASH-Associated Hepatocellular Carcinoma. Cancer Res 73: 4722–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Patsouris D, Reddy JK, Muller M, Kersten S (2006) Peroxisome proliferator-activated receptor alpha mediates the effects of high-fat diet on hepatic gene expression. Endocrinology 147: 1508–1516. [DOI] [PubMed] [Google Scholar]
- 38. Shimano H, Yahagi N, Amemiya-Kudo M, Hasty AH, Osuga J, et al. (1999) Sterol regulatory element-binding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes. J Biol Chem 274: 35832–35839. [DOI] [PubMed] [Google Scholar]
- 39. Panchal SK, Ward L, Brown L (2012) Ellagic acid attenuates high-carbohydrate, high-fat diet-induced metabolic syndrome in rats. Eur J Nutr 52: 559–568. [DOI] [PubMed] [Google Scholar]
- 40. Falcon A, Doege H, Fluitt A, Tsang B, Watson N, et al. (2010) FATP2 is a hepatic fatty acid transporter and peroxisomal very long-chain acyl-CoA synthetase. Am J Physiol Endocrinol Metab 299: E384–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ren LP, Chan SM, Zeng XY, Laybutt DR, Iseli TJ, et al. (2012) Differing endoplasmic reticulum stress response to excess lipogenesis versus lipid oversupply in relation to hepatic steatosis and insulin resistance. PLoS One 7: e30816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Turner N, Kowalski GM, Leslie SJ, Risis S, Yang C, et al. (2013) Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia 56: 1638–1648. [DOI] [PubMed] [Google Scholar]
- 43. Derdak Z, Villegas KA, Harb R, Wu AM, Sousa A, et al. (2012) Inhibition of p53 attenuates steatosis and liver injury in a mouse model of non-alcoholic fatty liver disease. J Hepatol 58: 785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Handa P, Maliken BD, Nelson JE, Morgan-Stevenson V, Messner DJ, et al.. (2013) Reduced adiponectin signaling due to weight gain results in nonalcoholic steatohepatitis through impaired mitochondrial biogenesis. Hepatology. [DOI] [PMC free article] [PubMed]
- 45. Jump DB, Botolin D, Wang Y, Xu J, Christian B, et al. (2005) Fatty acid regulation of hepatic gene transcription. J Nutr 135: 2503–2506. [DOI] [PubMed] [Google Scholar]
- 46. Galicia VA, He L, Dang H, Kanel G, Vendryes C, et al. (2010) Expansion of hepatic tumor progenitor cells in Pten-null mice requires liver injury and is reversed by loss of AKT2. Gastroenterology 139: 2170–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zeng N, Li Y, He L, Xu X, Galicia V, et al. (2011) Adaptive basal phosphorylation of eIF2alpha is responsible for resistance to cellular stress-induced cell death in Pten-null hepatocytes. Mol Cancer Res 9: 1708–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li Y, He L, Zeng N, Sahu D, Cadenas E, et al. (2013) Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) signaling regulates mitochondrial biogenesis and respiration via estrogen-related receptor alpha (ERRalpha). J Biol Chem 288: 25007–25024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jones DP, Go YM (2010) Redox compartmentalization and cellular stress. Diabetes Obes Metab 12 Suppl 2116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. He L, Hou X, Kanel G, Zeng N, Galicia V, et al. (2010) The critical role of AKT2 in hepatic steatosis induced by PTEN loss. Am J Pathol 176: 2302–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liao Y, Hung MC (2010) Physiological regulation of Akt activity and stability. Am J Transl Res 2: 19–42. [PMC free article] [PubMed] [Google Scholar]
- 52. Yuan M, Pino E, Wu L, Kacergis M, Soukas AA (2012) Identification of Akt-independent regulation of hepatic lipogenesis by mammalian target of rapamycin (mTOR) complex 2. J Biol Chem 287: 29579–29588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shimomura I, Shimano H, Korn BS, Bashmakov Y, Horton JD (1998) Nuclear sterol regulatory element-binding proteins activate genes responsible for the entire program of unsaturated fatty acid biosynthesis in transgenic mouse liver. J Biol Chem 273: 35299–35306. [DOI] [PubMed] [Google Scholar]
- 54. Miyazaki M, Dobrzyn A, Elias PM, Ntambi JM (2005) Stearoyl-CoA desaturase-2 gene expression is required for lipid synthesis during early skin and liver development. Proc Natl Acad Sci U S A 102: 12501–12506. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effects of PTENf/f and HFD on PTEN signaling. (A) Western blotting analysis of PTEN, pSer473Akt and total Akt using whole cell extracts isolated from chow/HFD-fed Alb-Cre and PTENf/f mice. (B) Quantification of PTEN expression. (C) Quantification of Akt phosphorylation. Data are means± SEM as analyzed by two-way ANOVA with a Bonferroni post hoc analysis (Alb-Cre group compared to PTENf/f group). Means with a common superscript letter are significantly different (N = 3 mice/group (***p<0.001)).
(TIF)
Antibodies used for Western blotting in this study.
(XLSX)
Primers used for this study.
(XLSX)