

Abstract
Pancreas transplantation is a therapeutic option for patients with type 1 diabetes. Advances in immunosuppression have reduced immunological failures, and these are usually categorized as chronic rejection. Yet studies in our cohort of pancreas transplant recipients identified several patients in whom chronic islet autoimmunity led to recurrent diabetes, despite immunosuppression that prevented rejection. Recurrent diabetes in our cohort is as frequent as chronic rejection, and thus is a significant cause of immunological graft failure. Our studies demonstrated islet autoimmunity by the presence of autoantibodies and autoreactive T-cells, which mediated β-cell destruction in a transplantation model. Biopsy of the transplanted pancreas revealed variable degrees of β-cell loss, with or without insulitis, in the absence of pancreas and kidney transplant rejection. Additional research is needed to better understand recurrent disease and to identify new treatment regimens that can suppress autoimmunity, as in our experience this is not effectively inhibited by conventional immunosuppression.
Keywords: Type 1 diabetes, pancreas transplantation, recurrent diabetes, autoimmunity, GAD65, autoantibodies
Introduction
Over 23,000 patients with type 1 diabetes (T1D) have received a pancreas transplant in the United States (1). Most transplant recipients receive organs from deceased donors. The majority (74%) received simultaneous pancreas-kidney transplants (SPK); pancreas after kidney transplants (PAK) and pancreas transplants alone (PTA) have been performed less frequently. The University of Miami Pancreas Transplant (PT) Program was established in 1990. As of January 2011, the program has performed over 400 pancreas transplants. Approximately 95% were SPK, 4% were PAK and 1% PTA transplants. All patients had been determined to have T1D by clinical history, insulin dependence and C-peptide determination (less than 0.1 ng/ml stimulated, predominantly by Sustacal Challenge Test). All patients have had components of diabetic triopathy (neuropathy, nephropathy, and/or retinopathy). Most had gastroparesis and reduced hypoglycemia awareness. From a technical standpoint, the pancreas transplants have been performed with portal vein-systemic venous drainage (bypassing the liver) and with duodenal transplant-bladder exocrine drainage (all but 2 recipients). The latter technique allows for measurement of urine amylase, and avoids enteric contamination at the time of pancreas transplantation. The immunosuppression has evolved over the past 2 decades, and currently includes induction antibody therapy with an anti-CD25 monoclonal antibody and Thymoglobulin (2), tacrolimus (levels 5 to 7 ng/ml), low-dose steroids, and either rapamycin (levels 5 to 7 ng/ml) or mycophenolate mofetil. Unpublished estimates of our current 10-year survival rates are 78% patient, 94% pancreas (death-censored) and 76% kidney (death-censored).
The ongoing follow-up of about 200 SPK patients transplanted at the University of Miami during the last two decades shows that approximately 15% of our recipients will eventually return to the clinic with hyperglycemia, many of them requiring the reinstitution of insulin therapy. Among these patients, the apparent causes of hyperglycemia include: 1) chronic rejection of the pancreas (5–6%); 2) post-transplant diabetes mellitus (PTDM, 6–7%), with insulin resistance secondary to obesity, weight gain, and/or medications (3;4); 3) late pancreas transplant thrombosis (rare); and 4) Type 1 Diabetes Recurrence (T1DR, 5–6%).
Recurrence of T1D after pancreas transplantation
Recurrence of autoimmunity after PT was originally described in twins and HLA-identical siblings from the Minnesota series (5–7), who received no or mild immunosuppression. Other studies contributed evidence that recurrence of islet autoimmunity can occur regardless of HLA sharing (8) and despite immunosuppression (9–11), and can be another important cause of immunological failure, both in pancreas and islet cell transplantation (5–9;12–19). Diabetes recurrence was <10% in a large series of recipients of deceased donor grafts given immunosuppression sufficient to prevent rejection in the 1980s (5–7;12). Studies of autoantibodies have also shown an association of autoimmunity with graft loss in immunosuppressed recipients (13–17), albeit these earlier studies did not have biopsy data and did not assess circulating autoreactive T cells. T1DR has not been traditionally considered a common cause of hyperglycemia after transplantation, given the assumption that immunosuppression that prevents rejection also suppresses autoimmunity.
In our cohort, patients with T1DR typically present with new onset hyperglycemia in the context of falling C-peptide with normal/unchanged creatinine and persistent/unchanged levels of urine amylase (an indicator of exocrine pancreas transplant function). Presentation has varied from 2½ to over 10 years following transplantation. On occasion, this has been accompanied by significant weight gain/obesity. Thus, while patients who develop T1DR may also have clinical features of PTDM, the hyperglycemia appears to be a β-cell-specific process and to be largely independent of rejection. Indeed, both kidney (same donor) and pancreatic exocrine function (urine amylase) remain unchanged in patients who develop T1DR. In our cohort, we have evaluated pancreas transplant recipient’s stored sera for the presence of T1D-associated autoantibodies, specifically anti-GAD65, anti-IA-2 and anti-ZnT8. Among most patients with T1DR, autoantibodies appear to be a clear risk factor for T1DR, both from ours (20) and earlier studies (17;21). In our cohort, autoantibodies usually become detectable during the first 3–5 years following transplantation, and patients develop hyperglycemia within another 3–5 years; however, these intervals may vary, and individual patients may exhibit shorter or longer intervals.
We have obtained pancreas transplant biopsies (a cubic cm of the tail) through surgery in most patients with clinical evidence of T1DR, to confirm diagnosis and guide possible therapeutic strategies (10). While percutaneous biopsies yielded insufficient material, extensive examination of the open biopsies allowed us to demonstrate insulitis and/or β-cell loss, with T-cell (CD3, CD4, CD8) and B-cell (CD20) infiltrates, as well as variable insulin staining (10). In the infiltrates, T cells (Fig. 1) were clearly more abundant than B cells. The severity of insulitis was variable. In these biopsies, we have not detected rejection in either the kidney or the pancreas transplants. Furthermore, the peripheral blood of most of these patients has been evaluated for T-cells specific for diabetes-related autoantigens, and found to contain autoreactive CD4 and CD8 T-cells (10;22). In selected patients, we could also identify autoreactive T cells in pancreas transplant tissue or in pancreas transplant lymph nodes, which corresponded to those detected in the circulation (10;23). In one patient, GAD65-specific autoreactive CD4 T-cells expressed the same V-beta type and CDR3 sequence in the blood and pancreas transplant lymph node (10). Thus, we have been able to identify the cardinal features of autoimmune recurrence in these patients, including recurrent hyperglycemia, autoantibodies preceding the hyperglycemia, biopsy demonstrating insulitis (not rejection) with variable insulin staining, and autoreactive T-cells (CD4/CD8).
Figure 1. Insulitis in the pancreas transplant biopsy from a patient with T1DR.
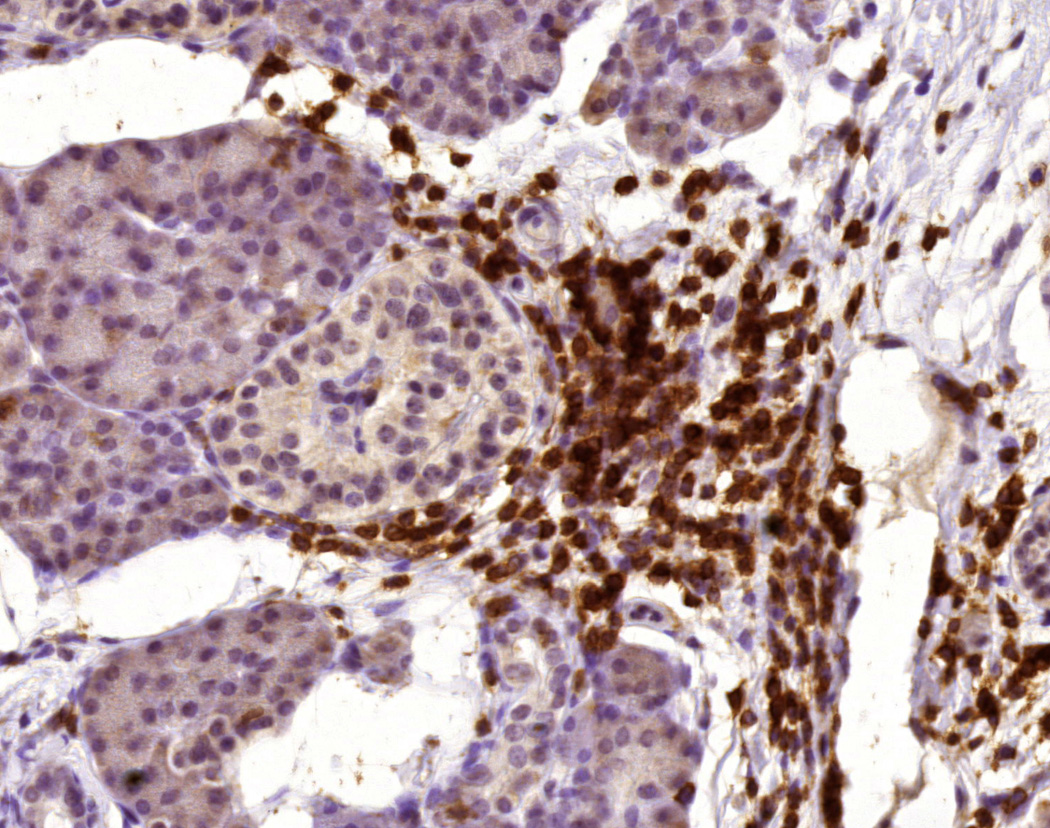
The figure shows a T cell infiltrate surrounding a pancreatic islet, as demonstrated by staining for CD3 (brown color; counterstained with hematoxylin, acquired with a 40× lens).
These investigations in patients have led to several observations: 1) the identification of insulin-positive, ductal cells in the pancreas transplant of most patients with T1DR, 2) therapeutic intervention, for those SPK recipients with T1DR in whom we found insulitis and residual insulin staining in the pancreas transplant biopsy, and who also had residual c-peptide secretion; or, re-transplantation of the pancreas in the absence of circulating C-peptide and of residual insulin staining in the islets of the pancreas transplant; 3) the demonstration of the cytotoxic effect of SPK patients’ GAD65-specific CD4 T-cells on human β-cells, in-vivo, when co-transplanted in immunodeficient mice and, finally 4) the generation of evidence suggesting the autoantigen specific T-cells are likely to have memory cell properties. These findings are summarized in the following paragraphs.
1. Insulin Protein and Proliferation in Ductal Cells of Patients with T1DR
T1D is an autoimmune disease resulting in the destruction of pancreatic β-cells and insulin dependence. However, residual insulin secretion is often detected at disease onset and marginal amounts of C-peptide can occur many years after diagnosis (24). Evidence for transdifferentiation and regeneration have been reported in various experimental conditions (25–30). Pancreatic cells with features of ductal and β-cells in pancreatic ducts have been identified by electron microscopy (31). Growing evidence suggests that ductal cells or precursors in the ducts may also be involved in β-cell regeneration (26); for example, human ductal cells transplanted into immunodeficient mice differentiate into new β-cells (32). Rare insulin-positive cells in pancreatic ducts were reported in the pancreas of patients with long-standing T1D (30;33). There is growing evidence that pancreatic tissue damage may trigger regenerative and remodeling mechanisms that may contribute to β-cell neogenesis (26;34).
We could examine pancreas transplant biopsies from 9 SPK patients with (n=6) or without (n=3) recurrent autoimmunity and pancreas biopsies from non-diabetic organ donors using immunohistochemistry and immunofluorescence (35). Numerous cytokeratin-19+ (CK-19) pancreatic ductal cells stained for insulin among the SPK recipients with T1DR. These cells also stained for the transcription factor pancreatic-duodenal homeobox-1 (Pdx-1), which is implicated in pancreatic development and β-cell differentiation (36). Between 33–90% of the ductal cells examined stained for insulin, with 17–95% of the ducts having insulin+CK-19+ cells; most, although not all, ductal cells stained for insulin in those ducts that contained insulin+CK-19+ cells. This suggested that, at least in some patients, these phenomena were quite extensive. The patient with the most severe β-cell destruction and complete loss of C-peptide secretion at the time of biopsy was the one with the highest number of ducts containing insulin+CK-19+ cells. In this patient, insulin+CK-19+Pdx-1+ cells also stained for the Ki-67 antigen, indicating proliferation. Ki-67+ β-cells could not be detected within the islets in any of the pancreas transplant biopsies examined. Some insulin+ cells within the ducts were CK-19- and also stained for chromogranin A, suggesting further endocrine differentiation. Insulin-positive cells were rarely noted in the pancreas transplant ducts in 3 SPK patients without islet autoimmunity, or in normal pancreata, and when present they did not co-stain for CK-19, e.g. they did not appear to have ductal origin. There was a patient who had evidence of possibly early autoimmunity who rarely had insulin+ cells in the ducts, and these did not stain for CK-19. This patient was normoglycemic, his biopsy did not show β-cell loss and only minimal insulitis, while he had recently developed autoantibodies and we detected autoreactive T cells in the circulation and pancreas/pancreas transplant lymph nodes (23). Thus, the presence of hyperglycemia and islet inflammation may be critical for triggering insulin synthesis in ductal cells. The link to chronic inflammation is also suggested by reports of similar phenomena in chronic autoimmune pancreatitis (37) and in several experimental models in which inflammation was a factor (38–41).
In conclusion, our findings suggest that ductal cells may participate in β-cell regenerative processes occurring in the transplanted human pancreas, in the context of hyperglycemia and recurrent autoimmunity. These may be critical stimuli to trigger pancreas remodeling mechanisms in the adult. Dissecting the mechanisms involved these remodeling processes could lead to therapeutic exploitation (42).
2. Treatment for T1DR
a. In patients with persistent C-peptide and biopsy-demonstrated insulin staining
We have reported 2 instances of intervening therapeutically with immunosuppressive antibodies (polyclonal and monoclonal) in SPK recipients diagnosed with T1DR, where there was evidence of persistent C-peptide circulating in the serum in response to a mixed meal challenge, and the PT biopsy was notable for an islet infiltrate and the presence of insulin staining (10). The first such patient developed hyperglycemia 5 years after SPK transplantation. Retrospectively, he had GAD65 and IA-2 autoantibodies persisting following transplantation with rising levels prior to hyperglycemia. There was also evidence of circulating CD4+ T-cells specific for GAD65 in the peripheral blood just prior to treatment, about a year after the return of hyperglycemia. The therapy included anti-CD25 monoclonal antibody (daclizumab, 1 mg/kg × 2 doses) and Thymoglobulin (polyclonal antibody, 1 mg/kg × 5 doses). Subsequently, levels of autoantibodies fluctuated, T-cells with autoantigen specificity where undetectable for approximately 1 year, and C-peptide levels initially rose over the next 6 months. However, the patient remained on insulin, and C-peptide levels fell to undetectable levels after the auto-reactive CD4 T-cells returned to the peripheral blood later on follow-up (10).
The second patient became hyperglycemic 9½ years after SPK transplantation. In this patient, anti-GAD65 and anti-IA-2 autoantibodies became detectable after about 6 years after transplant, followed by hyperglycemia 3.5 years later. In this patient, CD8+ T-cells reactive to IGRP - an islet cell auto-antigen (43;44) - were identified in the peripheral blood prior to immunosuppressive therapy (10). Drawing upon our previous experience, and the ultimate lack of efficacy with a T-cell only designed approach, in this instance a single dose of rituximab (375 mg/m2, anti-CD20, a marker on B-cells, monoclonal antibody) was added to the previous regimen of daclizumab and Thymoglobulin anticipating an effect on B-cells and possibly autoantibodies. The autoantibody levels again fluctuated, the CD8+ T-cells fell during the period of 1 year; the peripheral C-peptide levels initially increased, and yet this patient remained dependent on insulin injections; eventually, C-peptide levels became undetectable after the autoreactive CD8+ T-cells returned to the peripheral blood (10).
b. Re-transplantation of the pancreas in a patient with loss of both C-peptide and insulin staining
We have reported one instance of re-transplantation of the pancreas following T1DR, based on total loss of C-peptide response in the context of a PT biopsy (obtained at the time of re-transplantation) and the complete absence of insulin staining in the islets; there also was minimal insulitis, suggesting that the active phase of recurrent disease had passed (10). This patient received a second PT approximately 1 year after developing hyperglycemia, after 5 years of euglycemic PT function. Levels of auto-antibodies, both GAD65 and ZnT8 rose approximately 3 months prior to hyperglycemia and were gradually falling at the time of re-transplantation. CD4 T-cells specific for GAD65 were present at the time of re-transplantation, before induction immunosuppression was given. Induction immunosuppression included daclizumab, Thymoglobulin and rituximab; we also performed plasmapheresis in an attempt to reduce autoantibody levels. GAD65 autoantibodies were unaffected by plasmapheresis, but the levels of ZnT8 autoantibodies appear to have been reduced. Levels of both autoantibodies fell over the next year, although GAD65 autoantibodies rebounded soon after. Following re-transplantation, the patient became euglycemic for close to 3 years, but then was noted to express a similar autoimmune pattern to that which appeared just prior to his previous episode of T1DR. Levels of GAD65 and ZnT8 autoantibody rose sharply just ahead of the return to hyperglycemia and furthermore GAD65-specific CD4 T-cells also became detectable in the peripheral blood. Subsequently, C-peptide levels fell to undetectable levels (10). While the biopsy of the second pancreas transplant revealed a significant cellular infiltrate consistent with acute and chronic rejection, it is important that recrudescence of these autoimmune responses preceded the loss of the second pancreas transplant.
3. In vivo effect of GAD65-specific CD4 T-cells
GAD65-specific CD4 T-cell responses were obtained after in vitro culture with GAD65 peptide of peripheral blood cells from two of the above patients (10). Significant responses were noted, and we were able to purify these autoreactive T cells using class II tetramers, so that we had pure populations of CD4 T cells reacting against the GAD65 autoantigen. These cells were then co-transplanted with human islet cells from a different donor (HLA-mismatched) under the kidney capsule of an immunodeficient mouse. Controls included mice receiving human islets alone and mice receiving the same human islets along with irrelevant T-cells from the same patients (in one case, T cells reacting with an influenza peptide; in the other, polyclonal cells that had not responded to a negative control antigen). After approximately 10 to 16 days, the injected kidneys were recovered and the tissue stained with hematoxylin and eosin, as well as double stained for insulin and glucagon. In these experiments, the human islets alone and the human islets with irrelevant T-cells demonstrated good islet architecture and the expected pattern and abundance of insulin and glucagon staining. Sections from the transplants that received GAD65-specific autoreactive CD4 T-cells revealed severely disrupted islet architecture and much reduced insulin staining. In one of these experiments, recipient mice had had diabetes induced by streptozotocin prior to receiving the transplant; diabetes was reversed in the mice that received islets alone or islet and irrelevant T-cells while the mouse that received islets plus the autoreactive CD4 T-cells never returned to normoglycemia (10). Taken together, these experimental data with autoreactive T cells from patients who had developed T1DR demonstrate that such lymphocytes are capable of mediating damage to β-cells.
4. Evidence for return of memory T-cells in T1DR
It has been hypothesized that the PT portion of an SPK transplant may mimic an antigen booster immunization, with recurrent T1DR an anticipated outcome. Since long-standing T1D is characterized by a specific lack of islet cells, it is possible that the transplant may induce a recall memory response, reactivating memory cells that possibly have been quiescent since the original onset of T1D many years prior. Memory cells have been linked to autoimmunity (45). A role for memory cells is suggested in both spontaneous T1D (46–48) and in recurrence of hyperglycemia in islet cell transplant recipients (49). Our preliminary studies of TCR clonotypes in two of our patients (10;22) revealed that GAD65 autoreactive CD4 T cells expressing the same V-beta chains (5.1 and 9) and identical or similar CDR3 sequences reappeared after immunosuppression. The persistence of autoreactive T cells against the same autoantigen, expressing the same or closely related TCR V beta chains, is consistent with a memory response associated with recurrent autoimmunity in our patients with T1DR.
Conclusion
Our ongoing studies provide evidence that recurrent autoimmunity is an important cause of diabetes in SPK recipients, usually after several years of pancreas transplant function. In our series, the frequency of T1DR is similar to that of chronic rejection, and even some patients who resemble PTDM have evidence of islet autoimmunity. Thus, islet autoimmunity appears to be a factor in long term function of pancreas transplant grafts. Our experience in treating patients with T1DR using rational combinations of immunosuppression, the use of which is being implemented in clinical trials for T1D (50), achieved only a transient depletion of these autoreactive T-cells, marginal effects on autoantibodies and possibly temporary preservation of C-peptide secretion. Building on literature and our own findings, we are pursuing studies to formally test the association of memory, autoreactive T-cell responses with T1DR and to determine key functional features of the lymphocytes associated with this condition. This knowledge may allow the development of novel therapeutic designs to intervene in the disease process in a more effective fashion. Ultimately, our studies are raising awareness of this important cause of diabetes following pancreas transplantation, and it is hoped that multiple pancreas transplant centers will begin monitoring and studying recurrent islet autoimmunity. We also advocate that such studies could be more powerful and informative if multiple transplant centers could coordinate their efforts and share data, patient samples, protocols and reagents, believing that collaboration will facilitate a better understanding of the causal factors of T1DR and hopefully lead to more effective therapies.
Acknowledgments
Studies by the authors reviewed in this manuscript were supported by grants from the National Institutes of Health (5RO1-DK-070011 and AI-50864), the American Diabetes Association (1-09-RA-413, 1-05-RA-105), the Juvenile Diabetes Research Foundation (JDRF 1-2005-257), and the Diabetes Research Institute Foundation, Hollywood, FL. F.V. was supported by a JDRF Postdoctoral Research Fellowship (3-2008-32). We acknowledge support from the Diabetes Research Institute Cell Transplant Center in providing islets for our studies (NIH grant 5-U01-DK-070460-07).
Contributor Information
George W. Burke, III, Department of Surgery, Division of Transplantation, Leonard Miller School of Medicine, University of Miami, 1801 NW 9th Avenue, Miami FL 33136, Tel. 305-355-5000 Fax 305-355-5134 gburke@med.miami.edu.
Francesco Vendrame, Diabetes Research Institute, Leonard Miller School of Medicine, University of Miami, 1450 NW 10th Avenue, Miami, FL 33136 USA, Tel. 305-243-5353 Fax 305-243-4404 fvendrame@med.miami.edu.
Antonello Pileggi, Diabetes Research Institute and Department of Surgery, Leonard Miller School of Medicine, 1450 NW 10th Avenue, Miami, FL 33136 USA, Tel. 305-243-2924 Fax 305-243-4404 apileggi@med.miami.edu.
Gaetano Ciancio, Departments of Urology and Surgery, Division of Transplantation, Leonard Miller School of Medicine, University of Miami, 1801 NW 9th Avenue, Miami FL 33136, Tel. 305-355-5000 Fax 305-355-5134 gciancio@med.miami.edu.
Helena Reijonen, Benaroya Research Institute, 1201 Ninth Avenue, Seattle, WA 98101, Tel. 206-223-8813 Fax 206-223-7638 reijonen@benaroyaresearch.org.
Alberto Pugliese, Diabetes Research Institute, Department of Medicine, Division of Diabetes, Endocrinology and Metabolism, and Department of Microbiology and Immunology, Leonard Miller School of Medicine, 1450 NW 10th Avenue, Miami, FL 33136 USA, Tel. 305-243-5348 Fax 305-243-4404 apuglies@med.miami.edu.
References
- 1.White SA, Shaw JA, Sutherland DE. Pancreas transplantation. Lancet. 2009;373(9677):1808–1817. doi: 10.1016/S0140-6736(09)60609-7. [DOI] [PubMed] [Google Scholar]
- 2.Sageshima J, Ciancio G, Gaynor JJ, et al. Addition of anti-CD25 to thymoglobulin for induction therapy: delayed return of peripheral blood CD25-positive population. Clinical Transplantation. 2011;25(2):E132–E135. doi: 10.1111/j.1399-0012.2010.01360.x. [DOI] [PubMed] [Google Scholar]
- 3.Neidlinger N, Singh N, Klein C, et al. Incidence of and risk factors for posttransplant diabetes mellitus after pancreas transplantation. Am. J. Transplant. 2010;10(2):398–406. doi: 10.1111/j.1600-6143.2009.02935.x. [DOI] [PubMed] [Google Scholar]
- 4.Dean PG, Kudva YC, Larson TS, Kremers WK, Stegall MD. Posttransplant diabetes mellitus after pancreas transplantation. Am. J. Transplant. 2008;8(1):175–182. doi: 10.1111/j.1600-6143.2007.02018.x. [DOI] [PubMed] [Google Scholar]
- 5.Sutherland DE, Sibley R, Xu XZ, et al. Twin-to-twin pancreas transplantation: reversal and reenactment of the pathogenesis of type I diabetes. Trans. Assoc. Am. Physicians. 1984;97:80–87. [PubMed] [Google Scholar]
- 6.Sibley RK, Sutherland DE, Goetz F, Michael AF. Recurrent diabetes mellitus in the pancreas iso- and allograft. A light and electron microscopic and immunohistochemical analysis of four cases. Laboratory Investigation. 1985;53(2):132–144. [PubMed] [Google Scholar]
- 7.Sutherland DE, Goetz FC, Sibley RK. Recurrence of disease in pancreas transplants. Diabetes. 1989;38(S1):85–87. doi: 10.2337/diab.38.1.s85. [DOI] [PubMed] [Google Scholar]
- 8.Tyden G, Reinholt FP, Sundkvist G, Bolinder J. Recurrence of autoimmune diabetes mellitus in recipients of cadaveric pancreatic grafts. N. Engl. J. Med. 1996;335(12):860–863. doi: 10.1056/NEJM199609193351205. [DOI] [PubMed] [Google Scholar]
- 9.Santamaria P, Nakhleh RE, Sutherland DE, Barbosa JJ. Characterization of T lymphocytes infiltrating human pancreas allograft affected by isletitis and recurrent diabetes. Diabetes. 1992;41(1):53–61. doi: 10.2337/diab.41.1.53. [DOI] [PubMed] [Google Scholar]
- 10. Vendrame F, Pileggi A, Laughlin E, et al. Recurrence of type 1 diabetes after simultaneous pancreas-kidney transplantation, despite immunosuppression, is associated with autoantibodies and pathogenic autoreactive CD4 T-cells. Diabetes. 2010;59(4):947–957. doi: 10.2337/db09-0498. * This article is the original source for much of the data reviewed in this article.
- 11. Ishida-Oku M, Iwase M, Sugitani A, et al. A case of recurrent type 1 diabetes mellitus with insulitis of transplanted pancreas in simultaneous pancreas-kidney transplantation from cardiac death donor. Diabetologia. 2010;53(2):341–345. doi: 10.1007/s00125-009-1593-3. *This article identified another case of recurrent diabetes in an immunosuppressed patient.
- 12.Sibley RK, Sutherland DE. Pancreas transplantation. An immunohistologic and histopathologic examination of 100 grafts. Am. J. Pathol. 1987;128(1):151–170. [PMC free article] [PubMed] [Google Scholar]
- 13.Bosi E, Bottazzo GF, Secchi A, et al. Islet cell autoimmunity in type I diabetic patients after HLA-mismatched pancreas transplantation. Diabetes. 1989;38(S1):82–84. doi: 10.2337/diab.38.1.s82. [DOI] [PubMed] [Google Scholar]
- 14.Esmatjes E, Rodriguez-Villar C, Ricart MJ, et al. Recurrence of immunological markers for type 1 (insulin-dependent) diabetes mellitus in immunosuppressed patients after pancreas transplantation. Transplantation. 1998;66(1):128–131. doi: 10.1097/00007890-199807150-00022. [DOI] [PubMed] [Google Scholar]
- 15.Petruzzo P, Andreelli F, McGregor B, et al. Evidence of recurrent type I diabetes following HLA-mismatched pancreas transplantation. Diabetes Metab. 2000;26(3):215–218. [PubMed] [Google Scholar]
- 16.Thivolet C, Abou-Amara S, Martin X, et al. Serological markers of recurrent beta cell destruction in diabetic patients undergoing pancreatic transplantation. Transplantation. 2000;69(1):99–103. doi: 10.1097/00007890-200001150-00018. [DOI] [PubMed] [Google Scholar]
- 17.Braghi S, Bonifacio E, Secchi A, et al. Modulation of humoral islet autoimmunity by pancreas allotransplantation influences allograft outcome in patients with type 1 diabetes. Diabetes. 2000;49(2):218–224. doi: 10.2337/diabetes.49.2.218. [DOI] [PubMed] [Google Scholar]
- 18.Hilbrands R, Huurman VA, Gillard P, et al. Differences in baseline lymphocyte counts and autoreactivity are associated with differences in outcome of islet cell transplantation in type 1 diabetic patients. Diabetes. 2009;58(10):2267–2276. doi: 10.2337/db09-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huurman VA, Hilbrands R, Pinkse GG, et al. Cellular islet autoimmunity associates with clinical outcome of islet cell transplantation. PLoS ONE. 2008;3(6):e2435. doi: 10.1371/journal.pone.0002435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diamantopoulos S, Allende, Martin-Pagola A, et al. Recurrence of type 1 diabetes (T1DR) after simultaneous pancreas-kidney (SPK) transplantation is associated with islet cell autoantibody conversion. Acta Diabetologica. 2007;44(S1):S13. [Google Scholar]
- 21.Sundkvist G, Tyden G, Karlsson FA, Bolinder J. Islet autoimmunity before and after pancreas transplantation in patients with Type I diabetes mellitus. Diabetologia. 1998;41(12):1532–1533. doi: 10.1007/s001250051102. [DOI] [PubMed] [Google Scholar]
- 22.Laughlin E, Burke G, Pugliese A, Falk B, Nepom G. Recurrence of autoreactive antigen-specific CD4+ T cells in autoimmune diabetes after pancreas transplantation. Clinical Immunology. 2008;128(1):23–30. doi: 10.1016/j.clim.2008.03.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reijonen H, Geubtner K, Allende G, et al. Identification of islet-autoantigen specific CD4+ T-cells in the pancreatic lymph nodes and pancreas of a pancreas-kidney transplant patient with recurrence of autoimmunity. Diabetes. 2006;55(S1):88A. [Google Scholar]
- 24.Tsai EB, Sherry NA, Palmer JP, Herold KC. The rise and fall of insulin secretion in type 1 diabetes mellitus. Diabetologia. 2006;49:261–270. doi: 10.1007/s00125-005-0100-8. [DOI] [PubMed] [Google Scholar]
- 25.Smukler SR, Arntfield ME, Razavi R, et al. The adult mouse and human pancreas contain rare multipotent stem cells that express insulin. Cell Stem Cell. 2011;8(3):281–293. doi: 10.1016/j.stem.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 26.Bonner-Weir S, Li WC, Ouziel-Yahalom L, et al. Beta-cell growth and regeneration: replication is only part of the story. Diabetes. 2010;59(10):2340–2348. doi: 10.2337/db10-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keenan HA, Sun JK, Levine J, et al. Residual insulin production and pancreatic beta-cell turnover after 50 years of diabetes: Joslin Medalist Study. Diabetes. 2010;59(11):2846–2853. doi: 10.2337/db10-0676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meier JJ, Butler AE, Saisho Y, et al. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. 2008;57(6):1584–1594. doi: 10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Butler PC, Meier JJ, Butler AE, Bhushan A. The replication of beta cells in normal physiology, in disease and for therapy. Nat. Clin. Pract. Endocrinol. Metab. 2007;3(11):758–768. doi: 10.1038/ncpendmet0647. [DOI] [PubMed] [Google Scholar]
- 30.Meier JJ, Bhushan A, Butler AE, Rizza RA, Butler PC. Sustained beta cell apoptosis in patients with long-standing type 1 diabetes: indirect evidence for islet regeneration? Diabetologia. 2005;48(11):2221–2228. doi: 10.1007/s00125-005-1949-2. [DOI] [PubMed] [Google Scholar]
- 31.Melmed RN, Benitez CJ, Holt SJ. Intermediate cells of the pancreas. I. Ultrastructural characterization. J. Cell Sci. 1972;11(2):449–475. doi: 10.1242/jcs.11.2.449. [DOI] [PubMed] [Google Scholar]
- 32.Yatoh S, Dodge R, Akashi T, et al. Differentiation of affinity-purified human pancreatic duct cells to beta-cells. Diabetes. 2007;56(7):1802–1809. doi: 10.2337/db06-1670. [DOI] [PubMed] [Google Scholar]
- 33.Meier JJ, Ritzel RA, Maedler K, Gurlo T, Butler PC. Increased vulnerability of newly forming beta cells to cytokine-induced cell death. Diabetologia. 2005;49:83–89. doi: 10.1007/s00125-005-0069-3. [DOI] [PubMed] [Google Scholar]
- 34.Juhl K, Bonner-Weir S, Sharma A. Regenerating pancreatic beta-cells: plasticity of adult pancreatic cells and the feasibility of in-vivo neogenesis. Curr. Opin. Organ Transplant. 2010;15(1):79–85. doi: 10.1097/MOT.0b013e3283344932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin-Pagola A, Sisino G, Allende G, et al. Insulin protein and proliferation in ductal cells in the transplanted pancreas of patients with type 1 diabetes and recurrence of autoimmunity. Diabetologia. 2008;51(10):1803–1813. doi: 10.1007/s00125-008-1105-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jonsson J, Ahlgren U, Edlund T, Edlund H. IPF1, a homeodomain protein with a dual function in pancreas development. Int. J. Dev. Biol. 1995;39(5):789–798. [PubMed] [Google Scholar]
- 37.Tanaka S, Kobayashi T, Nakanishi K, et al. Evidence of primary beta-cell destruction by T-cells and beta-cell differentiation from pancreatic ductal cells in diabetes associated with active autoimmune chronic pancreatitis. Diabetes Care. 2001;24(9):1661–1667. doi: 10.2337/diacare.24.9.1661. [DOI] [PubMed] [Google Scholar]
- 38.Bottino R, Criscimanna A, Casu A, et al. Recovery of endogenous beta-cell function in nonhuman primates after chemical diabetes induction and islet transplantation. Diabetes. 2009;58(2):442–447. doi: 10.2337/db08-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang GS, Rosenberg L, Scott FW. Tubular complexes as a source for islet neogenesis in the pancreas of diabetes-prone BB rats. Lab Invest. 2005;85(5):675–688. doi: 10.1038/labinvest.3700259. [DOI] [PubMed] [Google Scholar]
- 40.Kauri LM, Wang GS, Patrick C, et al. Increased islet neogenesis without increased islet mass precedes autoimmune attack in diabetes-prone rats. Lab Invest. 2007;87(12):1240–1251. doi: 10.1038/labinvest.3700687. [DOI] [PubMed] [Google Scholar]
- 41.Xu X, D'Hoker J, Stange G, et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell. 2008;132(2):197–207. doi: 10.1016/j.cell.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 42.Xia B, Zhan XR, Yi R, Yang B. Can pancreatic duct-derived progenitors be a source of islet regeneration? Biochemistry Biophysics Res. Commun. 2009;383(4):383–385. doi: 10.1016/j.bbrc.2009.03.114. [DOI] [PubMed] [Google Scholar]
- 43.Jarchum I, Nichol L, Trucco M, Santamaria P, DiLorenzo TP. Identification of novel IGRP epitopes targeted in type 1 diabetes patients. Clinical Immunology. 2008;127(3):359–365. doi: 10.1016/j.clim.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Unger WW, Pinkse GG, Mulder-van der KS, et al. Human clonal CD8 autoreactivity to an IGRP islet epitope shared between mice and men. Ann. N. Y. Acad. Sci. 2007;1103:192–195. doi: 10.1196/annals.1394.024. [DOI] [PubMed] [Google Scholar]
- 45.King C, Ilic A, Koelsch K, Sarvetnick N. Homeostatic expansion of T cells during immune insufficiency generates autoimmunity. Cell. 2004;117(2):265–277. doi: 10.1016/s0092-8674(04)00335-6. [DOI] [PubMed] [Google Scholar]
- 46.Viglietta V, Kent SC, Orban T, Hafler DA. GAD65-reactive T cells are activated in patients with autoimmune type 1a diabetes. J. Clin. Invest. 2002;109(7):895–903. doi: 10.1172/JCI14114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Monti P, Scirpoli M, Rigamonti A, et al. Evidence for in vivo primed and expanded autoreactive T cells as a specific feature of patients with type 1 diabetes. J. Immunol. 2007;179(9):5785–5792. doi: 10.4049/jimmunol.179.9.5785. [DOI] [PubMed] [Google Scholar]
- 48.Monti P, Heninger AK, Bonifacio E. Differentiation, expansion, and homeostasis of autoreactive T cells in type 1 diabetes mellitus. Curr. Diab. Rep. 2009;9(2):113–118. doi: 10.1007/s11892-009-0020-y. [DOI] [PubMed] [Google Scholar]
- 49. Monti P, Scirpoli M, Maffi P, et al. Islet transplantation in patients with autoimmune diabetes induces homeostatic cytokines that expand autoreactive memory T cells. J. Clin. Invest. 2008;118(5):1806–1814. doi: 10.1172/JCI35197. *This article provides evidence for a role of memory cells in patients with islet cell transplants.
- 50.Matthews JB, Staeva TP, Bernstein PL, Peakman M, von HM. Developing combination immunotherapies for type 1 diabetes: recommendations from the ITN-JDRF Type 1 Diabetes Combination Therapy Assessment Group. Clin. Exp. Immunol. 2010;160(2):176–184. doi: 10.1111/j.1365-2249.2010.04153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]