

Abstract
Patients with Hurler or Hunter syndrome typically have moderate to severe growth deficiencies despite therapy with allogeneic hematopoietic stem cell transplantation and/or enzyme replacement therapy. It is unknown whether treatment with recombinant human growth hormone (hGH) can improve growth in these children. The objectives of this study were to determine the effects of hGH on growth, bone mineral density (BMD), and body composition in children with Hurler or Hunter syndrome enrolled in a longitudinal observational study. The difference in annual change in outcomes between hGH treated and untreated subjects was estimated by longitudinal regression models that adjusted for age, Tanner stage, and sex where appropriate. We report on 23 participants who completed at least 2 annual study visits (10 [43%] treated with hGH): Hurler syndrome (n=13) average age of 9.8±3.1 years (range 5.3-13.6 years; 54% female) and Hunter syndrome (n=10) average age of 12.0±2.7 years (range 7.0-17.0 years; 0% female). As a group, children with Hurler or Hunter syndrome treated with hGH had no difference in annual change in height (growth velocity) compared to those untreated with hGH. Growth velocity in hGH treated individuals ranged from -0.4 to 8.1 cm/yr and from 0.3 to 6.6 cm/yr in the untreated individuals. Among children with Hunter syndrome, 100% (N=4) of those treated but only 50% of those untreated with hGH had an annual increase in height standard deviation score (SDS). Of the individuals treated with hGH, those with GHD had a trend towards higher annualized growth velocity compared to those without GHD (6.5±1.9 vs. 3.5±2.1; p=.050). Children treated with hGH had greater annual gains in BMD and lean body mass. In conclusion, although as a group we found no significant difference in growth between individuals treated versus not treated with hGH, individual response was highly variable and we are unable to predict who will respond to treatment. Thus, a trial of hGH may be appropriate in children with Hurler or Hunter syndrome, severe short stature, and growth failure. However, efficacy of hGH therapy should be evaluated after 1 year and discontinued if no increase in growth velocity or height SDS. Finally, the long-term benefits of changes in body composition with hGH treatment in this population are unknown.
Keywords: Mucopolysaccharidosis, Hurler syndrome, Hunter syndrome, growth, growth hormone, body composition
1. Introduction
Hurler syndrome (MPS IH) and Hunter syndrome (MPS II) are lysosomal storage diseases due to deficiency of alpha-L-iduronidase and iduronate-2-sulfatase respectively; each of these enzymes is required for normal degradation of glycosaminoglycan (GAG). Without these enzymes, GAG accumulates throughout the body, resulting in multi-system disease affecting bone, skeletal muscle, heart, lungs, liver and frequently cognitive development [1,2]. Despite treatment with hematopoietic cell transplantation (HCT) for Hurler syndrome or enzyme replacement therapy (ERT) for Hunter syndrome, a moderate to severe short state is characteristic of both Hurler and Hunter syndromes [3–5].
Some children with Hurler or Hunter syndrome and short stature receive treatment with recombinant human growth hormone (hGH) in clinical practice, despite the paucity of data supporting this practice. These treatment decisions rely on a large body of literature showing that hGH has been used safely and successfully to improve growth and increase adult height in children with short stature; including those who are growth hormone deficient [6,7] and those who are not (i.e. Turner syndrome, small for gestational age [SGA], idiopathic short stature [ISS]) [8–11]. Besides the augmentation of growth, treatment with hGH in children with Prader-Willi Syndrome, Turner syndrome, born SGA, and with growth hormone deficiency (GHD) has been shown to increase lean body mass (LBM), bone mineral density (BMD) and exercise performance, and decrease percent body fat [12–17]. However, we are aware of no studies of hGH treatment in Hunter syndrome, and only one retrospective study of hGH treatment in Hurler syndrome, showing a modest improvement in growth velocity over the first year of treatment [18].
There has been concern that treating children with MPS with hGH will worsen skeletal disproportionality based on a report in achondroplasia [19], however another study of hGH treatment in achondroplasia did not find worsening skeletal disproportionality [20]. In addition, it is possible that the hip dysplasia in MPS increases the risk of a known rare complication of hGH treatment – slipped capital femoral epiphysis (SCFE) – as we have previously reported in a small group of children with Hurler syndrome treated with hGH after HCT [18].
Therefore, the objectives of this prospective observational study were to 1) determine if children with MPS treated with hGH have significantly higher annual growth velocity vs. those not treated, 2) evaluate the safety of hGH treatment in MPS, and 3) determine if children with MPS treated with hGH have greater annual gains in LBM, BMD and less gains in body fat % vs. those not treated.
2. Material and methods
2.1. Participants
Potential participants were recruited locally, through advertisements in patient advocacy newsletters, clinicaltrials.gov, and from patients with Hurler syndrome treated with HCT at our institution. Eligibility requirements were age ≥5 years and ≤16 years of age, a confirmed diagnosis of Hurler or Hunter syndrome, and ability to travel to the study center for annual evaluations. Exclusion criteria were pregnancy or participation in a therapeutic clinical trial. All participants with Hurler syndrome had previously been treated with HCT. All participants with Hunter syndrome started on ERT prior to study enrollment. Pubertal Tanner stage (20) was assessed by a trained study physician. GH deficiency was defined as a peak GH level less than 10 ng/ml during arginine and clonidine GH stimulation testing [21]. The University of Minnesota Institutional Review Board approved the study protocol.
2.2. Anthropometrics
Sitting and standing heights were measured by wall-mounted stadiometer (without shoes) to the nearest 0.1 cm and the average of three measurements taken. Upper to lower body ratio was calculated as sitting height/(standing height-sitting height). Weight was measured by electronic scale to the nearest 0.1 kg. Body mass index (BMI) was calculated by weight (kg) divided by height (m) squared. Height, weight, and BMI percentiles and standard deviation scores (SDS, also called height-for-age Z scores) were calculated using the SAS program from the Centers for Disease Control [http://www.cdc.gov/nccdphp/dnpao/growthcharts/resources/sas.htm, accessed Dec 2012].
2.3. Bone density and body composition measurements
Dual-energy x-ray absorptiometry (DXA) total body and lumbar spine scans were performed using a Lunar Prodigy scanner (pediatric software version 9.3; General Electric Medical Systems, Madison, WI). BMD (g/cm2) was measured for the total body excluding head and the lumbar spine. Whole body and lumbar spine BMD Z-scores (adjusted for age, gender and ethnicity) were calculated from the GE Lunar Prodigy database. Lumbar spine region of interest was L1-L4; vertebrae were excluded if BMD Z-score for an individual vertebra was 1 SD greater than any other vertebrae and 2 or more vertebrae were required for inclusion in analyses. Body fat (%) and LBM (kg) were determined from whole body scans.
2.4. Statistical analysis
Children were included in the analysis only if they had at least 2 annual study visits, so that annual changes in study outcomes could be estimated. During the observational period, individuals treated with hGH for at least 6 months were included in the hGH treated group; those treated for less than 6 months were excluded (N=2). Descriptive statistics are presented as means ± standard deviation for continuous variables and as numbers and percents for nominal variables. Student's t-test and Fisher tests were used to analyze cross-sectional differences between groups at last evaluation (Table 1). LBM, body fat %, total body BMD, and lumbar spine BMD were compared between groups by multivariable linear regression adjusted for age, sex, height and Tanner stage [Table 1]). Mixed-effects longitudinal regression was used to compare average annual change in growth velocity, height SDS, sitting height, and upper to lower body ratio in hGH treated vs. untreated subjects adjusted for age, sex (Hurler syndrome analysis only), and Tanner stage (Table 2); a random intercept for each patient was used to model the correlation between repeated measurements of the same subject. The difference in annual change between hGH treated and untreated subjects was estimated by the difference in age-slope between hGH groups, which was given by the interaction term between age and hGH treatment status. A similar mixed-effects model was used to compare average annual change in LBM, body fat %, total body and lumbar spine BMD in hGH treated vs. untreated subjects adjusted for age, sex (Hurler syndrome analysis only), Tanner stage, and height for LBM, total body and lumbar spine BMD, and for age, sex (Hurler syndrome analysis only), and Tanner stage for body fat % (Tables 2 and 3). Fisher's exact test was used to analyze differences in adverse events between hGH treated vs. untreated subjects (Table 4). Height values were plotted relative to percentiles from the CDC 2000 growth charts [22].
Table 1.
Population characteristics at study entry.
| hGH treatment (n=10) | No hGH treatment (n=13) | P value Treated vs. untreated | |
|---|---|---|---|
| Diagnosis, n (%) | |||
| Hurler syndrome | 6 (60) | 7 (54) | 1.000 |
| Hunter syndrome | 4 (40) | 6 (46) | |
| Age, yrs | 11±3 | 10±3 | 0.436 |
| Sex, %F | 4 (40) | 3 (23) | 0.650 |
| Race, % black | 0 (0) | 0 (0) | - |
| Tanner stage | |||
| 1 | 3 (30) | 9 (69) | |
| 2-3 | 2 (20) | 1 (8) | 0.262 |
| 4-5 | 5 (50) | 3 (23) | |
| GH deficient, yes | 4 (40) | 0 (0) | 0.024 |
| Height, SDS* | -2.4±1.6 | -2.0±1.8 | 0.575 |
| BMI, %* | 84±14 | 65±20 | 0.022 |
| LBM, kg† | 23±7 | 21±6 | 0.033 |
| Body fat, %† | 27±15 | 25±12 | 0.616 |
| Total body BMD, gm/cm2† | 0.73±0.08 | 0.73±0.07 | 0.798 |
| Lumbar spine BMD, gm/cm2† | 0.66±0.12 | 0.68±0.15 | 0.588 |
Mean ± SD or n(%) are presented. hGH=human growth hormone; GH=growth hormone; SDS=standard deviation score; BMI=body mass index; LBM=lean body mass; BMD=bone mineral density.
Relative to age and sex derived from CDC growth data.
Adjusted for age, sex, height and Tanner stage.
Table 2.
Annual change in height and height SDS in hGH vs. no hGH treatment groups.
| hGH treatment – no hGH treatment | P value | |
|---|---|---|
| Hurler syndrome (n=15) † | ||
| Growth velocity, cm/year | -0.3 (0.4) | .436 |
| Height SDS, SDS/year | -0.07 (0.05) | .168 |
| Sitting height, cm/year | 1.2 (1.7) | .466 |
| Upper to lower body ratio | 0.05 (0.13) | .694 |
| Hunter syndrome (n=10) †† | ||
| Growth velocity, cm/year | 0.5 (1.0) | .642 |
| Height SDS, SDS/year | 0.3 (0.2) | .127 |
| Sitting height, cm/year | 1.5 (0.8) | .110 |
| Upper to lower body ratio | 0.01 (0.01) | .511 |
Difference between groups estimate (SE). hGH=human growth hormone.
Adjusted for age, sex, and Tanner stage.
Adjusted for age and Tanner stage.
Table 3.
Annual change in body composition in hGH vs. no hGH treatment groups.
| hGH treatment – no hGH treatment | P value | |
|---|---|---|
| Hurler syndrome (n=15)† | ||
| LBM, kg/year | 0.4 (0.2) | .034 |
| Body fat, %/year* | 0.3 (0.7) | .665 |
| Total body BMD, gm/cm3/year | .005 (.005) | .371 |
| Lumbar spine BMD, gm/cm3/year | .005 (.014) | .749 |
| Hunter syndrome (n=10)†† | ||
| LBM, kg/year | 0.8 (0.5) | .155 |
| Body fat, %/year* | -1.9 (1.9) | .333 |
| Total body BMD, gm/cm2/year | .019 (.009) | .071 |
| Lumbar spine BMD, gm/cm2/year | .036 (.014) | .033 |
Difference between groups estimate (SE). hGH=human growth hormone; LBM=lean body mass; BMD=bone mineral density.
Hurler outcomes adjusted for age, height, Tanner stage and sex.
Hunter outcomes adjusted for age, height, and Tanner stage.
Body fat not adjusted for height.
Table 4. Frequency of adverse events in hGH treated subjects.
| hGH treatment N =10 | No hGH treatment N =13 | |
|---|---|---|
| Worsening ejection fraction? | 0 | 2 (15) |
| Worsening/new obstructive sleep apnea? | 1 (10) | 1 (8) |
| Worsening/new intracranial hypertension? | 1 (10) | 0 |
| Worsening/new kyphosis or scoliosis? | 0 | 1 (8) |
| Slipped capital femoral epiphysis (SCFE)? | 0 | 0 |
| If taking GH – injection site reactions? | 0 | - |
| Development of diabetes? | 0 | 0 |
N(%) with adverse event presented.
No significant differences by Fisher's exact test
3. Results
3.1. Participants
33 participants with Hurler or Hunter syndrome were enrolled; 23 were included in the analysis, with mean follow-up of 1.7 years (range 1.0 – 2.3 years) (Table 1). There were 13 participants with Hurler syndrome, average age at study entry of 9.8±3.1 years (range 5.3-13.6 years; 54% female), and 10 participants with Hunter syndrome, average age 12.0±2.7 years (range 7.0-17.0 years; 0% female). Four (31%) participants with Hurler syndrome had received treatment with ERT peritransplant. Three (23%) participants with Hurler syndrome had been treated with total body irradiation (TBI) prior to HCT. All of the participants with Hunter syndrome were receiving treatment with ERT for the duration of the study and had begun ERT an average of 4.0 years (range 1.1-7.6 years) prior to study enrollment.
Of 23 participants, 10 (43%) were receiving treatment with hGH from their local physicians for at least 6 months (6 Hurler syndrome; 4 Hunter syndrome). Four of the participants receiving hGH treatment had been diagnosed with GHD (2 Hurler syndrome; 2 Hunter syndrome). HGH dose was 0.34±0.10 mg/kg/wk (range 0.18-0.49 mg/kg/wk) and duration 3.6±1.4 years (range 1.4-5.7 years).
3.2. Growth
Participant growth data are shown superimposed on Center for Disease Control 2000 growth charts in Figure 1. Changes in annual growth outcomes between participants treated versus not treated with hGH are detailed in Table 2. Of note, there was no difference in growth velocity for participants with Hurler or Hunter syndrome treated versus not treated with hGH. Growth velocity was however quite variable; in hGH treated individuals growth velocity ranged from -0.4 to 8.1 cm/yr and from 0.3 to 6.6 cm/yr in the untreated individuals. For individuals with Hurler syndrome, there was no effect of history of TBI, peritransplant ERT, or age at HCT on annual change in height (cm) or height SDS between those treated versus not treated with hGH. Of the individuals treated with hGH, those with GHD had a trend towards higher annualized growth velocity compared to those without GHD (6.5±1.9 vs. 3.5±2.1; p=.050).
Fig. 1.
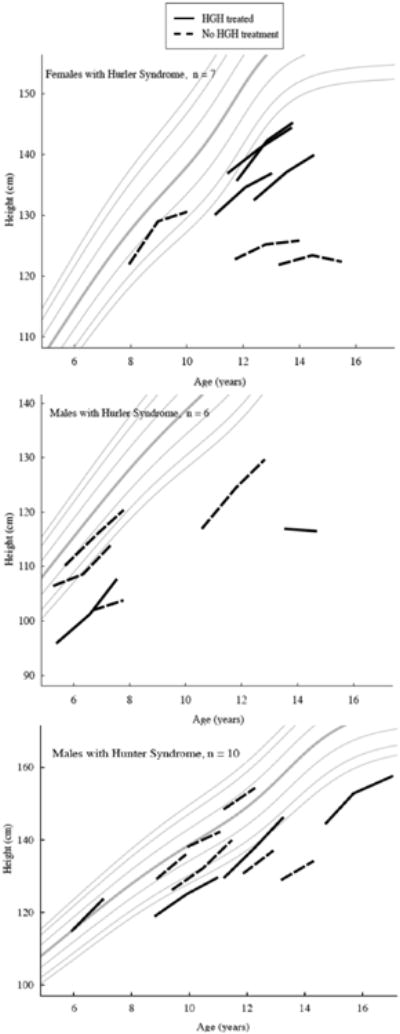
Change in height by gender for hGH treated (solid line) vs. not treated (dashed line) with Hurler syndrome and Hunter syndrome plotted on CDC 2000 growth charts.
For all participants, change in height SDS for those treated with hGH ranged from -0.7 to 0.8 SDS compared to -0.7 to 0.3 in those not treated with hGH. All 4 (100%) participants with Hunter syndrome treated with hGH compared to 3 (50%) participants with Hunter syndrome not treated with hGH had an annual increase in height SDS. No participant with Hurler syndrome had an annual increase in height SDS. There was no difference in age, duration of hGH treatment, or hGH dose between those with an annual increase in height SDS and those without an annual increase in height SDS.
3.3. Bone density and body composition
Detailed comparison of annual changes in bone density and body composition between participants treated with hGH versus those not treated with hGH divided by diagnosis is shown in Table 3. Participants with Hunter syndrome treated with hGH had a significantly greater annual change in lumbar spine BMD (p=.03) and a trend towards increased annual change in total body BMD (p=.07) compared to untreated participants. Changes in LBM in participants with Hurler syndrome treated vs. not treated with hGH were significantly higher (p=.03); the remaining changes in body composition were no different between groups (Table 3).
3.4. Safety concerns
There was no difference in annual change in upper to lower body ratio (Table 2) and no significant difference in adverse events described in Table 4 between treatment groups. However, one individual with Hunter syndrome treated with hGH developed worsening intracranial hypertension (IH) requiring intraventricular shunt placement 4.7 years after starting hGH. The IH was diagnosed by headache that did not resolve with discontinuation of hGH, enlarged ventricles with evidence of transependymal flow on brain MRI, and a central spinal fluid (CSF) opening pressure 3 days after discontinuation of hGH of 35 cm. Opening pressure prior to start of hGH was 24 cm and 1 year after starting hGH treatment was 24.8 cm. Estimated difference in annual change in bone age in hGH treated vs. not treated participants with Hurler and Hunter syndromes was 0.1±0.2 (p=.755) and -0.6±0.3 (p=.147) respectively.
4. Discussion
In this prospective observational study we found no difference in mean annual growth velocity in the cohorts of children with Hurler or Hunter syndrome treated versus not treated with hGH. However, there was a higher proportion (100%) of children with Hunter syndrome treated with hGH compared to not treated (50%) with hGH that had an annual increase in height SDS. This is particularly significant in that previous studies have shown progressive growth failure (i.e., decreasing height SDS over time) in children with Hurler and Hunter syndromes [3–5]. Finally, GH deficiency was common in our study population and we speculate related to HCT treatment and/or GAG deposition in the pituitary [23–25].
There are likely multiple factors contributing to the apparent lack of response in growth velocity for children with Hurler or Hunter syndrome treated with hGH. Abnormalities of the growth plate, such as chondrocyte and osteoblast abnormalities, including GAG accumulation, resulting in disorganized epiphyseal elongation and mineralization [26–30], may inhibit the efficacy of hGH treatment. In addition, we have previously shown that children with Hurler syndrome who received total body irradiation after HCT did not respond to treatment with hGH [18] likely due to radiation induced damage to the growth plate [31–33]; although we did not find that TBI was a significant confounder to response to hGH in the current study. Finally, the variability in growth measurements, hGH dosing and duration, combined with a relatively small sample size limited our ability to determine statistical significance.
HGH has been shown to have beneficial skeletal effects. Similar to our findings of increased annual change in BMD in MPS participants treated with hGH, treatment with hGH has been shown to increase bone mineral density and improve peak bone mass accrual in children with GH deficiency, cerebral palsy, and children born small-for-gestational-age [34–37]. This effect on bone is likely mediated though the increased production of insulin-like growth factor-1 (IGF-1) which has been shown in both humans and animal models to stimulate bone turnover in favor of bone formation resulting in increased bone mineral density [34,38–40].
We hypothesized an increased annual change in LBM would be observed in participants with MPS treated with hGH, based on previous reports in other pediatric populations treated with hGH. A positive effect of hGH on body composition is independent of whether a child is GH deficient; children with Turner syndrome, Prader-Willi syndrome, idiopathic short stature and born SGA who are not GH deficient have been shown to have improvements in LBM as well as decreased body fat [11–13,16,17]. In our study, participants treated with hGH had a higher average increase in LBM of 0.4-0.8 kg/year. Changes in body composition could have long-term benefits for bone and cardiovascular health.
Increased growth velocity, with or without treatment with hGH, is associated with progression or onset of scoliosis [41,42]. Thus, there has been concern that treatment of children with syndromes associated with scoliosis (e.g. Prader-Willi syndrome) will result in worsened abnormal spinal curvatures. However, a randomized, controlled clinical trial of hGH in children with Prader-Willi syndrome found no significant different in progression of scoliosis by X-ray measurement in children treated vs. those not treated with hGH [43]. Although we did not have direct measurements of spinal curvature in our population (we relied on parental report), it is reassuring that we too did not find any difference in progression or new onset of scoliosis or kyphosis between individuals with MPS treated versus not treated with hGH.
One child with Hunter syndrome in our study developed worsening IH during treatment with hGH. IH is a known complication of Hunter syndrome [44–46]. IH is also a rare side effect of treatment with hGH in any population. One review of 57,968 children treated with hGH in the KIGS (Pfizer International Growth Study database) found the overall incidence of IH to be 27.7 per 100,000 treatment years, with a slightly higher incidence found in children with Turner syndrome and Prader-Willi syndrome (56.4 and 68.3 respectively) and no cases reported in children with ISS [47]. The National Cooperative Growth Study (NCGS) found 61 cases of IH in 54,996 children treated with hGH [48]. The typical onset of IH in children treated with hGH is approximately 0.01 to 1.3 years from initiation of hGH treatment, however 22% of cases were reported more than 2 years after initiation of hGH treatment in the NCGS study [47,48]. The development of IH in our participant treated with hGH was thought most likely due to Hunter syndrome and not hGH treatment, given the presence of mild IH prior to initiation of hGH treatment, the late onset of progression of IH, and persistence after discontinuation of hGH. However, it is impossible to exclude hGH treatment as the stimulus for worsening IH.
This study is limited by being a retrospective observational study in design, and by reliance on the use of parental reports for adverse events. However, we do not think this is a significant limitation, given that all of the participants are followed at least annually for known complications of their disease, which typically includes x-rays for kyphoscoliosis and hip dysplasia, an echocardiogram for cardiac function and valvular disease, brain MRI for ventricular size, and sleep studies for obstructive sleep apnea. Thus, it seems unlikely that significant adverse events known to be associated with hGH treatment were not appreciated. In addition, we have no long-term data on the effect of hGH in this population.
5. Conclusions
In conclusion, we found no difference in annual growth velocity in children with Hurler or Hunter syndrome treated with hGH compared to those not treated with hGH in this prospective observational study; individuals with GHD responded better to treatment with hGH than those without GHD. HGH treatment in Hurler or Hunter syndrome may improve body composition and bone mineral density; however, the long-term implications of this are unknown. Finally, we found no safety concerns related to hGH treatment in children with Hurler or Hunter syndrome except for 1 patient with Hunter syndrome with worsening of IH 4.7 years after starting hGH treatment.
Based on our findings, and the variability in response to hGH treatment in these cohorts of children with Hunter or Hurler syndrome, we think a trial of hGH treatment for 1 year in children with Hurler or Hunter syndrome, short stature and growth failure, with or without GHD, with discontinuation of treatment after 1 year if no improvement in growth velocity or change in height SDS, is appropriate. Monitoring before and during treatment with hGH for IH is recommended. Finally, we found that GH deficiency was common in individuals with either Hurler or Hunter syndrome and therefore recommend that individuals should be screened for this annually and treated with hGH if found to be deficient.
Highlights.
We examined safety and efficacy of growth hormone (hGH) treatment in MPS
Growth velocity was not significantly different in hGH treated individuals
All children with Hunter syndrome treated with hGH had an increase in height SDS
Individuals treated with hGH had greater gains in bone density and lean body mass.
No safety concerns were identified except one child with intracranial hypertension
Acknowledgments
The authors would like to thank all the study participants and their parents, as well as the study coordinator Jane Kennedy, RN. Research reported in this publication was supported in part by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (NIH) under Award Number K23AR057789, the National Institutes of Neurological Disorders and Stroke (NINDS)/Diabetes and Digestive and Kidney (NIDDK) of the NIH under Award Number U54NS065768, a grant from the University of Minnesota Pediatric Foundation, and by the National Center for Advancing Translational Sciences of the NIH Award Number UL1TR000114. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
LEP has previously received research funding (drug only) from Genentech and has provided consulting support to and received grant support from Genzyme. BSM is a consultant for ENDO Pharmaceuticals, Genentech, Ipsen, Novo Nordisk and Sandoz and receives grant/research support from ENDO Pharmaceuticals, Eli Lilly, Ipsen, Novo Nordisk and Sandoz. CBW has been a consultant and recipient of grants from Actelion, Amicus, BioMarin, Genzyme, Shire, and Synageva.
Abbreviations
- MPS
Mucopolysaccharidosis
- HCT
hematopoietic stem cell transplantation
- ERT
enzyme replacement therapy
- hGH
recombinant human growth hormone
- BMD
bone mineral density
- SDS
standard deviation score
- GAG
glycosaminoglycan
- SGA
small for gestational age
- GHD
growth hormone deficiency
- LBM
lean body mass
- SCFE
slipped capital femoral epiphysis
- BMI
Body mass index
- TBI
total body irradiation
- IH
intracranial hypertension
- CSF
central spinal fluid
Footnotes
Conflicts of interest: WT and PO have no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
William Thomas, Email: thoma003@umn.edu.
Paul Orchard, Email: orcha001@umn.edu.
Chester B. Whitley, Email: whitley@umn.edu.
Bradley S. Miller, Email: mille685@umn.edu.
References
- 1.Whitley CB. McKusick's Heritable Disorders of Connective Tissue. Mosby; St. Louis: 1993. The mucopolysaccharidoses; pp. 367–500. [Google Scholar]
- 2.Neufeld E. The Metabolic and Molecular Basis of Inherited Disease. McGraw-Hill; New York: 2001. The Mucopolysaccharidoses; pp. 3421–3452. [Google Scholar]
- 3.Polgreen LE, Tolar J, Plog M, Himes JH, Orchard PJ, Whitley CB, et al. Growth and endocrine function in patients with Hurler syndrome after hematopoietic stem cell transplantation. Bone Marrow Transplant. 2008;41:1005–11. doi: 10.1038/bmt.2008.20. [DOI] [PubMed] [Google Scholar]
- 4.Gardner CJ, Robinson N, Meadows T, Wynn R, Will A, Mercer J, et al. Growth, final height and endocrine sequelae in a UK population of patients with Hurler syndrome (MPS1H) J Inherit Metab Dis. n.d;34:489–97. doi: 10.1007/s10545-010-9262-8. [DOI] [PubMed] [Google Scholar]
- 5.Jones SA, Parini R, Harmatz P, Giugliani R, Fang J, Mendelsohn NJ. The effect of idursulfase on growth in patients with Hunter syndrome: Data from the Hunter Outcome Survey (HOS) Mol Genet Metab. 2013 doi: 10.1016/j.ymgme.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 6.Blethen SL, Baptista J, Kuntze J, Foley T, LaFranchi S, Johanson A. Adult height in growth hormone (GH)-deficient children treated with biosynthetic GH. The Genentech Growth Study Group. J Clin Endocrinol Metab. 1997;82:418–420. doi: 10.1210/jcem.82.2.3734. [DOI] [PubMed] [Google Scholar]
- 7.Cutfield W, Lindberg A, Albertsson Wikland K, Chatelain P, Ranke MB, Wilton P. Final height in idiopathic growth hormone deficiency: the KIGS experience. KIGS International Board. Acta Paediatr Suppl. 1999;88:72–75. doi: 10.1111/j.1651-2227.1999.tb14356.x. [DOI] [PubMed] [Google Scholar]
- 8.Argente J, Gracia R, Ibanez L, Oliver A, Borrajo E, Vela A, et al. Improvement in growth after two years of growth hormone therapy in very young children born small for gestational age and without spontaneous catch-up growth: results of a multicenter, controlled, randomized, open clinical trial. J Clin Endocrinol Metab. 2007;92:3095–101. doi: 10.1210/jc.2007-0078. [DOI] [PubMed] [Google Scholar]
- 9.Bolar K, Hoffman AR, Maneatis T, Lippe B. Long-Term Safety of Recombinant Human Growth Hormone in Turner Syndrome. J Clin Endocrinol Metab. 2007 doi: 10.1210/jc.2007-1723. [DOI] [PubMed] [Google Scholar]
- 10.Hintz RL, Attie KM, Baptista J, Roche A. Effect of growth hormone treatment on adult height of children with idiopathic short stature. Genentech Collaborative Group. N Engl J Med. 1999;340:502–7. doi: 10.1056/NEJM199902183400702. [DOI] [PubMed] [Google Scholar]
- 11.Leschek EW, Rose SR, Yanovski JA, Troendle JF, Quigley CA, Chipman JJ, et al. Effect of growth hormone treatment on adult height in peripubertal children with idiopathic short stature: a randomized, double-blind, placebo-controlled trial. J Clin Endocrinol Metab. 2004;89:3140–8. doi: 10.1210/jc.2003-031457. [DOI] [PubMed] [Google Scholar]
- 12.Schweizer R, Martin DD, Schonau E, Ranke MB. Muscle function improves during growth hormone therapy in short children born small for gestational age: results of a peripheral quantitative computed tomography study on body composition. J Clin Endocrinol Metab. 2008;93:2978–83. doi: 10.1210/jc.2007-2600. [DOI] [PubMed] [Google Scholar]
- 13.Ari M, Bakalov VK, Hill S, Bondy CA. The effects of growth hormone treatment on bone mineral density and body composition in girls with turner syndrome. J Clin Endocrinol Metab. 2006;91:4302–5. doi: 10.1210/jc.2006-1351. [DOI] [PubMed] [Google Scholar]
- 14.Lanes R, Gunczler P, Esaa S, Weisinger JR. The effect of short- and long-term growth hormone treatment on bone mineral density and bone metabolism of prepubertal children with idiopathic short stature: a 3-year study. Clin Endocrinol (Oxf) 2002;57:725–30. doi: 10.1046/j.1365-2265.2002.01614.x. [DOI] [PubMed] [Google Scholar]
- 15.Widdowson WM, Gibney J. The effect of growth hormone replacement on exercise capacity in patients with GH deficiency: a metaanalysis. J Clin Endocrinol Metab. 2008;93:4413–7. doi: 10.1210/jc.2008-1239. [DOI] [PubMed] [Google Scholar]
- 16.de Lind van Wijngaarden RFA, Siemensma EPC, Festen DAM, Otten BJ, van Mil EGAH, Rotteveel J, et al. Efficacy and safety of long-term continuous growth hormone treatment in children with Prader-Willi syndrome. J Clin Endocrinol Metab. 2009;94:4205–4215. doi: 10.1210/jc.2009-0454. [DOI] [PubMed] [Google Scholar]
- 17.Myers SE, Carrel AL, Whitman BY, Allen DB. Sustained benefit after 2 years of growth hormone on body composition, fat utilization, physical strength and agility, and growth in Prader-Willi syndrome. J Pediatr. 2000;137:42–9. doi: 10.1067/mpd.2000.105369. [DOI] [PubMed] [Google Scholar]
- 18.Polgreen LE, Plog M, Schwender JD, Tolar J, Thomas W, Orchard PJ, et al. Short-term growth hormone treatment in children with Hurler syndrome after hematopoietic cell transplantation. Bone Marrow Transplant. 2009;44:279–85. doi: 10.1038/bmt.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanaka N, Katsumata N, Horikawa R, Tanaka T. The comparison of the effects of short-term growth hormone treatment in patients with achondroplasia and with hypochondroplasia. Endocr J. 2003;50:69–75. doi: 10.1507/endocrj.50.69. [DOI] [PubMed] [Google Scholar]
- 20.Hertel NT, Eklöf O, Ivarsson S, Aronson S, Westphal O, Sipilä I, et al. Growth hormone treatment in 35 prepubertal children with achondroplasia: a five-year dose-response trial. Acta Paediatr. 2005;94:1402–1410. doi: 10.1111/j.1651-2227.2005.tb01811.x. [DOI] [PubMed] [Google Scholar]
- 21.Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: summary statement of the GH Research Society. GH Research Society. J Clin Endocrinol Metab. 2000;85:3990–3. doi: 10.1210/jcem.85.11.6984. [DOI] [PubMed] [Google Scholar]
- 22.Kuczmarski RJ, Ogden CL, Guo SS, Grummer-Strawn LM, Flegal KM, Mei Z, et al. 2000 CDC Growth Charts for the United States: methods and development. Vital Health Stat. 2002;11:1–190. [PubMed] [Google Scholar]
- 23.Bakker B, Oostdijk W, Bresters D, Walenkamp MJ, Vossen JM, Wit JM. Disturbances of growth and endocrine function after busulphan-based conditioning for haematopoietic stem cell transplantation during infancy and childhood. Bone Marrow Transplant. 2004;33:1049–56. doi: 10.1038/sj.bmt.1704481. [DOI] [PubMed] [Google Scholar]
- 24.Huma Z, Boulad F, Black P, Heller G, Sklar C. Growth in children after bone marrow transplantation for acute leukemia. Blood. 1995;86:819–24. [PubMed] [Google Scholar]
- 25.Nagashima K, Endo H, Sakakibara K, Konishi Y, Miyachi K, Wey JJ, et al. Morphological and biochemical studies of a case of mucopolysaccharidosis II (Hunter's syndrome) Acta Pathol Jpn. 1976;26:115–32. doi: 10.1111/j.1440-1827.1976.tb03297.x. [DOI] [PubMed] [Google Scholar]
- 26.Abreu S, Hayden J, Berthold P, Shapiro IM, Decker S, Patterson D, et al. Growth plate pathology in feline mucopolysaccharidosis VI. Calcif Tissue Int. 1995;57:185–90. doi: 10.1007/BF00310256. [DOI] [PubMed] [Google Scholar]
- 27.Monroy MA, Ross FP, Teitelbaum SL, Sands MS. Abnormal osteoclast morphology and bone remodeling in a murine model of a lysosomal storage disease. Bone. 2002;30:352–9. doi: 10.1016/s8756-3282(01)00679-2. [DOI] [PubMed] [Google Scholar]
- 28.Nuttall JD, Brumfield LK, Fazzalari NL, Hopwood JJ, Byers S. Histomorphometric analysis of the tibial growth plate in a feline model of mucopolysaccharidosis type VI. Calcif Tissue Int. 1999;65:47–52. doi: 10.1007/s002239900656. [DOI] [PubMed] [Google Scholar]
- 29.Russell C, Hendson G, Jevon G, Matlock T, Yu J, Aklujkar M, et al. Murine MPS I: insights into the pathogenesis of Hurler syndrome. Clin Genet. 1998;53:349–61. doi: 10.1111/j.1399-0004.1998.tb02745.x. [DOI] [PubMed] [Google Scholar]
- 30.Silveri CP, Kaplan FS, Fallon MD, Bayever E, August CS. Hurler syndrome with special reference to histologic abnormalities of the growth plate. Clin Orthop Relat Res. 1991:305–11. [PubMed] [Google Scholar]
- 31.Frisk P, Arvidson J, Gustafsson J, Lönnerholm G. Pubertal development and final height after autologous bone marrow transplantation for acute lymphoblastic leukemia. Bone Marrow Transplantation. 2004;33:205–210. doi: 10.1038/sj.bmt.1704324. [DOI] [PubMed] [Google Scholar]
- 32.Bakker B, Massa GG, Oostdijk W, Van Weel-Sipman MH, Vossen JM, Wit JM. Pubertal development and growth after total-body irradiation and bone marrow transplantation for haematological malignancies. Eur J Pediatr. 2000;159:31–7. doi: 10.1007/s004310050006. [DOI] [PubMed] [Google Scholar]
- 33.Couto-Silva AC, Trivin C, Esperou H, Michon J, Baruchel A, Lemaire P, et al. Final height and gonad function after total body irradiation during childhood. Bone Marrow Transplant. 2006;38:427–32. doi: 10.1038/sj.bmt.1705455. [DOI] [PubMed] [Google Scholar]
- 34.Ali O, Shim M, Fowler E, Greenberg M, Perkins D, Oppenheim W, et al. Growth hormone therapy improves bone mineral density in children with cerebral palsy: a preliminary pilot study. J Clin Endocrinol Metab. 2007;92:932–7. doi: 10.1210/jc.2006-0385. [DOI] [PubMed] [Google Scholar]
- 35.Kandemir N, Gonc EN, Yordam N. Responses of bone turnover markers and bone mineral density to growth hormone therapy in children with isolated growth hormone deficiency and multiple pituitary hormone deficiencies. J Pediatr Endocrinol Metab. 2002;15:809–16. doi: 10.1515/jpem.2002.15.6.809. [DOI] [PubMed] [Google Scholar]
- 36.Saggese G, Baroncelli GI, Bertelloni S, Barsanti S. The effect of long-term growth hormone (GH) treatment on bone mineral density in children with GH deficiency. Role of GH in the attainment of peak bone mass. J Clin Endocrinol Metab. 1996;81:3077–83. doi: 10.1210/jcem.81.8.8768878. [DOI] [PubMed] [Google Scholar]
- 37.Willemsen RH, Arends NJ, Bakker-van Waarde WM, Jansen M, van Mil EG, Mulder J, et al. Long-term effects of growth hormone (GH) treatment on body composition and bone mineral density in short children born small-for-gestational-age: six-year follow-up of a randomized controlled GH trial. Clin Endocrinol (Oxf) 2007;67:485–92. doi: 10.1111/j.1365-2265.2007.02913.x. [DOI] [PubMed] [Google Scholar]
- 38.DiGirolamo DJ, Mukherjee A, Fulzele K, Gan Y, Cao X, Frank SJ, et al. Mode of growth hormone action in osteoblasts. J Biol Chem. 2007;282:31666–74. doi: 10.1074/jbc.M705219200. [DOI] [PubMed] [Google Scholar]
- 39.Ernst M, Froesch ER. Growth hormone dependent stimulation of osteoblast-like cells in serum-free cultures via local synthesis of insulin-like growth factor I. Biochem Biophys Res Commun. 1988;151:142–7. doi: 10.1016/0006-291x(88)90570-0. [DOI] [PubMed] [Google Scholar]
- 40.Zhao G, Monier-Faugere MC, Langub MC, Geng Z, Nakayama T, Pike JW, et al. Targeted overexpression of insulin-like growth factor I to osteoblasts of transgenic mice: increased trabecular bone volume without increased osteoblast proliferation. Endocrinology. 2000;141:2674–82. doi: 10.1210/endo.141.7.7585. [DOI] [PubMed] [Google Scholar]
- 41.Docquier PL, Mousny M, Jouret M, Bastin C, Rombouts JJ. Orthopaedic concerns in children with growth hormone therapy. Acta Orthop Belg. 2004;70:299–305. [PubMed] [Google Scholar]
- 42.Wever DJ, Tønseth KA, Veldhuizen AG, Cool JC, van Horn JR. Curve progression and spinal growth in brace treated idiopathic scoliosis. Clin Orthop Relat Res. 2000:169–179. doi: 10.1097/00003086-200008000-00023. [DOI] [PubMed] [Google Scholar]
- 43.de Lind van Wijngaarden RFA, de Klerk LWL, Festen DAM, Duivenvoorden HJ, Otten BJ, Hokken-Koelega ACS. Randomized controlled trial to investigate the effects of growth hormone treatment on scoliosis in children with Prader-Willi syndrome. J Clin Endocrinol Metab. 2009;94:1274–1280. doi: 10.1210/jc.2008-1844. [DOI] [PubMed] [Google Scholar]
- 44.Manara R, Priante E, Grimaldi M, Santoro L, Astarita L, Barone R, et al. Brain and spine MRI features of Hunter disease: frequency, natural evolution and response to therapy. J Inherit Metab Dis. 2011;34:763–780. doi: 10.1007/s10545-011-9317-5. [DOI] [PubMed] [Google Scholar]
- 45.Sawaf S, Mayatepek E, Hoffmann B. Neurological findings in Hunter disease: Pathology and possible therapeutic effects reviewed. Journal of Inherited Metabolic Disease. 2008;31:473–480. doi: 10.1007/s10545-008-0878-x. [DOI] [PubMed] [Google Scholar]
- 46.Wraith JE, Beck M, Giugliani R, Clarke J, Martin R, Muenzer J, et al. Initial report from the Hunter Outcome Survey. Genet Med. 2008;10:508–516. doi: 10.1097/gim.0b013e31817701e6. [DOI] [PubMed] [Google Scholar]
- 47.Darendeliler F, Karagiannis G, Wilton P. Headache, Idiopathic Intracranial Hypertension and Slipped Capital Femoral Epiphysis during Growth Hormone Treatment: A Safety Update from the KIGS Database. Hormone Research. 2007;68:41–47. doi: 10.1159/000110474. [DOI] [PubMed] [Google Scholar]
- 48.Bell J, Parker KL, Swinford RD, Hoffman AR, Maneatis T, Lippe B. Long-term safety of recombinant human growth hormone in children. J Clin Endocrinol Metab. n.d;95:167–77. doi: 10.1210/jc.2009-0178. [DOI] [PubMed] [Google Scholar]