

Abstract
GABAA receptors, the major mediators of fast inhibitory neuronal transmission, are heteropentameric glycoproteins assembled from a panel of subunits, usually including α and β subunits with or without a γ2 subunit. The α1β2γ2 receptor is the most abundant GABAA receptor in brain. Co-expression of γ2 with α1 and β2 subunits causes conformational changes, increases GABAA receptor channel conductance, and prolongs channel open times. We reported previously that glycosylation of the three β2 subunit glycosylation sites, N32, N104 and N173, was important for α1β2 receptor channel gating. Here, we examined the hypothesis that steric effects or conformational changes caused by γ2 subunit co-expression alter the glycosylation of partnering β2 subunits. We found that co-expression of γ2 subunits hindered processing of β2 subunit N104 N-glycans in HEK293T cells. This γ2 subunit-dependent effect was strong enough that a decrease of γ2 subunit expression in heterozygous GABRG2 knockout (γ2+/−) mice led to appreciable changes in the endoglycosidase H (endo H) digestion pattern of neuronal β2 subunits. Interestingly, as measured by flow cytometry, γ2 subunit surface levels were decreased by mutating each of the β2 subunit glycosylation sites. The β2 subunit mutation N104Q also decreased GABA potency to evoke macroscopic currents and reduced conductance, mean open time and open probability of single channel currents. Collectively, our data suggested that γ2 subunits interacted with β2 subunit N-glycans and/or subdomains containing the glycosylation sites, and that γ2 subunit co-expression-dependent alterations in the processing of the β2 subunit N104 N-glycans were involved in altering the function of surface GABAA receptors.
Introduction
N-linked glycosylation occurs in approximately two-thirds of proteins, is important for protein biogenesis and function (1) and may be disrupted in disease-causing mutations (2). In the endoplasmic reticulum (ER), N-glycan precursors are co-translationally transferred to protein glycosylation sites. These attached N-glycans are subjected to intensive processing in the ER and Golgi apparatus and are conferred with endo H resistance by Golgi-resident enzymes (3). Usually, endo H resistance of N-glycans correlates with trafficking of a glycoprotein beyond the Golgi apparatus. However, the presence of an endo H-sensitive N-glycan does not necessarily indicate ER retention of glycoproteins because subunit folding and/or assembly can sterically hinder N-glycan processing (4).
GABAA receptors, the predominant mediators of inhibitory synaptic transmission in brain, are involved in nearly every aspect of brain activity. The receptors are pentamers assembled from combinations of nineteen subunit subtypes (α1-6, β1-3, γ1-3, δ, ε, θ, π and ρ1-3) (5). Subunit composition influences channel properties of GABAA receptors profoundly. For instance, αβγ receptors generally have larger conductance, longer mean open time, and different kinetic properties than αβ receptors (6–8). Despite the well-established functional differences between α1β2 and α1β2γ2 channels, the underlying bases for these differences remain incompletely understood.
We demonstrated previously that the β2 subunit extracellular N-terminal domain contains three N-linked glycosylation sites: N32, N104 and N173 (9). These glycosylation sites are especially well positioned to alter inter-subunit interactions at the γ-β or β-α interface. Molecular modeling predicts that a very short segment including the first glycosylation site of β3 (10) and β2 subunits (not shown) interacts strongly with a crucial sequence (residues 83–90 of the γ2 subunit (11)) that allows oligomerization of the β and γ subunits. Moreover, β3 subunit residue N33 (10) forms a direct salt bridge with the γ2 subunit residue R82 that transduces GABA binding at the α-β interface into channel opening (12). Similarly, the second β2 subunit glycosylation site is immediately across from the γ2 subunit region that is homologous to the GABA receptor “C loop” at the subunit interface. Finally, the β2 subunit residue N173 lies within loop 7 (the “signature cys loop”) that interacts with the linker sequence between transmembrane domains 2 and 3 and transduces ligand binding into channel activation. Given that N-linked glycosylation could regulate channel gating of GABAA receptors (9), we hypothesized that the functional effects of γ2 subunit incorporation might involve altered N-glycan processing of partnering α1 or β2 subunits. We found that there was indeed a complex interdependency between β2 subunit glycosylation and γ2 surface expression. First, γ2 subunit co-expression changed endo H digestion patterns of partnering β2 subunits by preventing N104 N-glycans from acquiring endo H resistance in the Golgi apparatus. Moreover, a comparison of endo H digestion patterns of β2 subunits obtained from wild-type and heterozygous GABRG2 knockout mice (γ2+/−) revealed that prevention of N-glycan processing occurred in vivo and depended on expression levels of γ2 subunits. Conversely, surface expression of γ2 subunits was affected by β2 subunit glycosylation and mutating any of the three β2 subunit glycosylation sites decreased surface levels of α1β2γ2S receptors. We also found that β2 subunit glycosylation site mutations decreased GABA potency, and that the N104Q mutation, specifically, decreased conductance and mean open time of single-channel currents. These results advance our understanding of how protein glycosylation helps regulate the expression and function of GABAA receptors in a subunit-specific fashion.
Experimental Procedures
DNA constructs
Complementary DNA (cDNA) encoding the human GABAA receptor α1, α3, β2, γ2S or γ2L subunit polypeptide, was inserted into a pcDNA3.1(+) vector. The FLAG epitope, DYKDDDDK, was introduced between the 8th and 9th amino acids of the mature α1 subunit polypeptide or between the 4th and 5th amino acids of the mature β2 or γ2S subunit polypeptide (13). Three β2 subunit glycosylation sites, N32, N104 and N173, counting from the first methionine of the immature polypeptide, were individually mutated to glutamine using the QuikChange Site-Directed Mutagenesis Kit (Stratagene).
Cell culture and transfection
Human embryonic kidney cells (HEK293T) were incubated at 37°C in 5% CO2/95% air and grown in Dulbecco’s Modified Eagle Medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen) plus 100 IU/ml each of penicillin and streptomycin (Invitrogen). Altogether, 1 μg each of α1/3, β2 and γ2 subunit plasmids were mixed with 9 μl of FuGENE 6 transfection reagent (Roche Applied Science) and incubated with about 4 × 105 cells in a 60 mm diameter culture dish. For binary α1β2 subunit co-expression, empty vector was added so that the same total mass of DNA was transfected for all subunit combinations. In experiments using different sizes of culture dishes, the DNA-FuGENE 6 mixture volumes were scaled up or down proportionally to the surface areas. Cells were used for experiments forty-eight hours after transfection. For whole cell recording, plasmids were introduced into cells using the modified calcium phosphate precipitation method (6). To select for positively transfected cells, pHook-1 (Invitrogen) plasmids in the amount of one sixth of the total amount of subunit plasmids or 10 ng of GFP plasmids was co-transfected. Cells expressing pHook were selected using immunomagnetic beads one day after transfection, and were recorded the next day after selection (14).
Endoglycosidase digestions
Membrane or surface biotinylated proteins from HEK293T cells, or immunopurified protein complexes from neonatal mice were subjected to endo H or peptide N-glycosidase-F (PNGase F) digestion (New England BioLab) at 37°C for 3 hours. All procedures using mice were approved by the Vanderbilt Institutional Committee for Animal Use and Care.
Western blots
Transfected HEK293T cells were broken by freeze-and-thaw cycles. The cytoplasmic fraction was then separated by centrifugation. The pellet containing the membrane fraction was extracted using radio-immune precipitation assay (RIPA) buffers, which contained 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1–2% NP-40, 0.25-0.5% sodium deoxycholate and protease inhibitor cocktail (Sigma-Aldrich). Insoluble components were removed by centrifugation at 16,000× g for 30 minutes. The supernatants were either subjected to further glycosidase treatment or directly subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins in gels were transferred to polyvinylidene fluoride (PVDF) membranes (Millipore).
A monoclonal anti-GABAA receptor α1 subunit antibody (final concentration 5 μg/ml; clone: BD24, Millipore) was used to detect α1 subunits, and a monoclonal anti-GABAA receptor β2/3 subunit antibody (4 μg/ml; clone: 62-3G1, Millipore) or polyclonal rabbit anti-GABAA receptor β2 subunit cytoplasmic loop antibody (0.2 μg/ml; Millipore) was used to detect control (wild-type subunits in the control condition) or mutant human β2 subunits. Polyclonal rabbit anti-GABAA receptor γ2 subunit antibodies (0.8 μg/ml; Alomone) were used to detect γ2 subunits. Following incubation with primary antibodies, secondary goat anti-mouse or anti-rabbit IgG heavy and light chain antibodies conjugated with horseradish peroxidase were used at a 1:10,000X dilution (Jackson ImmunoResearch laboratories) for visualization of specific bands in enhanced chemiluminescent detection system (Amersham Biosciences).
The signals were collected in a digital ChemImager (Alpha Innotech). The integrated density volumes (IDV; pixel intensity × mm2) were then calculated using the FluorChem 5500 software. Adjusted IDVs (normalized to loading control Na+/K+-ATPase IDVs) of mutant and partnering subunits were expressed as % of adjusted IDVs of corresponding control subunits. Data were expressed as mean ± S.D.
Immunoprecipitation
Generation of mutant mice with γ2 subunit deficiency (γ2+/−) has been described previously (15). Wild-type (γ2+/+), and heterozygous (γ2+/−) and homozygous (γ2−/−) mutant mice were obtained by intercrossing heterozygous mice, but only γ2+/+ and γ2+/− mice were used. Postnatal day 1 or day 2 mice were deeply anesthetized with isoflurane and decapitated under a protocol approved by Vanderbilt University’s Institutional Animal Care and Use Committee. The whole brain excluding cerebellum from each of the neonatal mice was extracted separately using RIPA buffer. Insoluble components were removed by centrifugation at 16,000× g for 30 minutes. GABAA receptor protein complexes from each brain extract were immunoprecipitated using 10 μg monoclonal anti-GABAA receptor β2/3 subunit antibodies. The immunoprecipitated protein complexes were further subjected to endoglycosidase digestions or directly resolved by SDS-PAGE.
Surface biotinylation
HEK293T cell surface proteins were biotinylated with membrane impermeable reagent sulfo-NHS-SS-biotin (1 mg/ml, Thermo Scientific) in phosphate buffered saline containing 0.1 mM CaCl2 and 1 mM MgCl2 (PBS+CM) at 4°C for 1 hour. After incubation, the biotin was quenched with 0.1 M glycine in PBS+CM. Following washes with PBS+CM, cells were lysed in RIPA buffer supplemented with protease inhibitors (Sigma-Aldrich). After centrifugation to pellet cellular debris, the biotin-labeled plasma membrane proteins were pulled down by streptavidin beads (Thermo Scientific) at 4°C overnight.
Surface expression measurement using flow cytometry
Measurement of surface expression of GABAA receptor subunits using flow cytometry has been described previously (13). Briefly, cells were trypsinized and then resuspended in FACS buffer (phosphate buffered saline, PBS supplemented with 2% FBS and 0.05% sodium azide). Following washes with FACS buffer, cells were incubated with anti-FLAG IgG directly conjugated with R-Phycoerythrin (PE, 1:50 dilution, Martek) for 1 hour. Cells were then washed with FACS buffer and fixed with 2% paraformaldehyde. The surface fluorescence intensity of each cell was measured using a LSR II (BD Biosciences).
The acquired data were analyzed using FlowJo 7.1 (Treestar, Inc.). Mean fluorescence of the viable population, which excluded 7-amino-actinomycin D (7-AAD; Invitrogen), of a given experimental condition was obtained. To account for cell auto-fluorescence and non-specific staining, mean fluorescence of mock transfected cells (transfected with empty vector, pcDNA3.1(+)) were subtracted from that obtained in each experimental condition. For comparison among various experimental conditions, the mock-subtracted mean fluorescence value for each experimental condition was expressed as a percentage of that with control subunit co-expression. Data were expressed as mean ± S.D.
Recording and analysis of macroscopic GABA evoked currents
As described before (16), voltage-clamp whole cell recordings were performed on transfected HEK293T cells bathed in an external solution consisting of 142 mM NaCl, 8 mM KCl, 6 mM MgCl2, 1 mM CaCl2, 10 mM glucose, and 10 mM HEPES (pH 7.4, 320–330 mOsm). The glass electrodes were pulled from thin-walled borosilicate capillary glass (Fisher) on a P-2000 Quartz Micropipette Puller (Sutter Instruments), fire-polished to a resistance of 1–2 MΩ on an MF-830 Micro Forge (Narishige), and filled with an internal solution consisting of 153 mM KCl, 1 mM MgCl2, 5 mM EGTA, 10 mM HEPES, and 2 mM MgATP (pH 7.3, 310 mOsm). The Cl− equilibrium potential across the cell membrane was close to zero with the combination of external and internal solutions. Cells were voltage-clamped at −20 mV using an Axopatch 200A amplifier (Axon Instrument). GABA was applied to the lifted cells using a rapid perfusion system (open tip exchange times < 700 μs) consisting of multibarrel square glass connected to a Perfusion Fast-Step (Warner Instruments), which was controlled by Clampex 9.0 (Axon Instrument).
Inward GABA-evoked macroscopic currents were low-pass filtered at 2 kHz and digitized at 5–10 kHz using Digidata 1322A. Their peak amplitudes and 10–90% rise time were analyzed using Clampfit 9.0 (Axon Instruments). Desensitization and deactivation time courses were fitted using the Levenberg-Marquardt least squared error method in the form: Σ Aie(−t/τi) + C, where Ai and τi are the relative amplitude and the time constant of the ith component, respectively; t is the time; C is the residual current at the end of fitting. Deactivation time courses were then evaluated by weighted time constant calculated with the follow: (Σ Ai τi)/(Σ Ai). Data were expressed as mean ± S.E.
Single-channel recording and data analysis
Single-channel currents were recorded in cell-attached configuration as described previously (9, 17). Cell-attached single-channel currents were recorded from HEK293T cells bathed in external solution containing 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM glucose, and 10 mM HEPES (pH 7.4). Glass electrodes were pulled from thick-walled borosilicate capillary glass (World Precision Instruments) on a P-2000 Quartz Micropipette Puller (Sutter Instruments) and fire-polished to a resistance of 10–20 MΩ on an MF-830 Micro Forge (Narishige) before use. During recording, 1 mM GABA was present in the electrode solution containing 120 mM NaCl, 5 mM KCl, 10 mM MgCl2, 0.1 mM CaCl2, 10 mM glucose, and 10 mM HEPES (pH 7.4) (18). The electrode potential was held at +80 mV.
Single channel currents were amplified and low-pass filtered at 2 kHz using an Axopatch 200B amplifier, digitized at 20 kHz using Digidata 1322A, and saved using pCLAMP 9 (Axon Instruments). Data were analyzed using TAC 4.2 (Bruxton Corporation). Open and closed events were analyzed using the 50% threshold detection method. All events were carefully checked visually before being accepted. Only patches showing no overlaps of simultaneous openings were accepted. Open and closed time histograms as well as amplitude histograms were generated using TACFit 4.2. Single-channel amplitudes (i) were calculated by fitting all-point histograms with single- or multi-Gaussian curves. The difference between the fitted “closed” and “open” peaks was taken as i. Duration histograms of the mean open time and mean closed time were fitted with exponential components in the form: Σ (Ai/τi) exp(−t/τi), where Ai and τi are the relative area and the time constant of the ith component, respectively, and t is the time. The number of components required to fit the duration histograms was increased until an additional component did not significantly improve the fit (7). The mean open time and the mean closed time were then calculated as follow: Σ Ai τi (7, 17). The open probability (Popen) within clusters of burst openings (10 ms) of single channel activity was calculated as the mean of fractions of time that a channel is open within clusters. Data were expressed as the mean ± S.E.M.
Statistical analysis
Unless otherwise specified, one-way ANOVA with Tukey’s post hoc test was used to determine if there were significant differences among different transfection conditions.
Results
Co-expression of γ2S subunits with α1 and β2 subunits changed endo H digestion patterns of β2, but not α1, subunits
Co-expression of ternary αβγ proteins creates receptors with markedly different functional properties from binary αβ containing receptors. Since oligomerization of subunits to form polymeric proteins can hinder N-glycan processing at certain positions (4) and disrupted glycosylation produces profound changes in receptor expression and function, we wanted to test the hypothesis that inclusion of γ2 subunits into GABAA receptors alters N-glycan processing of α1 and β2 subunits. Toward that end, we compared the endo H digestion patterns of α1 and β2 subunits with co-expression of α1β2 or α1β2γ2S subunits in HEK293T cells (Figure 1). Endo H removes high-mannose, but not complex, N-glycans from glycoproteins, while PNGase F removes all N-glycans from glycoproteins. The endo H resistance of a given N-glycan indicates its exposure to and processing by Golgi-resident enzymes. Relative to the mobility of the same subunits treated with PNGase F, subunits that migrated more slowly and at higher molecular masses following treatment with endo H were classified as “endo H resistant”. By comparing mobility of endo H resistant subunits to those of undigested and PNGase F treated subunits, we evaluated the hindrance of N-glycan processing caused by subunit folding and/or receptor assembly.
Figure 1. Endo H digestion patterns of β2 subunits with α1β2 subunit co-expression were different from those with α1β2γ2S subunit co-expression.
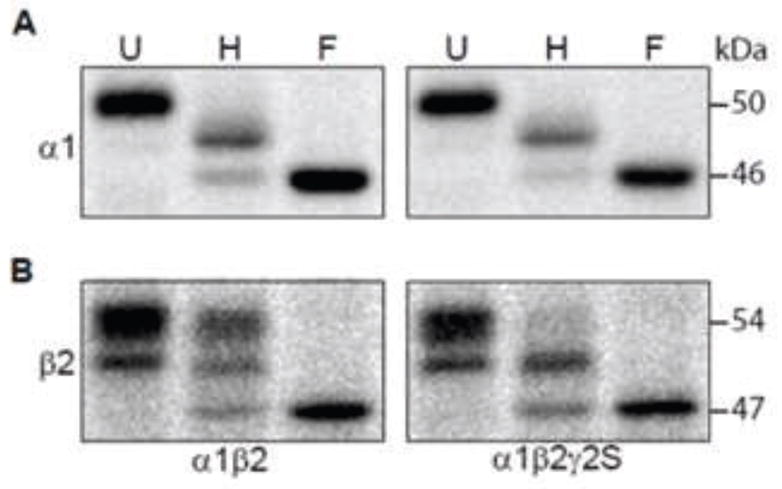
A. RIPA buffer extracted proteins from HEK293T cells with co-expression of binary α1β2 subunits or ternary α1β2γ2S subunits were undigested (U) or digested with endo H (H) or PNGase F (F) endoglycosidase and then were subjected to SDS-PAGE. Resolved proteins were probed with an anti-α1 subunit antibody. After endo H digestion, subunits migrating at the same mass as that of subunits digested with PNGase F (46 kDa) were considered endo H-sensitive, while those migrating more slowly (> 46 kDa) were considered endo H resistant. Combinations of subunit co-expression are indicated along the bottom of the figure. B. The same as A, but proteins were probed with an anti-β2/3 subunit monoclonal antibody. After endo H digestion, subunits migrating more slowly than those at 47 kDa were considered endo H resistant.
The α1 and β2 subunits contain two and three N-linked glycosylation sites, respectively, and all of the sites were glycosylated in HEK293T cells (9, 19). Consistent with previous reports (13, 20), with co-expression of α1β2 or α1β2γ2S subunits, untreated α1 subunits migrated at 51 kDa, and endo H digestion patterns were similar and each showed two bands: one endo H resistant band migrating at 48 kDa (post Golgi processed subunits with one mature and one immature glycan) and one endo H-sensitive band migrating at 46 kDa (ER resident subunits with two immature glycans) (Figure 1A). Since there was no band migrating at 51 kDa following endo H treatment (expected for both sites having mature glycans), processing of one of the two N-glycans of α1 subunits in the Golgi apparatus was hindered in a γ2S subunit-independent manner.
Glycosylation of the three β2 glycosylation is relatively complex. We have shown previously that the glycan occupancy and enzymatic processing may be different at each of the three potential glycosylation sites. More specifically, we found that a significant portion of the subunits do not actually have a glycan at the N32 site. In untreated protein samples from αβ transfected cells, this produced an upper (54 kDa) triply glycosylated band (N32, N104 and N173 sites) and a lower (51 kDa) doubly glycosylated band (N104 and N173 sites). In addition, the N173 site was incompletely processed within the Golgi and was therefore endo H-sensitive. Following endo H treatment, the subunits from cells transfected with binary αβ subunits have 3 bands (Figure 1B). The uppermost band (53 kDa) represents subunits with 2 mature glycans (N32 & N104), the middle band (50 kDa) is due to proteins with only a single, fully processed N104 glycan and the faint lowest band represents subunits with no mature glycans. The endo H digestion pattern of β2 subunits, however, was altered by γ2 subunit co-expression. Similar to binary αβ subunit expressing cells, with α1β2γ2S subunit co-expression the untreated β2 subunits ran as two separate bands at 53 and 51 kDa. However, following endo H treated membrane β2 subunits showed only one endo H resistant band migrating at 51 kDa consistent with only a single, fully processed glycan (Figure 1B). Thus processing of one of the three N-glycans of β2 subunits in the Golgi apparatus (glycan at either the N32 or N104 site) was hindered in a γ2S subunit-dependent manner, demonstrating that co-expression of γ2S subunits altered the endo H digestion pattern of β2, but not α1, subunits.
The endo H digestion pattern of β2 subunits from neonatal γ2+/− knockout mice that have a γ2 subunit deficiency was distinguishable from that of β2 subunits from wild-type mice
With α1β2γ2 subunit co-expression, insufficient γ2 subunit levels resulted in expression of both binary and ternary receptors (21). Consistent with this report, we observed that a small fraction of endo H-digested β2 subunits migrated at 53 kDa when plasmids encoding α1, β2 and γ2S subunits were co-transfected into HEK293T cells at a 1:1:0.5 ratio (data not shown). To confirm the physiological relevance of these findings, we sought to determine if a similar change in glycosylation pattern occurred in the brain. Heterozygous γ2+/− mice have normal numbers of GABA binding sites but decreased numbers of benzodiazepine binding sites, suggesting a relative decrease of γ2 subunit-containing receptors (15). To determine whether the decreased γ2 subunit incorporation in vivo resulted in altered β2 subunit glycosylation, the endo H digestion pattern of β2 subunits from neonatal wild-type mice was compared to that from mutant γ2+/− mice with lower γ2 subunit expression levels (15). However, given that α3, but not α1, subunits are mainly expressed in cortex and thalamus at early developmental stages (22), we first examined whether γ2S subunits also led to disappearance of the 53 kDa fraction of endo H-digested β2 subunits with α3β2γ2S subunit co-expression in HEK293T cells (Supplemental Figure 1). When co-expressed with α3 subunits, undigested β2 subunits showed the same mobility (migrating at 54 and 51 kDa) as when they were co-expressed with α1 subunits. Similarly, endo → surface β2 subunits with α3β2 subunit co-expression migrated as two bands at 53 and 50 kDa, while those with α3β2γ2S subunit co-expression migrated at 51 and 47 kDa (Supplemental Figure 1). Thus, the γ2S subunit-mediated hindrance of β2 subunit N-glycan processing occurred in both α1 and α3 subunit-containing GABAA receptors.
Using a monoclonal antibody against both β2 and β3 subunits, β2 and β3 subunits and their associated proteins were then immunoprecipitated from wild-type or mutant mice, and the levels and mobilities of β2 and γ2 subunits were evaluated by a combination of SDS-PAGE and Western blots using polyclonal antibodies against β2 or γ2 subunits (Figure 2). The β2 subunits purified from neonatal mice mainly migrated at 52 and 50 kDa, and the γ2 subunits mainly migrated at 47 and 43 kDa (Figure 2A). Consistent with a previous report (15), heterozygous GABRG2 deficiency led to about a 23% reduction of γ2 subunits associated with β2/3 subunits relative to the wild-type condition (Figure 2B). In proteins isolated from wild-type mouse brain, endo H treatment shifted neuronal β2 subunits from 52 and 50 kDa to 49 and 47 kDa (Figure 2C). The molecular mass shifts were similar to those of endo H-digested β2 subunits with α1β2γ2S subunit co-expression in HEK293T cells. In addition, β2 subunits from heterozygous GABRG2 knockout mice had an appreciable endo H resistant third band at 52 kDa, which was at the expense of reduction of the endo H-sensitive band at 47 kDa (Figure 2C, D). These findings suggested that the decrease of γ2 subunit level could be quantified by the increase of the 52 kDa fraction of endo H-digested β2 subunits in the brain. It is worth mentioning that mobilities of neuronal β2 subunits were slightly difference from those of subunits purified from HEK293T cells. Further investigation is required to determine whether the difference was due to absent glycosylation at N173 or to different processing of N-glycans attached to the three glycosylation sites.
Figure 2. The endo H digestion pattern of β2 subunits from heterozygous GABRG2 -knockout mice was different from that of wild-type mice.

A. Whole brains without cerebella of wild-type (γ2+/+) or GABRG2 heterozygous-knockout (γ2+/−) mice were extracted with RIPA buffer. GABAA receptor complexes were immunoprecipitated using the monoclonal anti-β2/3 subunit antibody. Purified protein complexes were resolved as duplicates using SDS-PAGE and subjected to western blotting. Each of the duplicates was probed by anti-β2 subunit cytoplasmic loop antibodies (upper panel) or by anti-γ2 subunit antibodies (lower panel). B. The integrated density volumes (IDV; pixel intensity × mm2) of β2 or γ2 subunits from A were calculated and normalized to the corresponding subunit IDV of an arbitrarily chosen wild-type condition The ratio of γ2 subunit IDV versus β2 subunit IDV for each condition was then calculated and then normalized to the γ2:β2 ratio of protein from wild-type animals in each experiment. The plot shows the pooled ratios of the γ2+/+ (grey bar) and γ2+/− groups (checkered bar). C. The GABAA receptor protein complexes from wild-type (upper panel; γ2+/+) or γ2 subunit heterozygous knockout (lower panel; γ2+/−) mice immunoprecipitated by the monoclonal anti-β2/3 subunit antibody were undigested (U) or digested with endo H (H) or PNGase F (F) endoglycosidase. After being resolved by SDS-PAGE, β2 subunits were stained by anti-β2 subunit cytoplasmic loop antibodies. D. IDVs of endo H-digested β2 subunits migrating at 52, 49 and 47 from γ2+/+ (grey bars) or γ2+/− (checkered bars) mice were calculated. The IDV of each band was expressed as % of the sum of the three bands. Data were presented as mean ± S.D. *, ** and *** indicate p < 0.05, p < 0.01 and p < 0.001 relative to wild-type conditions.
Co-expression of γ2S subunits caused a fraction of surface β2 subunits to be endo H-sensitive
The endo H digestion of whole cell extracts of HEK293T cells or neonatal mouse brain suggested that co-assembly with γ2 subunits hindered β2 subunit N-glycans from acquiring endo H resistance at some glycosylation site(s). We sought to determine whether a small fraction of the β2 subunits remained “endo H-sensitive” despite being trafficked beyond the Golgi apparatus. Of course, another source of endo H-sensitive proteins are the recently translated intracellular proteins that have not had sufficient time to complete processing and trafficking through the ER and Golgi apparatus. To help differentiate these two sources of endo H-sensitive proteins, we isolated GABAA receptors from the cell surface and subjected them to endo H digestion (Figure 3).
Figure 3. A fraction of N-glycans attached to the N104 sites of surface β2 subunits with co-expression of α1 and γ2S subunits was endo H-sensitive.
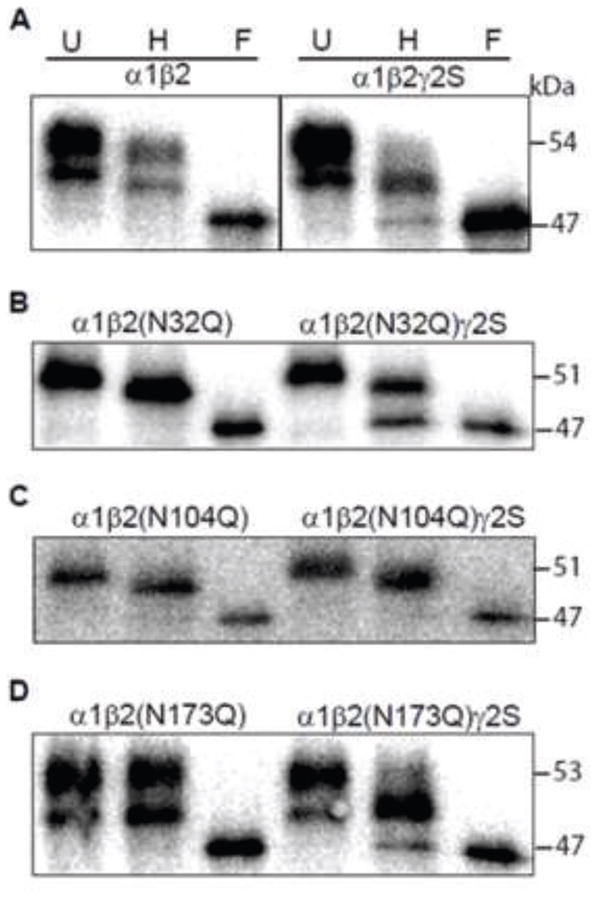
A. Surface proteins of HEK cells with binary subunit co-expression (control α1 and β2 subunits; left panel) or ternary subunit co-expression (control α1, β2 and γ2S subunits; right panel) were biotinylated and pulled down with streptavidin beads. The purified surface proteins were undigested (U) or digested with endo H (H) or PNGase F (F) endoglycosidase. Digested proteins were resolved by SDS-PAGE and probed with anti-β2 subunit cytoplasmic loop antibodies. B–D. The same as in A, but the β2 subunits contained an Asn to Gln mutation of N32 (B), N104 (C) or N173 (D) glycosylation site. The combinations of subunit co-expression are indicated along the top of the panels.
Previously we demonstrated that co-expression of α1β2 subunits produces surface β2 subunits with an immature glycan at N173 and that a portion of the surface β2 subunits also have no glycan at all at N32. This results in two bands in untreated samples, and a small downward shift of both bands following endo H digestion (9). A similar pattern was found in the present study for β2 subunits assembled in surface α1β2 receptors (Figure 3A). It is worth noting that, unlike with whole cell lysates (Figure 1), while both surface β2 subunit bands were shifted to lower masses following endo H treatment (to 53 and 50 kDa), they remained higher than those of the PNGase F treated β2 subunits (47 kDa). These results suggested that surface β2 subunit N32 glycans (only in the 53 kDa fraction) and N104 glycans (in the 53 and 50 kDa fractions) underwent complete processing within the Golgi apparatus when β2 subunits were co-expressed in surface binary α1β2 receptors (Figure 3A, left panel). On the other hand, in surface ternary α1β2γ2S receptors, the endo H-digested β2 subunits migrated as two bands at 50 and 47 kDa (Figure 3A, right panel). The presence of the endo H-sensitive 47 kDa band suggested that co-expression of γ2 subunits hindered processing of N104 N-glycans because if co-expression of γ2S subunits hindered processing of only those N-glycans attached to the N32 glycosylation sites, the endo H-digested surface β2 subunits would migrate as a single band at 50 kDa (see above).
An intact N104 glycosylation site was required for γ2S subunit-dependent hindrance of β2 subunit N-glycan processing
To further determine the site at which N-glycans were affected by incorporation of γ2S subunits, we compared endo H digestion patterns of β2 subunits containing an Asn to Gln mutation at one of the three N-linked glycosylation sites (i.e. N32Q, N104Q or N173Q) in surface receptors from cells co-expressing binary α1β2 or ternary α1β2γ2S subunits (Figure 3B–D). Mutating the N32 glycosylation site with either α1β2(N32Q) or α1β2(N32Q)γ2S subunit co-expression converted the pair of bands seen in untreated surface β2(N32Q) subunits to a single band migrating at 51 kDa, again suggesting that N32 was glycosylated inefficiently (Figure 3B). Moreover, in surface binary α1β2(N32Q) receptors, this band was shifted to 50 kDa following endo H treatment. In contrast, in cells co-expressing α1β2(N32Q)γ2S subunits, endo H treatment of the β2(N32Q) subunits produced two bands migrating at 50 and 47 kDa. Taking into account that N173 glycans remained endo H-sensitive, these findings suggested that co-expression of α1β2(N32Q)γ2S subunits produced a mixed population of β2(N32Q) subunits in which some N104 glycans were processed within the Golgi apparatus, while others were not.
In support of the hypothesis that an intact N104 site was required for the γ2S subunit induced differences, endo H treated surface β2(N104Q) subunits migrated primarily as one 50 kDa band when co-expressed either with both α1 and γ2S subunits or with α1 subunits alone (Figure 3C). Of note, the presence of a single 50 kDa band also suggested that the N104 mutation facilitated glycosylation at N32, because any β2(N104Q) subunits lacking N32 glycans should migrate at 48 kDa. The reason for this is unclear, but it may be related to the fact that subunits lacking glycans at both N104 and N32 glycosylation sites were rapidly degraded and rarely assembled into receptors (8). Furthermore, the 1 kDa shift seen in β2(N104Q) subunits after endo H digestion suggested that N-glycan processing at one of the remaining glycosylation sites, presumably N173, was hindered with both binary α1β2(N104Q) and ternary α1β2(N104Q)γ2S subunit co-expression.
Finally, mutation of the N173 site did not block the γ2S subunit effect (Figure 3D). With co-expression of binary α1β2(N173Q) subunits, undigested surface β2(N173Q) subunits migrated as two bands at 53 and 50 kDa, and this pattern was unaffected by endo H treatment. In contrast, with co-expression of ternary α1β2(N173Q)γ2S subunits, while surface β2(N173Q) subunits also migrated as two bands at 53 and 50 kDa, endo H treatment shifted the two bands to 51 and 47 kDa. The presence of the 47 kDa band in endo H-treated β2(N173Q) subunits from surface receptors from α1β2(N173Q)γ2S subunit transfected cells suggested the existence of a population of β2(N173Q) subunits in which the N32 sites were not glycosylated and N104 N-glycans were incompletely processed within the ER and/or Golgi apparatus. Taken together, these results suggested that co-expression of γ2S subunits did not change endo H sensitivity of surface β2 subunit N32 and N173 N-glycans. Rather, γ2S subunit co-expression hindered processing of the glycan group attached to the N104 site of β2 subunits.
It is worth noting that the enrichment of surface proteins from HEK293T cells with binary α1β2 subunit co-expression was done in parallel with that from cells with ternary α1β2γ2S subunit co-expression. Since there was no apparent fraction of surface β2(N32Q) or β2(N173Q) subunits with binary subunit co-expression that had a band migrating at 47 kDa, the observed endo H-sensitive fraction of the mutant surface β2 subunits with ternary subunit co-expression was not likely due to the labeling of intracellular subunits.
Intact β2 subunit glycosylation sites were important for surface expression of γ2S subunits
Previously we showed that glycosylation of N32 was not required for α1β2 receptor surface expression (9). However, it is possible that glycosylation of N32 was important for surface expression of α1β2γ2S receptors. This hypothesis is supported by preliminary molecular modeling that predicts that β2 subunit residues 30–31 interact strongly with γ2 subunit residues 82–90, which are crucial for γ-β intersubunit communication (11, 12) (data not shown). Consequently, there may be appreciable surface expression of binary α1β2(N32Q) receptors in HEK293T cells transfected with α1β2(N32Q)γ2S subunits. The resultant mixture of binary α1β2(N32Q) and ternary α1β2(N32Q)γ2S receptors may explain why the processing of N104 N-glycans of surface β2(N32Q) subunits was not fully blocked by γ2S subunits. To examine this hypothesis, we used flow cytometry of cells transfected with ternary α1β2γ2S subunits to quantify surface levels of individual mutant β2 subunits containing an Asn to Gln mutation of one of the three N-linked glycosylation sites and compared these with “control” β2 subunits (Figure 4). We and others have found that flow cytometry with unpermeabilized cells is an efficient way to selectively measure transfected subunit surface expression, without contamination from the intracellular proteins (13, 23). To monitor surface levels of individual subunits, FLAG epitopes were introduced into α1, β2 and γ2S subunits and only one of the three subunits contained a FLAG tag when co-expressed. Thus, for each experimental condition, such as to evaluate effects of the N32Q mutation, three experimental conditions were used, i.e. α1FLAGβ2(N32Q)γ2S, α1β2(N32Q)FLAGγ2S and α1β2(N32Q)γ2SFLAG subunit co-transfection. Surface levels of the FLAG-tagged partnering α1 and γ2S subunits and FLAG-tagged mutant β2 subunits were then expressed as percentages of their corresponding “control” subunit levels, i.e. α1FLAG subunits with α1FLAGβ2γ2S, β2FLAG subunits with α1β2FLAGγ2S, and γ2FLAG subunits with α1β2γ2SFLAG subunit co-expression (Figure 4B).
Figure 4. Glycosylation site mutations of β2 subunits altered surface levels of partnering subunits.

A. Flow cytometry was used to determine surface expression levels of ternary α1β2γ2S GABAA receptors containing mutated, glycosylation-deficient β2 subunits. Surface levels of FLAG-tagged subunits were analyzed using PE-conjugated monoclonal anti-FLAG antibody (M2 clone) and plotted as fluorescence intensity histograms. The x-axis indicates the fluorescence intensity in arbitrary units (note the log scale), and the y-axis indicates the percentage of the maximum cell count. Representative distributions obtained from mock transfected cells (unfilled histograms) were overlaid with each experimental distribution (filled histograms). Upper panels indicate FLAG intensities on the cell surface from FLAG-tagged α1 subunits (α1FLAG) co-expressed with non-tagged control or mutant β2 subunits, and with non-tagged γ2S subunits. Middle panels indicate FLAG intensities on the cell surface from FLAG-tagged β2 subunits (β2FLAG), with or without glycosylation site mutations, co-expressed with non-tagged α1 and γ2S subunits. Lower panels indicate FLAG intensities on the cell surface from FLAG-tagged γ2S subunits (γ2SFLAG) co-expressed with non-tagged α1 subunits and with non-tagged control or mutant β2 subunits. B. Surface α1FLAG (black bars), β2FLAG (grey bars) and γ2SFLAG (white bars) subunit levels were quantified in each condition and expressed as percentages of cell surface levels of control subunits. The glycosylation site mutations are indicated along the bottom of the figure. Data are presented as mean ± S.D. *, **, and *** indicate p < 0.05, p < 0.01 and p < 0.001, respectively, relative to control subunit co-expression.
We previously found that the N104Q mutation severely suppressed the α1 and β2 subunit levels on the surface when expressed as binary receptors. The N104Q mutation in ternary receptors caused a similar, albeit less dramatic, reduction to 82% and 59% of corresponding control subunit surface levels of partnering α1 and γ2S subunits. The N32Q and N173Q β2 subunit mutations decreased partnering γ2S subunit surface levels to 84% and 76% of control subunit surface levels, respectively (Figure 4B). In contrast, the N32Q mutation significantly increased surface levels of partnering α1 subunits to 118% of control levels (Figure 4B). Though not statistically significant, N32Q, N104Q and N173Q mutation caused corresponding mutant β2 subunits to 120%, 82% and 120% of control levels. Together, these data suggested that binary α1β2(N32Q) receptors were expressed on the cell surface at the expense of ternary α1β2(N32Q)γ2S receptor surface expression when the N32 glycosylation sites were mutated. Further, the presence of surface binary α1β2(N32Q) receptors may explain the significant portion of the β2(N32Q) subunits that had endo H resistant N104 glycans with α1β2(N32Q)γ2S subunit co-expression.
N104 glycosylation regulated gating of α1β2γ2L channels
We next explored the functional consequences of blocking individual glycosylation sites using electrophysiological recordings. For these experiments we used the long form of the γ2 subunit (γ2L) to allow us to compare the current findings to previous work from our laboratory. Given that γ2S and γ2L subunits only differ by eight amino acids in the TM3-4 cytoplasmic loop, it is not surprising that we found that both isoforms had identical effects on glycosylation of amino acids at the β2 extracellular N-terminus (data not shown). As discussed above, co-expression of γ2 subunits appeared to affect N-glycan processing most prominently at the N104 site. Based on the homology model of the GABAA receptor β2 subunit, the N32 and N104 glycosylation sites are spatially adjacent and located on the top of the complementary side of the extracellular domain facing the γ-β interface (10). This region is homologous to the β-α interface where GABA binding occurs. A ring of similar intersubunit interfaces around the pentamer (i.e. at the β-α, α-γ and γ-β interfaces) have been hypothesized to be involved in transducing GABA binding at the β-α interface into conformational changes of the whole pentameric receptor complex. For example, the epilepsy associated γ2(R82Q) mutation lies within the γ2 subunit sequence that is homologous to a portion of the GABA binding pocket of the β subunit at the β-α interface. While the γ2(R82Q) subunit mutation lies at the γ-β interface and therefore would not be predicted to directly mediate GABA-binding, this mutation has been shown to severely disrupt GABA evoked channel activation by apparently long-distance conformational changes that affect GABA binding (12). We hypothesized that lacking the large glycan group at N32 or N104 could very well also severely impair signal transduction at GABA receptors. To address this possibility, we recorded steady-state single channel currents evoked by 1 mM GABA from cells expressing α1β2γ2L, α1β2(N32Q)γ2L, α1β2(N104Q)γ2L or α1β2(N173Q)γ2L subunits (Figure 5A). The amplitude of single channel currents recorded from cells expressing α1β2(N104Q)γ2L subunits, 1.55 pA, was significantly smaller than those expressing control α1β2γ2L and mutant α1β2(N32Q)γ2L or α1β2(N173Q)γ2L subunits, which were 2.32 pA, 2.15 and 2.14 pA, respectively (Figure 5B, Table 1). The N104Q mutation also decreased the mean open time of single channel openings from 5.3 ms to 2.4 ms (Figure 5C). To better understand how the N104Q mutation decreased the mean open time, open duration histograms of single channel currents recorded from cells expressing α1β2γ2L or α1β2(N104Q)γ2L subunits were compared (Table 1). Each open duration histogram was fitted best with a sum of three exponential functions. Analysis by ANOVA with a Dunnett’s post-test revealed that the N104Q mutation decreased the time constant of the intermediate open states (τO2) and the relative contribution of the long open state (AO3). Changes of exponential components suggested that the N104Q mutation decreased the mean open time by destabilizing the intermediate open states and shifted the proportion of long open state to the other two states. Furthermore, previous work has shown that activation of ternary α1β3γ2L receptors tends to produce clusters with multiple openings (7). Since analysis of individual openings could therefore miss important features of channel function, we next determined whether the overall mean open probability was affected by blocking glycosylation of individual glycosylation sites. The open probability of single channel currents recorded from cells expressing control α1β2γ2L subunits was 0.6, and the N32Q, N104Q and N173Q mutations each significantly decreased it to 0.4 (Figure 5D). Thus, single channel analyses of ternary receptors revealed that each of the three β2 glycans plays a pivotal role in channel gating. Importantly, abolishing glycosylation of the N104 residue distinctively impaired both gating efficiency and amplitude of individual single channel openings.
Figure 5. Disrupted glycosylation at the N104 residue channel conductance, mean open time, and open probability of α1β2γ2L receptor single channels.

A. Representative steady-state single-channel currents evoked by 1 mM GABA were obtained from cells co-expressing α1β2γ2L with either control β2 or one of the glycosylation deficient β2(NxQ) mutants. Each of the traces was a continuous 400 ms recording. Cells were voltage-clamped at +80 mV. Openings were downward. B–D. Amplitudes of single channel openings (B), means of the open durations (C), and open probabilities of cluster openings (D) of single channels recorded from these cells were plotted. Data are presented as mean ± S.E. * and ** indicated p < 0.05 and p < 0.01, respectively, relative to control α1β2γ2L receptor currents.
Table 1.
Effects of β2 subunit glycosylation-site mutations on α1β2γ2L receptor channel function
| control β2 | β2(N32Q) | β2(N104Q) | β2(N173Q) | |
|---|---|---|---|---|
| Whole-cell currentsa | ||||
| 1mM GABA | ||||
| Imax[1mM] (pA) | 3668 ± 546 | 3257 ± 277 | 1996 ± 640 | 1921 ± 583 |
| rise time (ms) | 1.8 ± 0.2 | 2.0 ± 0.4 | 2.1 ± 0.5 | 3.5 ± 0.5** |
| τdeactivation | 119 ± 20 | 105 ± 16 | 73 ± 23 | 46 ± 5* |
| 10μM GABA | ||||
| Imax[10μM] (%of Imax[1mM]) | 24.0 ± 3.3 | 10.0 ± 1.8*** | 2.7 ± 1.5***,† | 3.3 ± 1.2***, † |
| Single-channel currentsb | ||||
| i (pA) | 2.21 ± 0.10 | 2.15 ± 0.06 | 1.55 ± 0.15**, †† | 2.14 ± 0.10 |
| open probability | 0.59 ± 0.04 | 0.40 ± 0.04** | 0.40 ± 0.04** | 0.41 ± 0.07* |
| mean open time (ms) | 5.29 ± 1.03 | 3.31 ± 0.50 | 2.37 ± 0.29* | 4.03 ± 0.59 |
| τO1 (ms) | 0.92 ± 0.03 | 0.87 ± 0.07 | 0.79 ± 0.04‡ | 0.84 ± 0.05 |
| τO2 (ms) | 4.38 ± 0.47 | 2.66 ± 0.52 | 2.08 ± 0.22‡‡ | 3.89 ± 0.73 |
| τO3 (ms) | 6.55 ± 0.46 | 7.00 ± 0.84 | 5.28 ± 0.74 | 6.20 ± 0.80 |
| AO1 (%) | 11 ± 2 | 22 ± 5 | 13 ± 1 | 18 ± 2 |
| AO2 (%) | 73 ± 5 | 68 ± 5 | 85 ± 1 | 67 ± 1 |
| AO3 (%) | 15 ± 3 | 11 ± 2 | 1 ± 1‡ | 14 ± 1 |
Kinetic parameters were obtained from macroscopic currents recorded from lifted cells, which were voltage-clamped at −20 mV and exposed with 1 mM GABA or 10 μM GABA for 4 seconds. Imax, Iresidual and τdeactivation refer to peak current amplitudes, residual current amplitudes at the end of GABA applications and weighted deactivation time constants, respectively. Values reported are mean ± S.E.
Kinetic parameters of single-channel currents in attached-cell patches held at +80 mM with 1 mM GABA in the glass-electrodes were obtained. The i refers to current amplitudes of single-channel currents; the τs and As refer to the time constants and fractions of the three exponential components (O1, O2 and O3), which best represent the distributions of the single channel openings. Values reported are mean ± S.E.
indicate p < 0.05, p < 0.01 and p < 0.001, respectively, relative to control conditions by ANOVA with Tukey’s post hoc tests.
indicate p < 0.05 and p < 0.01, respectively, relative to α1β2(N32Q)γ2L receptor currents by ANOVA with Tukey’s post hoc tests.
indicate p < 0.05 and p < 0.01, respectively, relative to control parameters by t-tests.
Since the flow cytometry results suggested that blockade of one or more glycosylation sites might cause increased surface expression of binary αβ receptors, we specifically compared the current single channel current results (α1β2(NxQ)γ2L) with previously acquired data from cells transfected with only α1 and glycosylation deficient β2 subunits (α1β2(NxQ))(9). Typically, binary α1β2 receptors have small single channel conductances, and short mean open times comprised primarily of brief (O1) openings. We found previously that mutant binary α1β2(NxQ) receptors had similar properties, but even briefer mean open times. In contrast, ternary α1β2γ2L receptors had much larger single channel conductances, and longer mean open times with the vast majority of openings occurring as clusters of longer open events (O2 & O3). We found that the single channel properties of ternary α1β2(N32Q)γ2L and α1β2(N173Q)γ2L receptors could easily be distinguished from binary α1β2(N32Q) and α1β2(N173Q) receptors, respectively. In each instance the single channel properties recorded from cells co-transfected with mutant β2(N32Q) or β2(N173Q) and α1γ2L subunits showed more than a two-fold greater current amplitude and mean open time than recorded from cells expressing α1β2(NxQ) subunits. Moreover, while essentially all of the openings of single channel currents recorded from cells expressing binary α1β2(N32Q) or α1β2(N173Q) subunits were due to brief (O1) events, the open time distributions recorded from ternary α1β2(N32Q)γ2L or α1β2(N173Q)γ2L subunits were dominated by the longer O2 and O3 events, with only a very small fraction of brief O1 events. In summary, while the protein expression results indicate a possible mixture of binary α1β2(N32Q) and ternary α1β2(N32Q)γ receptors expressed on the cell surface, the single channel data suggest that any surface binary αβ receptors make only a trivial contribution to the overall GABA evoked currents. However, it should also be noted that the steady-state recordings reported here may underestimate the expression of highly desensitizing binary αβ containing receptors (24).
Glycosylation of β2 subunits regulated GABA potency of α1β2γ2L receptors
We hypothesized that altering the glycan conformation at N32 and/or N104 might disrupt the normal subunit interactions responsible for coupling agonist binding to channel activation. While our single channel results provide insight into the steady state activation by saturating concentrations of GABA, altering the protein conformations at the γ-β subunit interface may also cause changes in the rates and/or GABA concentration-dependence of receptor activation. To test this hypothesis, we recorded the whole cell currents evoked by 1 mM (a near saturating GABA concentration for α1β2γ2 receptors) or 10 μM (near the GABA EC25 for α1β2γ2 receptors) GABA application to lifted HEK293T cells co-expressing α1β2γ2L, α1β2(N32Q)γ2L, α1β2(N104Q)γ2L or α1β2(N173Q)γ2L subunits (Figure 6). Unlike our previous findings with binary αβ receptors, we found that elimination of any single β2 subunit glycosylation site did not cause a robust reduction in α1β2γ2L current amplitudes evoked by 1 mM GABA, although there was a non-significant trend toward current amplitude reduction with β2(N104Q) and β2(N173Q) mutations (Figure 6A and D). On the other hand, peak amplitudes of 10 μM GABA currents normalized to the 1 mM current amplitudes recorded with expression of α1β2(N32Q)γ2L, α1β2(N104Q)γ2L or α1β2(N173Q)γ2L subunits were 10%, 3% and 3% of 1 mM currents, respectively, and were smaller than those recorded from cells expressing control α1β2γ2L subunits, which were 24% of 1 mM currents (Figure 6A, D). These results suggest that removal of any of the three glycosylation sites caused less efficient coupling between agonist binding and channel opening.
Figure 6. Glycosylation site mutations decreased potency of 10 μM GABA and changed macroscopic current kinetics.

A. Representative currents were obtained from cells co-expressing α1β2γ2L, α1β2(N32Q)γ2L, α1β2(N104Q)γ2L, or α1β2(N173Q)γ2L subunits in response to rapid, 4 sec application of 1 mM (black traces) or 10 μM (grey traces) GABA. The duration of GABA application was indicated by a black bar above the current traces. Cells were clamped at −20 mV. B. The GABA-evoked currents (1 mM) recorded from cells co-expressing α1β2γ2L (solid line) α1β2(N173Q)γ2L (dashed line) subunits were scaled to have the same peak size. The rise time of currents recorded from α1β2(N173Q)γ2L subunits was slower than that of control currents. C. The same as B, but the 1 mM GABA-evoked currents (1 mM) recorded from cells co-expressing α1β2γ2L or α1β2(N173Q)γ2L subunits were scaled to have the same size of residual current amplitudes at the end of a 4 s GABA application. The currents recorded from α1β2(N173Q)γ2L subunits reached the baseline faster than those recorded from α1β2γ2L subunits. D. Means of peak current amplitudes (Imax) recorded from cells co-expressing α1β2γ2L (black bars), α1β2(N32Q)γ2L (checkered bars), α1β2(N104Q)γ2L (white bars), or α1β2(N173Q)γ2L (hatched bars) subunits evoked by 1 mM GABA or by 10 μM GABA (expressed as % of Imax) were plotted. E, Rise time and weighted deactivation time constants of currents recorded from α1β2γ2L (black bars), α1β2(N32Q)γ2L (checkered bars), α1β2(N104Q)γ2L (white bars), or α1β2(N173Q)γ2L (hatched bars) subunits were plotted. Data are presented as mean ± S.E. *, ** and *** indicated p < 0.05, p < 0.01 and p < 0.001, respectively, relative to control α1β2γ2L receptor currents.
To explore further functional differences, we evaluated rise time, desensitization and deactivation rates of macroscopic currents evoked by 1 mM GABA application. In addition to the disrupted gating described above, we also found that the rise time of currents recorded from co-expressed α1β2(N173Q)γ2L subunits was 3.5 ms, which was significantly slower than those from α1β2γ2L, α1β2(N32Q)γ2L and α1β2(N104Q)γ2L subunits (1.8 ms, 2.0 ms and 2.1 ms, respectively) (Figure 6B, E). The extents of desensitization were not different from controls, but the deactivation time of currents recorded from co-expressed α1β2(N173Q)γ2L subunits was significantly faster than that of the wild-type currents (Figure 6C, E). The weighted deactivation time constants of currents recorded from co-expressed α1β2γ2L, α1β2(N32Q)γ2L, α1β2(N104Q)γ2L and α1β2(N173Q)γ2L subunits were 119 ms, 105 ms, 73 ms, and 46 ms, respectively. Collectively, while the β2 subunit N104Q mutation produced the greatest disruption of agonist-evoked activation, removal any of the three β2 subunit glycosylation sites impaired GABA potency and reduced single channel mean open time. In addition, the β2 subunit N173Q mutation resulted in currents with slower rise times and faster deactivation than controls. The molecular mechanism for these changes remains undetermined. However, the combination of reduced potency, slowed activation and faster deactivation raise the possibility that mutating N173 caused a long-distance change in GABA binding sites at β-α interface. While the β2 subunit N32Q mutation produced more modest changes, it is worth remembering that a significant fraction of wild-type α1β2γ2 receptors also include β2 subunits lacking N32 glycans, and therefore this analysis of mutant N32Q subunits likely underestimates the importance of N32 glycosylation on function of α1β2γ2 receptors.
Discussion
β2 subunit N-glycosylation affected GABAA receptor assembly and function
In this study, we identified complex interactions among β2 subunit N-glycosylation, γ2 subunit incorporation, and α1β2γ2 GABAA receptor function. Inactivating any of the three glycosylation sites reduced γ2 subunit incorporation, GABA potency and single channel open probability. However each glycan also had unique effects on GABAA receptor expression and function. In the ER lumen, standard “core” glycans are attached to the side chain nitrogen of asparagines that are in the glycosylation consensus sequon, Asn-Xaa-Ser/Thr (Xaa not Pro) (Bause, 1983; Imperiali and Shannon, 1991). However, sequons with threonine residues are more efficiently glycosylated than sequons containing serine residues (Kaplan et al. 1987), and N32 is in a serine-containing sequon. Thus, similar to α1β2 receptors, with expression of α1β2γ2 receptors the β2 subunit N32 glycosylation site was inefficiently glycosylated, but all glycans occupying that site acquired endo H resistance. The N32 residue lies in a short segment of the β2 subunit that participates in formation of the γ-β subunit interface with residues 83–90 of the γ2 subunit (11). Removal of this glycan reduced GABA potency and decreased single channel open probability. Some of these changes, along with the surface expression studies, are consistent with increased expression of binary α1β2 receptors on the cell surface. Surface expression of binary αβ receptors has been well-substantiated in recombinant expression systems, and may even play a modest role in conveying tonic inhibition in neurons(25). Moreover, the single channel open time kinetics argue that binary α1β2(N32Q) receptors were only a minor component in the overall GABA-evoked charge transfer in α1β2(N32Q)γ2 transfected cells. The high-mannose (immature) glycan at the N173 site also lies in a crucial segment of the N-terminus, the “signature cys loop” (loop7). Very close by within the same cys-loop is residue D170, which forms an intramolecular salt bridge with residue K239 in the β2 pre-M1 segment (26). This interaction is critical for coupling GABA binding to channel opening. Consequently, it is not surprising that mutation of the N173 glycosylation site had relatively modest effects on subunit expression and steady state single channel properties, but greatly disturbed the kinetic properties of receptor activation/deactivation. Activation and deactivation of GABAA receptors can shape the amplitude and duration of IPSCs, respectively, so the glycan at N173 could affect both amplitude and duration of IPSCs. In this way, glycosylation of N173 might play a role in net charge transfer of IPSCs and strength of neuronal transmission.
Somewhat unexpected was the finding that the N104Q glycosylation-deficient mutation had the greatest impact on receptor expression and function in both binary (9, 26) and ternary GABAA receptors. This particular residue does not reside in, or appear to directly interact with any well-defined GABA binding/transduction sequence. Nonetheless, loss of glycosylation at this site impaired overall protein expression and caused reduced single channel conductance, open probability and mean open time. We speculated previously that the N104 glycans of β2 subunits in binary αβ receptors may interact with adjacent α1 and β2 subunits (for the potential β2-β2-α1-β2-α1 counterclockwise stoichiometry when viewed from the synaptic cleft) and help form the α-β and β-β subunit interfaces. Given the current results that co-expression of γ2 subunits prevented N104 glycans from acquiring endo H resistance, we further hypothesize that the incorporation of γ2 subunits and the subsequent conformational change of GABAA receptors impaired complete enzymatic processing of N104 glycans.
Preliminary molecular modeling of α1β2γ2 receptor (the γ2-β2-α1-β2-α1 counterclockwise stoichiometry) showed that the N104 site on one of the β2 subunit proteins is immediately facing the adjacent γ subunit’s C loop (loop 10). There are a number of hydrophilic amino acids in this region, and one might hypothesize that the glycan moiety could form multiple hydrogen bonds, thereby serving as a structural interface between subunits. One potential mechanism is that the glycan itself may help form the γ-β and α-β subunit interface, perhaps by providing a series of hydrogen bonds between the β2 subunit, and some of the nearby γ2 sequences, such as those in the loop equivalent to the GABA-binding C loop (loop10). This possibility is supported by our finding that γ2 subunit levels were inversely correlated with β2 subunit N104 glycan maturity in both recombinant and native GABAA receptors; that is, γ2 subunit incorporation into the receptor pentamer appeared to impair processing of the N104 glycan. While speculative, the fact that inclusion of γ2 subunits blocks access to and/or complete processing by Golgi resident glycosidases is certainly consistent the N104 glycan being buried in the intersubunit interface.
β2 subunit N104 N-glycan processing was impaired specifically by γ2 subunit incorporation
It is generally accepted that most GABAA receptors contain at least two α and two β subunits and that the fifth position varies among receptor isoforms (13, 27). Thus, the fifth position may be occupied by an α or β subunit in binary α1β2 receptors and by a γ subunit in ternary α1β2γ2 receptors. However, other ternary receptors exist, most notably αβδ receptors. Interestingly, similar to co-expression of α1β2 subunits, co-expression of α1β2δ subunits in HEK293T cells yielded surface β2 subunits that mainly migrated at 53 and 50 kDa after endo H digestion (data not shown). These preliminary data suggested that processing of β2 subunit N104 N-glycans was hindered specifically by γ2 subunit co-expression and not simply by any non-β subunit occupying the fifth subunit position.
Incorporation of the γ2 subunit into α1β2γ2S receptors can be quantified using the relative expression of the endo H resistant 52 kDa fraction of neuronal β2 subunit protein
The distinct endo H digestion pattern of β2 subunits with α1β2γ2S subunit co-expression is potentially useful to probe the relative decrease of ternary α1/3β2γ2 receptor assembly in vivo. With a decrease of approximately 20% of γ2 subunit-containing receptors, we were able to observe an increase in the 52 kDa fraction of endo H-digested β2 subunits in neonatal mice (Figure 2). Several mutations in γ2 subunits associated with genetic epilepsy have been demonstrated to impair assembly of γ2 subunit-containing receptors (28). For instance, a mouse model confirmed that the γ2 subunit R82Q mutation decreased the surface level of γ2 subunits but left the surface level of α1 subunits unchanged (29). It is expected that the 52 kDa fraction of endo H-digested β2 subunits of the γ2(R82Q) mice should also be increased.
Because GABAA receptors are highly heterogeneous in brain (30), further investigation is necessary to determine whether the increase of the 52 kDa fraction of endo H-digested β2 subunits purified from γ2 subunit-deficient mouse brain was due to assembly of binary receptors or due to the compensatory expression of other ternary receptors, such as αβ2δ receptors. Additionally, although α3 subunits could replace α1 subunits to support the γ2 subunit-mediated hindrance of N104 N-glycan processing, whether other α subunit subtypes give similar results also will require further investigation.
The γ2 subunit-induced alterations in β2 subunit glycosylation were independent of subunit position
A question of interest regarding heterogeneity of β2 subunit N-linked glycosylation is whether hindrance of N104 N-glycan processing by γ2 subunits occurs on both of the two β2 subunits of α1β2γ2 receptors. With co-expression of α1β2γ2S subunits, endo H-digested surface β2 subunits mainly migrated at 47 and 51 kDa (Figure 3A, right panel). The lack of a prominent third band migrating at 53 kDa suggested that the majority of the surface β2 subunits had processed N-glycans attached to either N32 or N104, but not both sites. It is worth noting that the ratio of the 54 and 51 kDa fractions of undigested β2 subunits of the surface α1β2γ2 receptors subunits (Figure 3A, right panel, lane 1) was not apparently different from that of the 51 and 47 kDa fractions of endo H-digested subunits (Figure 3A, right panel, lane 2). Taking into account that only a subset of β2 subunit proteins have a glycan at N32, and the fact that the glycan at N173 is endo H-sensitive, this line of evidence suggested that the 51 but not the 47 kDa fraction of the endo H-digested β2 subunits of surface α1β2γ2 receptors carried the processed N-glycan at N32 site, and, more importantly, the hindrance of N104 N-glycan processing happened on both β2 subunits of a given α1β2γ2 receptor.
As previously mentioned, an α1 or β2 subunit is expected to occupy the fifth position in α1β2 receptor pentamers (31, 32). Consequently, the different β2 subunit digestion patterns with binary α1β2 and ternary α1β2γ2S receptors might reflect different N-glycan processing of β2 subunits taking the fifth position in α1β2 receptors. If this were true, endo H-digested surface β2 subunits from binary receptors would be a combination of two different digestion patterns. One digestion pattern might be the same as those from α1β2γ2 receptors that migrated at 51 and 47 kDa and the other from those β2 subunits taking the fifth position that migrated at 53 and 50 kDa. However, the endo H-digested surface β2 subunits of binary α1β2 receptors mainly migrated at 53 and 50 kDa without a visible band at 47 kDa. Given that the ratio of 53 to 50 kDa fractions of α1β2 receptors treated with endo H (Figure 3A, left panel, lane 2) was similar to the ratio of undigested 54 to 51 kDa fractions (Figure 3A, left panel, lane 1), there was no evidence suggesting hindrance of N-glycan processing of N32 and/or N104 in the Golgi apparatus. Thus, N-glycans of β2 subunits taking the fifth position were likely processed in the same way as those conjugated to the other two β2 subunits in α1β2 receptors.
It was intriguing that endo H-digested β2(N32Q) subunits of surface α1β2(N32Q)γ2 receptors migrated as two bands at 50 and 47 kDa but not a single band at 47 kDa. This observation suggested that there was a mixture of mature and immature glycans at the N104 glycosylation site. Given that the N32Q mutation increased surface α1 subunit levels at the expense of surface γ2 subunit levels, a mixed population of binary and ternary receptors is a potential cause for heterogeneity of endo H sensitivity. However, unlike wild-type receptors, we could not rule out the possibility that the N32Q mutation resulted in heterogeneous N104 N-glycan processing within a given α1β2(N32Q)γ2 receptor. For example, the N104 residue at the γ-β interface may have impaired glycan processing, while the N104 glycan at the α-β interface is fully processed.
Collectively, our results show that, similar to binary α1β2 receptors, glycosylation of the β2 subunit is an important regulator of ternary α1β2γ2 receptor expression and function. In addition, the present findings report a γ2 subunit-dependent impairment of N104 glycan processing. How inclusion of the γ2 subunit impairs glycan processing remains to be determined. Furthermore, we speculate that the N104 glycan may even help form part of the functional interface between the γ and β subunits, thereby providing a novel function of protein glycosylation in GABAA receptors.
Supplementary Material
Acknowledgments
We thank Dr. Michael Cooper for equipment use, Dr. Martin J. Gallagher for critical reading of this manuscript, and Yueli Zhang, Wangzhen Shen, Ningning Hu, Joseph B. Toplon, and Teniel Sonya Ramikie for technical assistance. This work was supported by National Institutes of Health research grants R01 NS33300 to R.L.M. and K08 NS045122 to A.H.L. This material is based upon work supported in part by the Veterans Health Administration 1I01BX001189 to A.H.L. The VA does not specifically endorse the findings reported here.
Footnotes
The authors have no conflicts of interest.
Reference List
- 1.Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta. 1999;1473:4–8. doi: 10.1016/s0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- 2.Tanaka M, Olsen RW, Medina MT, Schwartz E, Alonso ME, Duron RM, Castro-Ortega R, Martinez-Juarez IE, Pascual-Castroviejo I, Machado-Salas J, Silva R, Bailey JN, Bai D, Ochoa A, Jara-Prado A, Pineda G, Macdonald RL, Delgado-Escueta AV. Hyperglycosylation and reduced GABA currents of mutated GABRB3 polypeptide in remitting childhood absence epilepsy. Am J Hum Genet. 2008;82:1249–1261. doi: 10.1016/j.ajhg.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- 4.Cals MM, Guenzi S, Carelli S, Simmen T, Sparvoli A, Sitia R. IgM polymerization inhibits the Golgi-mediated processing of the μ-chain carboxy-terminal glycans. Mol Immunol. 1996;33:15–24. doi: 10.1016/0161-5890(95)00132-8. [DOI] [PubMed] [Google Scholar]
- 5.Whiting PJ, Bonnert TP, McKernan RM, Farrar S, Le Bourdelles B, Heavens RP, Smith DW, Hewson L, Rigby MR, Sirinathsinghji DJ, Thompson SA, Wafford KA. Molecular and functional diversity of the expanding GABAA receptor gene family. Ann N Y Acad Sci. 1999;868:645–653. doi: 10.1111/j.1749-6632.1999.tb11341.x. [DOI] [PubMed] [Google Scholar]
- 6.Angelotti TP, Macdonald RL. Assembly of GABAA receptor subunits: α1β1 and α1β1γ2S subunits produce unique ion channels with dissimilar single-channel properties. J Neurosci. 1993;13:1429–1440. doi: 10.1523/JNEUROSCI.13-04-01429.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fisher JL, Macdonald RL. Single channel properties of recombinant GABAA receptors containing γ2 or δ subtypes expressed with α1 and β3 subtypes in mouse L929 cells. J Physiol. 1997;505 (Pt 2):283–297. doi: 10.1111/j.1469-7793.1997.283bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haas KF, Macdonald RL. GABAA receptor subunit γ2 and δ subtypes confer unique kinetic properties on recombinant GABAA receptor currents in mouse fibroblasts. J Physiol. 1999;514 (Pt 1):27–45. doi: 10.1111/j.1469-7793.1999.027af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lo WY, Lagrange AH, Hernandez CC, Harrison R, Dell A, Haslam SM, Sheehan JH, Macdonald RL. Glycosylation of β2 subunits regulates GABAA receptor biogenesis and channel gating. J Biol Chem. 2010;285:31348–31361. doi: 10.1074/jbc.M110.151449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gurba KN, Hernandez CC, Hu N, Macdonald RL. GABRB3 mutation, G32R, associated with childhood absence epilepsy alters α1β3γ2L gamma-aminobutyric acid type A (GABAA) receptor expression and channel gating. J Biol Chem. 2012;287:12083–12097. doi: 10.1074/jbc.M111.332528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klausberger T, Fuchs K, Mayer B, Ehya N, Sieghart W. GABAA receptor assembly. Identification and structure of γ2 sequences forming the intersubunit contacts with α1 and β3 subunits. J Biol Chem. 2000;275:8921–8928. doi: 10.1074/jbc.275.12.8921. [DOI] [PubMed] [Google Scholar]
- 12.Goldschen-Ohm MP, Wagner DA, Petrou S, Jones MV. An epilepsy-related region in the GABAA receptor mediates long-distance effects on GABA and benzodiazepine binding sites. Mol Pharmacol. 2010;77:35–45. doi: 10.1124/mol.109.058289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lo WY, Botzolakis EJ, Tang X, Macdonald RL. A conserved Cys-loop receptor aspartate residue in the M3–M4 cytoplasmic loop is required for GABAA receptor assembly. J Biol Chem. 2008;283:29740–29752. doi: 10.1074/jbc.M802856200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greenfield LJ, Jr, Sun F, Neelands TR, Burgard EC, Donnelly JL, Macdonald RL. Expression of functional GABAA receptors in transfected L929 cells isolated by immunomagnetic bead separation. Neuropharmacology. 1997;36:63–73. doi: 10.1016/s0028-3908(96)00150-5. [DOI] [PubMed] [Google Scholar]
- 15.Gunther U, Benson J, Benke D, Fritschy JM, Reyes G, Knoflach F, Crestani F, Aguzzi A, Arigoni M, Lang Y, Bluethmann H, Mohler H, Luscher B. Benzodiazepine-insensitive mice generated by targeted disruption of the γ2 subunit gene of gamma-aminobutyric acid type A receptors. Proc Natl Acad Sci U S A. 1995;92:7749–7753. doi: 10.1073/pnas.92.17.7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lagrange AH, Botzolakis EJ, Macdonald RL. Enhanced macroscopic desensitization shapes the response of α4 subtype-containing GABAA receptors to synaptic and extrasynaptic GABA. J Physiol. 2007;578:655–676. doi: 10.1113/jphysiol.2006.122135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang X, Hernandez CC, Macdonald RL. Modulation of spontaneous and GABA-evoked tonic α4β3δ and α4β3γ2L GABAA receptor currents by protein kinase A. J Neurophysiol. 2010;103:1007–1019. doi: 10.1152/jn.00801.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lema GM, Auerbach A. Modes and models of GABAA receptor gating. J Physiol. 2006;572:183–200. doi: 10.1113/jphysiol.2005.099093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buller AL, Hastings GA, Kirkness EF, Fraser CM. Site-directed mutagenesis of N-linked glycosylation sites on the gamma-aminobutyric acid type A receptor α1 subunit. Mol Pharmacol. 1994;46:858–865. [PubMed] [Google Scholar]
- 20.Gallagher MJ, Shen W, Song L, Macdonald RL. Endoplasmic reticulum retention and associated degradation of a GABAA receptor epilepsy mutation that inserts an aspartate in the M3 transmembrane segment of the α1 subunit. J Biol Chem. 2005;280:37995–38004. doi: 10.1074/jbc.M508305200. [DOI] [PubMed] [Google Scholar]
- 21.Boileau AJ, Li T, Benkwitz C, Czajkowski C, Pearce RA. Effects of γ2S subunit incorporation on GABAA receptor macroscopic kinetics. Neuropharmacology. 2003;44:1003–1012. doi: 10.1016/s0028-3908(03)00114-x. [DOI] [PubMed] [Google Scholar]
- 22.Laurie DJ, Wisden W, Seeburg PH. The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III Embryonic and postnatal development. J Neurosci. 1992;12:4151–4172. doi: 10.1523/JNEUROSCI.12-11-04151.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Castaneda JT, Harui A, Kiertscher SM, Roth JD, Roth MD. Differential Expression of Intracellular and Extracellular CB2 Cannabinoid Receptor Protein by Human Peripheral Blood Leukocytes. J Neuroimmune Pharmacol. 2013 doi: 10.1007/s11481-012-9430-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haas KF, Macdonald RL. GABAA receptor subunit γ2 and δ subtypes confer unique kinetic properties on recombinant GABAA receptor currents in mouse fibroblasts. J Physiol. 1999;514 (Pt 1):27–45. doi: 10.1111/j.1469-7793.1999.027af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mortensen M, Smart TG. Extrasynaptic αβ subunit GABAA receptors on rat hippocampal pyramidal neurons. J Physiol. 2006;577:841–856. doi: 10.1113/jphysiol.2006.117952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kash TL, Dizon MJ, Trudell JR, Harrison NL. Charged residues in the β2 subunit involved in GABAA receptor activation. J Biol Chem. 2004;279:4887–4893. doi: 10.1074/jbc.M311441200. [DOI] [PubMed] [Google Scholar]
- 27.Tretter V, Ehya N, Fuchs K, Sieghart W. Stoichiometry and assembly of a recombinant GABAA receptor subtype. J Neurosci. 1997;17:2728–2737. doi: 10.1523/JNEUROSCI.17-08-02728.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Macdonald RL, Kang JQ, Gallagher MJ. Mutations in GABAA receptor subunits associated with genetic epilepsies. J Physiol. 2010;588:1861–1869. doi: 10.1113/jphysiol.2010.186999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan HO, Reid CA, Single FN, Davies PJ, Chiu C, Murphy S, Clarke AL, Dibbens L, Krestel H, Mulley JC, Jones MV, Seeburg PH, Sakmann B, Berkovic SF, Sprengel R, Petrou S. Reduced cortical inhibition in a mouse model of familial childhood absence epilepsy. Proc Natl Acad Sci U S A. 2007;104:17536–17541. doi: 10.1073/pnas.0708440104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olsen RW, Sieghart W. GABAA receptors: subtypes provide diversity of function and pharmacology. Neuropharmacology. 2009;56:141–148. doi: 10.1016/j.neuropharm.2008.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baumann SW, Baur R, Sigel E. Subunit arrangement of gamma-aminobutyric acid type A receptors. J Biol Chem. 2001;276:36275–36280. doi: 10.1074/jbc.M105240200. [DOI] [PubMed] [Google Scholar]
- 32.Boileau AJ, Pearce RA, Czajkowski C. Tandem subunits effectively constrain GABAA receptor stoichiometry and recapitulate receptor kinetics but are insensitive to GABAA receptor-associated protein. J Neurosci. 2005;25:11219–11230. doi: 10.1523/JNEUROSCI.3751-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.