

Abstract
Purpose
To investigate the impact of 5-formytetrahydrofolate on the activities of pralatrexate, as compared to methotrexate (MTX), in vitro.
Methods
Cells were exposed to (6S)5-formyltetrahydrofolate (5-formylTHF) for 24h, before or after a 6h exposure to antifolates following which the cellular accumulation and activities of the drugs were evaluated in HeLa cells.
Results
A 24h delay between a 6h exposure to antifolates and a subsequent 24h exposure to 4 μM 5-formylTHF sustained the full activities of both antifolates. A 72h interval was required between a single exposure of up to 4 μM 5-formylTHF and subsequent exposure to drugs to sustain activities of the antifolates. When cells were incubated with 4 μM 5-formylTHF for 24h weekly, for 4 weeks, there was no significant increase in the IC50 for pralatrexate, but the MTX IC50 increased 2.5-fold as compared to cells growing continuously in 25nM 5-formylTHF. This cyclical exposure to 5-formylTHF increased the cell folate pool by 16%, had no significant effect on the intracellular pralatrexate level, but decreased intracellular MTX by 15%. An extracellular concentration of methotrexate 50-fold higher than that of pralatrexate was required to achieve an intracellular level, and growth inhibition, comparable to that of pralatrexate.
Conclusions
Cyclical exposures to 5-formylTHF at levels in excess of what is achieved in most clinical “rescue” regimens do not affect pralatrexate accumulation nor antitumor activity in HeLa cells, in contrast to MTX. An important element in preserving pralatrexate activity is achieving a sufficient interval between exposure to 5-formylTHF and the next dose of antifolate.
Keywords: pralatrexate, methotrexate, (6S)5-formytetrahydrofolate, 5-formyltetrahydrofolate, leucovorin, rescue, reduced folate carrier, folylpolyglutamate synthetase
Introduction
Pralatrexate, the 10-propargyl-10-deaza analog of aminopterin, is a potent inhibitor of dihydrofolate reductase (DHFR) [1], the enzyme required for the maintenance of tetrahydrofolate (THF) cofactor pools within cells when 5,10-methylene THF is oxidized to dihydrofolate (DHF) during the synthesis of thymidylate mediated by thymidylate synthase [2,3]. Pralatrexate’s pharmacological advantage is based upon its superior properties relative to methotrexate (MTX) as a substrate for the reduced folate carrier (RFC) and folylpolyglutamate synthase (FPGS) resulting the rapid formation, and accumulation to high levels, of its polyglutamate derivatives that sustain suppression of its target enzyme for long intervals within cells [1,4]. Unlike its very high affinity for RFC, pralatrexate has a lower affinity for the proton-coupled folate transporter (PCFT) than MTX so that this carrier is unlikely to contribute to the transport of this agent into tumor cells. However, the low affinity for PCFT, the mechanism of intestinal absorption of folates and antifolates, would tent to increase the fecal excretion and clearance of this agent [4]. Pralatrexate is approved for the treatment of relapsed and refractory peripheral T-cell lymphoma (PTCL) and transformed mycosis fungoides (T-MF) [5,6,7] and is currently being evaluated for its efficacy in other malignancies [8,9].
Pralatrexate is ~10-fold more potent than MTX with continuous exposure, and the difference becomes much greater with a 6h exposure to the antifolates [4]. However, pralatrexate can be administered parenterally at a weekly dose of 30 mg/m2 with folic acid supplementation, comparable to the weekly dosing for MTX [10,5]. Based upon the relative potencies of pralatrexate and MTX, current pralatrexate regimens might be considered comparable to “high-dose” MTX without leucovorin (6(R,S)5-formylTHF) “rescue”. These observations indicate that pralatrexate has a greater degree of selectivity than MTX; nonetheless, mucosites is often dose-limiting for the drug [5]. Hence, studies have been initiated and planned to evaluate the impact of “rescue” regimens on the incidence and intensity of the mucosites associated with this agent [11,12]. The objective of the current study is to determine the extent to which 5-formyltetrahydrofolate alters the activity of pralatrexate, as compared to MTX, using the active “S” isomer (5-formylTHF) and an in vitro protocol that simulates elements of how this folate would be employed in the clinical setting.
Methods
Reagents
[3′,5′,7,9-3H](6S)5-formylTHF, [3′,5′,7-3H]MTX and generally labeled [3H]pralatrexate were obtained from Moravek Biochemicals (Brea, CA), The agents were purified as necessary, and purity monitored by liquid chromatography as described previously [13,4]. Pralatrexate was purified using a 5-mm OSD2 4.6 x 250 mm reversed-phase column (Waters Spherisorb) by isocratic elution with 100 mM sodium acetate pH 5.5 (solvent A) and 15% acetonitrile (solvent B). The mobile phase was delivered at 1 ml/min, reaching 100% solvent B in 30 min. Nonlabeled pralatrexate (Folotyn) and (6S)5-formylTHF (Fusilev) were provided by Spectrum Pharmaceuticals (Irvine, CA). MTX was obtained from Sigma-Aldrich (St. Louis, MO).
Cell lines
HeLa cells were maintained in folate-free RPMI 1640 medium supplemented with 10% dialyzed fetal bovine serum (Gemini Bio-Products, CA), 100 units/ml penicillin and 100 μg/ml streptomycin (Gibco Life Technologies, CA) at 37°C in a humidified atmosphere of 5% CO2. (6S)5-formylTHF (25 nM) was the folate source in the medium.
Experimental Design and Growth inhibition assays
Experiments were designed to simulate exposures to 5-formylTHF as it is administered in clinical regimens. The experiments evaluated the impact of 5-formylTHF administered after pralatrexate as in “rescue” along with its impact on the activity of the “next” dose of pralatrexate as would occur in a weekly regimen with this agent. The studies were performed within the context of a comparison with MTX at concentrations that produced comparable growth inhibition. The details of each type of experiment are described in the Results section and the legends to the figures. HeLa cells were seeded in 96-well plates at a density of 2 × 103 cells/well. At some point, pralatrexate or MTX was added to achieve a spectrum of concentrations. After 6h, the cells were washed then grown in drug-free medium for 3–5 days. The cells were then assayed by sulforhodamine B staining. Absorbance was measured at 540 nm with the VERSAmax plate reader (GE Intelligent Platforms, Charlottesville, VA).
Measurement of total cellular folate and antifolate levels
For determination of total cellular folates, HeLa cells were grown for a week in folate-free medium supplemented with GAT (0.2 mM glycine, 0.1 mM adenosine, and 0.01 mM thymidine) to deplete endogenous folates. The cells were then replated in medium supplemented with 25 nM [3H](6S)5-formylTHF. One portion was maintained in this medium, the other portion was exposed to 4 μM [3H](6S)5-formylTHF for 24h weekly. After 4 weeks (four, 7-day, cycles) the cells were washed in ice-cold HBS buffer (20 mM Hepes, 140 mM NaCl, 5 mM KCl, 2 mM MgCl2 and 5 mM dextrose; adjusted with 1 N NaOH to achieve a pH of 7.4) and then digested with 500 μl of 0.2 N NaOH at 65°C for 45 minutes. Twenty μl was used for protein determination by the bicinchoninic acid protein (BCA) assay (Pierce Chemical, IL), 400 μl for assessment of tritium on a liquid scintillation spectrometer.
For determination of cellular antifolate levels, HeLa cells were grown in medium supplemented with 25 nM (6S)5-formylTHF containing either 0.1 μM [3H]pralatrexate or 5μM [3H]MTX for 6h then washed twice in ice-cold HBS. These concentrations of antifolates approximated the level required to produce comparable growth inhibition (IC50’s). One portion of cells was assessed for total tritium and the other portion for total protein as described above. Intracellular radioactivity is expressed as picomoles of tritiated substrate per mg of protein.
Statistical analysis
The IC50 values were calculated from an analysis of growth inhibition (as percent of control growth) as a function of the log of the extracellular antifolate concentration. In some experiments, the data is plotted as the ratio of the IC50 of the cells exposed to 1 ≥ μM 5-formylTHF to cells grown in 25 nM 5-formylTHF as a function of the extracellular (6S)5-formylTHF concentration. Statistical comparisons were performed by the two-tailed Student’s paired t test. All statistical analyses utilized GraphPad Prism (version 6.0 for Windows, GraphPad Software).
Results
Analysis of 5-formylTHF “rescue” of HeLa cells as a function of time between exposure to antifolates and exposure to 5-formylTHF
On day 1, HeLa cells were incubated for 6h with a spectrum of pralatrexate or MTX concentrations. Twenty-four or 48h later, the cells were incubated with 1 μM 5-formylTHF for 24h following which the cells were grown without either agent but in presence of 25nM 5-formylTHF, representative of the “normal” blood folate level. The IC50 (the antifolate concentration at which growth was 50% that of cells not exposed to drugs) for pralatrexate (Fig. 1A) or MTX (Fig. 1B) was not affected by these exposures to 5-formylTHF. Comparing the two panels, it can be seen that pralatrexate was ~130-fold more potent than MTX under these conditions.
Figure 1. The impact of exposure to 5-formylTHF after exposure to antifolates on growth inhibition.
HeLa cells were exposed to pralatrexate (Panel A) or MTX (Panel B) for 6h at the indicated concentrations. After 24h (open square) or 48h (open triangle) the cells were incubated for 24h with 1 μM 5-formylTHF then grown in 25 nM 5-formylTHF for additional 4 and 3 days, respectively. Control cells (filled circle) were maintained continuously in 25 nM 5-formylTHF. Growth in the absence of drug is indicated as 100%. The vertical line intercepts the x axis at the concentration at which growth inhibition is 50% of the level of growth in the absence of drug (IC50). Data are the mean ± S.E.M from three independent experiments. Panel C: HeLa cells were incubated with a spectrum pralatrexate or MTX concentrations for 6h. After 24h, the cells were exposed to 5-formylTHF at the indicated concentrations for 24h and then grown in 25 nM 5-formylTHF for an additional 4 days. Control cells were maintained in 25 nM 5-formylTHF. The Y axis is the ratio of the IC50 of the cells exposed to 1 ≥ μM 5-formylTHF to cells grown in 25 nM 5-formylTHF. Cells were always washed after each manipulation. Data are the mean ± S.E.M from three independent experiments.
The impact of the 5-formylTHF concentration on “rescue”
Because a 24h interval was sufficient to sustain the cytotoxicity of MTX and pralatrexate at an extracellular concentration of 1 μM 5-formylTHF, studies evaluated the impact of increasing the 5-formylTHF concentration. On day 1 HeLa cells were incubated for 6h with pralatrexate or MTX, 24h later the cells were incubated with 1 to 16 μM 5-formylTHF for 24h following which the cells were grown for an additional 4 days in 25nM 5-formylTHF. As indicated in Figure 1C, the pralatrexate and MTX IC50 ratios (the ratio of the IC50 of the cells exposed to 1≥ μM 5-formylTHF to cells grown in 25 nM 5-formylTHF) were unchanged at 5-formylTHF concentrations up to 4 μM; the small increase in the pralatrexate IC50 at 4μM was not significant. Comparing the average IC50 of the lowest three with the average of the highest three 5-formylTHF concentrations, there was only a small increase in the pralatrexate IC50 (47±2 vs 71±2 nM, P=0.006) and the MTX IC50 (2.9±0.15 vs 3.8±0.2 μM, P=0.08).
An analysis of the “protective” potential of 5-formylTHF on the subsequent exposure to pralatrexate and MTX in HeLa cells
The previous experiments indicated that a single 6h incubation with pralatrexate or MTX followed 24h later by growth in up to 4 μM 5-formylTHF did not alter the activities of these antifolates. Other studies assessed the extent to which exposure to the folate prior to the exposure to the antifolates affects the antitumor activity. HeLa cells were exposed for 24h to 1 μM 5-formylTHF, then replated in medium with 25 nM 5-formylTHF 24h, 48h or 72h before a 6h incubation with pralatrexate or MTX. As indicated in Figure 2A, pralatrexate activity was decreased ~2-fold (IC50 increased from ~28 nM to ~60 nM) when cells were exposed to the drugs 24h or 48h after treatment with 5-formylTHF. MTX activity was comparably decreased (IC50 increased from ~3 μM to ~7 μM) under similar conditions (Fig. 2B). The activities of both drugs were fully restored when exposure to the antifolates was delayed until 72h after exposure to the 5-formylTHF.
Figure 2. Impact of the interval between exposure to 5-formylTHF followed by exposure to the antifolates on growth inhibition.

HeLa cells were exposed to 1 μM 5-formylTHF for 24 (open square), 48 (open triangle) or 72h (open circle) before exposure to pralatrexate or MTX for 6h at the indicated concentrations. The cells were then grown for additional 5 days in 25 nM 5-formylTHF. Control cells (filled circle) were maintained continuously in 25 nM 5-formylTHF. Growth in the absence of drug is indicated as 100%. The vertical line intercepts the x axis at the concentration at which growth inhibition is 50% of the level of growth in the absence of drug (IC50). Cells were always washed after each manipulation. Data are the mean ± S.E.M from three independent experiments.
Antifolate “protection” as function of the 5-formylTHF concentration
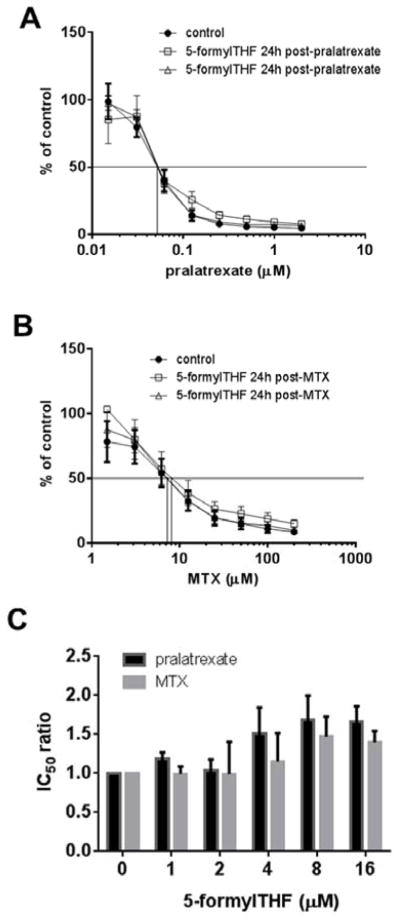
To determine the extent to which protection was dependent upon the 5-formylTHF concentration, HeLa cells were exposed to from 1 to 16 μM 5-formylTHF for 24h, then washed and incubated in medium with 25nM 5-formylTHF; 72h later the cells were incubated for 6h with pralatrexate or MTX then grown in absence of antifolate until the end of the assay. Figure 3A indicates that the IC50 ratio for pralatrexate was unchanged up to 8 μM 5-formylTHF, the small increase at 16 μM 5-formylTHF was not significant. Similarly, while there was a trend towards an increase in the IC50 ratio for MTX at the highest 5-formylTHF concentrations, this did not reach statistical significance.
Figure 3. Impact of a single or four-weekly exposures to 5-formylTHF, 72h prior to exposure to pralatrexate or MTX, on antifolate activities.

HeLa cells were exposed once (Panel A), or weekly X 4 (Panel B), for 24h to 5-formylTHF at the indicated concentrations. Seventy-two hours after the last exposure, the cells were exposed to a range of pralatrexate or MTX concentrations for 6h and then grown for additional 5 days in 25 nM 5-formylTHF. Control cells were maintained continuously in 25 nM 5-formylTHF. Panel C is a representation that illustrates the increase in IC50 ratio as a function of the extracellular concentration of 5-formylTHF. The inset amplifies this relationship over the 1–4 μM 5-formylTHF range. The IC50 ratio is described in the legend to Figure 2. Cells were always washed after each manipulation. Data are the mean ± S.E.M from three independent experiments.
Next, the cumulative effect of repeated exposures to 5-formylTHF on pralatrexate and MTX activities was assessed. HeLa cells were subjected to 24h incubations with 4μM 5-formylTHF weekly for four weeks. Seventy-two hrs after the last (4th) exposure, the cells were incubated with pralatrexate or MTX for 6h then replated with 25 nM 5-formylTHF for an additional 5 days. Figure 3B indicates that pralatrexate activity was minimally affected by exposures to 5-formylTHF. Figure 3C illustrates the slope of the IC50 ratio as a function of the extracellular concentration of 5-formylTHF. While the slope for pralatrexate is significant (p=0.03, Panel C), there was only a negligible increase in the IC50 ratio at 4 μM 5-formylTHF. The pattern for MTX was different as indicated in Figure 3B and 3C. The IC50 ratio increased as the 5-formylTHF concentration increased reaching a level >2.5-fold greater for cells growing in 4 μM as compared to 25 nM 5-formylTHF (P=0.005, Fig. 3C). The inset to Figure 3C amplifies the slope of the IC50 ratios within the 4 μM range. It can be seen that the slope of the MTX IC50 ratio as a function of the extracellular 5-formylTHF concentration exceeded that of pralatrexate by a factor of 10 (0.4 vs 0.04, P=0.01). Hence, loading cells with 5-formylTHF clearly impacts on the activity of MTX but has a negligible effect on pralatrexate activity particularly at concentrations of the folate to which cells are exposed in clinical rescue regimens (see Discussion).
Cumulative effect of 5-formylTHF on total cell folate
The impact of the multiple exposures to 5-formylTHF on total cell folate was assessed by first depleting cells of endogenous folates by growth in GAT for one week. The cells were then grown continuously with 25 nM [3H](6S)5-formylTHF except for a 24h exposure to 4 μM [3H](6S)5-formylTHF at the same specific activity, each week for four weeks; this was the concentration at which the difference between MTX and pralatrexate growth inhibition was maximal. The control cells were maintained in 25 nM [3H](6S)5-formylTHF. Figure 4,A indicates that there was a small (16%) but significant increase in the total folate pool (58.8 ± 1.0 vs 68.3±2.7 pmol/mg of protein respectively, P=0.02) after the fourth cycle.
Figure 4. Effect of 4 weekly exposures to 5-formylTHF on total intracellular folate and antifolate accumulation.

(Panel A) HeLa cells were grown for one week in GAT to deplete endogenous folates and then replated for 4 weeks in 25 nM [3H]5-formylTHF. Once a week for four weeks a portion of cells was treated for 24h with 4 μM [3H]5-formylTHF, another portion was maintained in 25 nM [3H]5-formylTHF. Seventy-two hours after the last exposure to [3H]5-formylTHF, total intracellular radioactivity was measured. (Panel B) HeLa cells were grown in 25 nM 5-formylTHF; once each week the cells were exposed for 24h to 4 μM 5-formylTHF. Control cell were maintained in 25 nM 5-formylTHF. Seventy-two hours after the last exposure to 5-formylTHF, the cells were exposed to 0.1 μM [3H]pralatrexate or 5 μM [3H]MTX for 6h and intracellular radioactivity measured. Cells were always washed after each manipulation. Data are the mean ± S.E.M from three independent experiments.
The impact of four weekly exposures to 5-formylTHF on pralatrexate and MTX accumulation
To evaluate the impact of four cycles of exposure to 5-formylTHF, as described above, on the accumulation of the antifolates, 72h after the 4th exposure to 5-formylTHF, cells were exposed to 0.1[3H]pralatrexate or 5 μM [3H]MTX for 6h following which the cells were analyzed for their intracellular antifolate content. As indicated in Figure 4B, the small decrease in the accumulation of [3H]pralatrexate (16.84±0.1 vs 15.16±1.3 pmol/mg of proteins) was not significant (p=0.4). The 15% decrease in accumulation of [3H]MTX (16.95±0.79 vs 14.44±0.64) was highly significant (p=0.004) in cells exposed to multiple cycle of 5-formylTHF as compared with the cells maintained in 25 nM 5-formylTHF. Under these conditions, >80% of intracellular antifolates were the polyglutamate forms. It is noteworthy that a 50-fold higher concentration of [3H]MTX (5 μM) was required to achieve a total intracellular antifolate level comparable to what was achieved with 0.1 μM [3H]pralatrexate over the 6h incubation, consistent with the more efficient transport and polyglutamation of the latter antifolate. These concentrations also reflected the differences in IC50’s for these agents.
Discussion
Pralatrexate has a much higher therapeutic index than MTX. However, the degree of toxicity is far less than might be expected from an agent that is so potent relative to MTX but can be administered weekly intravenously at a dose only slightly less than that of MTX [6]. While sharing the same target, DHFR, pralatrexate’s 8–10-fold higher affinity for RFC, and 10-fold greater catalytic activity mediated by FPGS, relative to MTX, results in the formation and accumulation of high levels of its active polyglutamate derivatives that are retained in tumor cells and produce sustained inhibition of its target enzyme [1,4]. This is illustrated by the observation that a 6h incubation with MTX required a ~50-fold higher extracellular concentration than pralatrexate to achieve a comparable intracellular level and growth inhibition. However, the mucosites associated with pralatrexate can be dose-limiting and an impediment to the use of this agent. This has raised the possibility that a leucovorin “rescue” regimen might improve the clinical utility of this agent [12]. Some patients have been treated with a “rescue” regimen (a single 50 mg dose 24h after pralatrexate) with amelioration of toxicity and, while anecdotal, antitumor activity was preserved at least in part [12].
Previous studies from this laboratory described the adverse impact of 5-formylTHF on the activities of a variety of antifolates [14,15]. The impact on pemetrexed activity was shown to be due to inhibition of the formation of polyglutamate derivatives of this agent required for inhibition of its target enzymes [14]. Recently, a similar phenomenon was observed for new generation GARFTase inhibitors that also have a high affinity for FPGS [16]. When the exposure to MTX is brief, its activity is very sensitive to the cellular folate pool. This reflects the much lower affinity of MTX for FPGS relative to pralatrexate and the much longer time required to accumulate its polyglutamate derivatives in cells [17,18,14]. It was important to understand the effect that 5-formyTHF might have on the activity of pralatrexate in order to minimize, as much as possible, a negative impact on its efficacy.
The natural (S) isomer of 5-formylTHF was utilized in the current study. The unnatural (D) isomer has essentially no biological activity; it is a very poor substrate for RFC [19] and what does enter cells cannot be metabolized. Hence, the concentrations of (6S)5-formylTHF employed in this study are chemically equivalent to half the concentration of the racemic mixture but equal to the biologically active fraction. The exposures to 5-formylTHF encompassed and far exceeded the concentration of this folate, and 5-methyltetrahydrofolate to which it is rapidly converted, achieved in “rescue” regimens in vivo. For instance, following a 50 mg intravenous dose of leucovorin, the same dose as in the case reports with pralatrexate described above [12], the peak (6S)5-formylTHF blood level was ~4.4 μM with a t½ of 32 min, the peak 5-methylTHF level was ~1.6 μM with a t½ of 224 min so that at 6h the former was ~0.02 μM and the latter was ~0.8 μM; by 12h the latter was ~0.2 μM [20]. When leucovorin was administered p.o. at 25 mg every 8h, the mean 5-methylTHF blood level was only ~0.7 μM at 48h [20]. Likewise, after an intravenous dose of 10 or 25 mg/m2 leucovorin, the peak 5-methylTHF blood levels were 0.23 and 0.6 μM, respectively, with an average t½ of 6.6h. With p.o. dosing, the peak 5-methylTHF levels were 0.32, 0.47 μM, respectively with an average t½ of 3.1h [21]. At these 5-methylTHF blood levels, there would be essentially no impact on pralatrexate activity.
A 24h interval between exposure to the antifolates and 5-formylTHF in the current study was sufficient to sustain full growth inhibition by MTX and pralatrexate to a 5-formylTHF concentration of 4μM with only a small (50%) increase in IC50 at 16 μM 5-formylTHF. On the other hand, when the impact of 5-formylTHF on the “next” dose of antifolate was considered, a 72h delay after exposure to 5-formylTHF was necessary to preserve antifolate activities. Even when the exposure to 5-formylTHF was repeated weekly for four weeks, there was no increase in the pralatrexate IC50 ratio at blood levels that exceeded those achieved in “rescue” regimens; however, the MTX IC50 increased to 2.5-fold as the 5-formylTHF concentration was increased to 4 μM. Hence, it would appear that a critical element in preserving antifolate activity is the delay between 5-formylTHF and the next dose of antifolate. Indeed, in a weekly regimen the delay would be 4 days if the “rescue” interval was restricted to 24h.
The rationale for high-dose MTX with leucovorin “rescue”, and its putative selectivity, is based upon elements at the tumor, cellular, and biochemical levels [22,3]. High concentrations of drug facilitate passive diffusion into the poorly perfused interstitium of solid tumors. The subsequent provision of much lower doses of leucovorin poorly penetrate tumors but are readily accessible to precursor cells of the intestine and bone marrow with their intact vascular system. Likewise, high concentrations of MTX would passively diffuse into tumor cells with impaired transport due to low expression or loss-of-function mutations of RFC. The low concentration of leucovorin during “rescue” which enters cells via the same transporter would have limited access to the tumor cells but would readily enter normal tissues with intact RFC. At the biochemical level, MTX polyglutamate derivatives are potent direct inhibitors of thymidylate synthase and AICAR transformylase [23,24,25]. This interferes with the utilization of 5,10-methyleneTHF and 10-formylTHF, respectively, formed by the interconversion of leucovorin [26]. However, utilization of these one-carbon donors in bone marrow and intestine is less impeded since only low levels of MTX polyglutamate derivatives accumulate in these cells [27,28,29,30]. Finally, the interconversion of leucovorin to dihydrofolate results in the displacement of the monoglutamate of MTX from DHFR reactivating tetrahydrofolate synthesis. However, this does not occur when high levels of MTX polyglutamates accumulate in tumor cells [27,31,32,26]. There is no information, as yet, as to the extent to which pralatrexate polyglutamates are direct inhibitors of tetrahydrofolate cofactor-requiring enzymes in cells.
There are preclinical in vivo data that support the concept that leucovorin “rescue” diminishes the toxicity, while preserving the activity, of MTX and aminopterin [33,34]. Leucovorin “rescue” clearly decreases the toxicity of high-dose MTX regimens; however, the extent to which the antitumor activity of MTX is diminished is uncertain. One clinical study compared standard-dose MTX (40 mg/m2 weekly for 8 weeks) with or without low-dose leucovorin “rescue” (10 mg/m2 p.o. every six hours, starting 24h after MTX, for four doses) in patients with squamous cell head and neck cancer. Leucovorin decreased drug toxicity but also diminished antitumor activity (response rate, 17.1 and 36.7%, respectively, P=0.047) [35]. Data in the current report indicate that there is a greater adverse impact of loading cells with folates on MTX than pralatrexate anti-tumor activities. Because of the high therapeutic index of pralatrexate relative to MTX, it may be that very modest doses of 5-formylTHF administered over short intervals will obviate the mucosites without an adverse impact on pralatrexate’s therapeutic efficacy, as suggested by the initial report described above [12].
Finally, the observations in this report are focused on one cell line. It is possible that the critical intervals between exposures to antifolates and 5-formylTHF, along with the magnitude of the suppressive effects of 5-formylTHF, may vary among cells of different origin. However, irrespective of these considerations, these studies indicate that the impact of “rescue” with intermittent exposures to 5-formylTHF will be less for pralatrexate than for MTX. Since, as indicated above, there is evidence that leucovorin “rescue” is associated with decreased efficacy of MTX, this represents an important advantage for this next-generation DHFR inhibitor.
Abbreviations
- (6S)5-formylTHF
(6S)5-formyltetrahydrofolate
- leucovorin
d,l,5-formyltetrahydrofolate
- AICART
phosphoribosylaminoimidazolecarboxamide formyltransferase
- DHFR
dihydrofolate reductase
- FPGS
folylpolyglutamate synthetase
- GARFT
glycinamide ribonucleotide formyltransferase
- TS
thymidylate synthase
- RFC
reduced folate carrier
- MTX
methotrexate
Footnotes
Conflict of Interest
This study was supported by a grant from Spectrum Pharmaceuticals, Inc (Irvine, CA).
Reference List
- 1.Sirotnak FM, DeGraw JI, Colwell WT, Piper JR. A new analogue of 10-deazaaminopterin with markedly enhanced curative effects against human tumor xenografts in mice. Cancer Chemother Pharmacol. 1998;42:313–318. doi: 10.1007/s002800050823. [DOI] [PubMed] [Google Scholar]
- 2.Goldman ID, Chattopadhyay S, Zhao R, Moran RG. The Antifolates: Evolution, New Agents in the Clinic, and How targeting delivery via specific membrane transporters is driving the development of a next generation of folate analogs. Curr Opin Investig Drugs. 2010;11:1409–1423. [PubMed] [Google Scholar]
- 3.Visentin M, Zhao R, Goldman ID. The antifolates. Hematol Oncol Clin North Am. 2012;26:629–648. doi: 10.1016/j.hoc.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Visentin M, Unal ES, Zhao R, Goldman ID. The membrane transport and polyglutamation of pralatrexate: a new-generation dihydrofolate reductase inhibitor. Cancer Chemother Pharmacol. 2013;72:597–606. doi: 10.1007/s00280-013-2231-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Connor OA, Horwitz S, Hamlin P, Portlock C, Moskowitz CH, Sarasohn D, Neylon E, Mastrella J, Hamelers R, Macgregor-Cortelli B, Patterson M, Seshan VE, Sirotnak F, Fleisher M, Mould DR, Saunders M, Zelenetz AD. Phase II-I-II study of two different doses and schedules of pralatrexate, a high-affinity substrate for the reduced folate carrier, in patients with relapsed or refractory lymphoma reveals marked activity in T-cell malignancies. J Clin Oncol. 2009;27:4357–4364. doi: 10.1200/JCO.2008.20.8470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Connor OA, Pro B, Pinter-Brown L, Bartlett N, Popplewell L, Coiffier B, Lechowicz MJ, Savage KJ, Shustov AR, Gisselbrecht C, Jacobsen E, Zinzani PL, Furman R, Goy A, Haioun C, Crump M, Zain JM, Hsi E, Boyd A, Horwitz S. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;%20(29):1182–1189. doi: 10.1200/JCO.2010.29.9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Foss F, Horwitz SM, Coiffier B, Bartlett N, Popplewell L, Pro B, Pinter-Brown LC, Shustov A, Furman RR, Haioun C, Koutsoukos T, O’Connor OA. Pralatrexate is an effective treatment for relapsed or refractory transformed mycosis fungoides: a subgroup efficacy analysis from the PROPEL study. Clin Lymphoma Myeloma Leuk. 2012;12:238–243. doi: 10.1016/j.clml.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 8.Weems G. [Accessed 23 November 2013];Study of Pralatrexate in Female Patients With Previously-treated Breast Cancer. 2013 http://www.clinicaltrials.gov/ct2/show/NCT01118624?term=pralatrexate&rank=16.
- 9.Weems G. [Accessed 21 November 2013];Study of Pralatrexate vs. Erlotinib for Non-Small Cell Lung Cancer After at Least 1 Prior Platinum-based Treatment. 2013 http://www.clinicaltrials.gov/ct2/show/NCT00606502?term=pralatrexate&rank=1.
- 10.Malik SM, Liu K, Qiang X, Sridhara R, Tang S, McGuinn WD, Jr, Verbois SL, Marathe A, Williams GM, Bullock J, Tornoe C, Lin SC, Ocheltree T, Vialpando M, Kacuba A, Justice R, Pazdur R. Folotyn (pralatrexate injection) for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma: U.S. Food and Drug Administration drug approval summary. Clin Cancer Res. 2010;16:4921–4927. doi: 10.1158/1078-0432.CCR-10-1214. [DOI] [PubMed] [Google Scholar]
- 11.Sawas A. Phase 1 Study of Fusilev to Prevent or Reduce Mucositis in Patients With Non-Hodgkin’s Lymphoma Receiving Folotyn 2013 [Google Scholar]
- 12.Koch E, Story SK, Geskin LJ. Preemptive leucovorin administration minimizes pralatrexate toxicity without sacrificing efficacy. Leuk Lymphoma. 2013;54:2448–2451. doi: 10.3109/10428194.2013.779688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao R, Babani S, Gao F, Liu L, Goldman ID. The mechanism of transport of the multitargeted antifolate, MTA-LY231514, and its cross resistance pattern in cell with impaired transport of methotrexate. Clin Cancer Res. 2000;6:3687–3695. [PubMed] [Google Scholar]
- 14.Zhao R, Gao F, Goldman ID. Marked suppression of the activity of some, but not all, antifolate compounds by augmentation of folate cofactor pools within tumor cells. Biochem Pharmacol. 2001;61:857–865. doi: 10.1016/s0006-2952(01)00532-9. [DOI] [PubMed] [Google Scholar]
- 15.Chattopadhyay S, Tamari R, Min SH, Zhao R, Tsai E, Goldman DI. Commentary: a case for minimizing folate supplementation in clinical regimens with pemetrexed based on the marked sensitivity of the drug to folate availability. Oncologist. 2007;12:808–815. doi: 10.1634/theoncologist.12-7-808. [DOI] [PubMed] [Google Scholar]
- 16.Desmoulin SK, Wang L, Polin L, White K, Kushner J, Stout M, Hou Z, Cherian C, Gangjee A, Matherly LH. Functional loss of the reduced folate carrier enhances the antitumor activities of novel antifolates with selective uptake by the proton-coupled folate transporter. Mol Pharmacol. 2012;82:591–600. doi: 10.1124/mol.112.079004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCloskey DE, McGuire JJ, Russell CA, Rowan BG, Bertino JR, Pizzorno G, Mini E. Decreased folylpolyglutamate synthetase activity as a mechanism of methotrexate resistance in CCRF-CEM human leukemia sublines. J Biol Chem. 1991;266:6181–6187. [PubMed] [Google Scholar]
- 18.Zhao R, Titus S, Gao F, Moran RG, Goldman ID. Molecular analysis of murine leukemia cell lines resistant to 5,10-ideazatetrahydrofolate identifies several amino acids critical to the function of folylpolyglutamate synthetase. J Biol Chem. 2000;275:26599–26606. doi: 10.1074/jbc.M002580200. [DOI] [PubMed] [Google Scholar]
- 19.Sirotnak FM, Chello PL, Moccio DM, Kisliuk RL, Combepine G, Gaumont Y, Montgomery JA. Stereospecificity at carbon 6 of formyltetrahydrofolate as a competitive inhibitor of transport and cytotoxicity of methotrexate in vitro. Biochem Pharmacol. 1979;28:2993–2997. doi: 10.1016/0006-2952(79)90599-9. [DOI] [PubMed] [Google Scholar]
- 20.Straw JA, Szapary D, Wynn WT. Pharmacokinetics of the diastereoisomers of leucovorin after intravenous and oral administration to normal subjects. Cancer Res. 1984;44:3114–3119. [PubMed] [Google Scholar]
- 21.Priest DG, Schmitz JC, Bunni MA, Stuart RK. Pharmacokinetics of leucovorin metabolites in human plasma as a function of dose administered orally and intravenously. J Natl Cancer Inst. 1991;83:1806–1812. doi: 10.1093/jnci/83.24.1806. [DOI] [PubMed] [Google Scholar]
- 22.Zhao R, Goldman ID. Resistance to antifolates. Oncogene. 2003;22:7431–7457. doi: 10.1038/sj.onc.1206946. [DOI] [PubMed] [Google Scholar]
- 23.Allegra CJ, Chabner BA, Drake JC, Lutz R, Rodbard D, Jolivet J. Enhanced inhibition of thymidylate synthase by methotrexate polyglutamates. J Biol Chem. 1985;260:9720–9726. [PubMed] [Google Scholar]
- 24.Allegra CJ, Hoang K, Yeh GC, Drake JC, Baram J. Evidence for direct inhibition of de novo purine synthesis in human MCF-7 breast cells as a principal mode of metabolic inhibition by methotrexate. J Biol Chem. 1987;262:13520–13526. [PubMed] [Google Scholar]
- 25.Allegra CJ, Drake JC, Jolivet J, Chabner BA. Inhibition of Folate-Dependent Enzymes by Methotrexate Polyglutamates. 1985:348–359. [Google Scholar]
- 26.Matherly LH, Barlowe CK, Phillips VM, Goldman ID. The effects of 4-aminoantifolates on 5-formyltetrahydrofolate metabolism in L1210 cells. J Biol Chem. 1987;262:710–717. [PubMed] [Google Scholar]
- 27.Fry DW, Anderson LA, Borst M, Goldman ID. Analysis of the role of membrane transport and polyglutamylation of methotrexate in gut and Ehrlich tumor in vivo as factors in drug sensitivity and selectivity. Cancer Res. 1983;43:1087–1092. [PubMed] [Google Scholar]
- 28.Fabre I, Fabre G, Goldman ID. Polyglutamylation, an important element in methotrexate cytotoxicity and selectivity in tumor versus murine granulocytic progenitor cells in vitro. Cancer Res. 1984;44:3190–3195. [PubMed] [Google Scholar]
- 29.Poser RG, Sirotnak FM, Chello PL. Differential synthesis of methotrexate polyglutamates in normal proliferative and neoplastic mouse tissues in vivo. Cancer Res. 1981;41:4441–4446. [PubMed] [Google Scholar]
- 30.Koizumi S, Curt GA, Fine RL, Griffin JD, Chabner BA. Formation of methotrexate polyglutamates in purified myeloid precursor cells from normal human bone marrow. J Clin Invest. 1985;75:1008–1014. doi: 10.1172/JCI111761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matherly LH, Fry DW, Goldman ID. Role of methotrexate polyglutamylation and cellular energy metabolism in inhibition of methotrexate binding to dihydrofolate reductase by 5-formyltetrahydrofolate in Ehrlich tumor cells in vitro. Cancer Res. 1983;43:2694–2699. [PubMed] [Google Scholar]
- 32.Matherly LH, Barlowe CK, Goldman ID. Antifolate polyglutamylation and competitive drug displacement at dihydrofolate reductase as important elements in leucovorin rescue in L1210 cells. Cancer Res. 1986;46:588–593. [PubMed] [Google Scholar]
- 33.Goldin A, Mantel N, Greenhouse SW, Venditti JM, Humphreys SR. Estimation of the antileukemic potency of the antimetabolite aminopterin, administered alone and in combination with citrovorum factor or folic acid. Cancer Res. 1953;13:843–850. [PubMed] [Google Scholar]
- 34.Goldin A, Venditti J, Kline I, Mantel N. Eradication of Leukaemic Cells (L1210) by Methotrexate adn Methotrexate plus citrovorum factor. Nature. 1966;212:1548–1550. doi: 10.1038/2121548a0. [DOI] [PubMed] [Google Scholar]
- 35.Browman GP, Goodyear MD, Levine MN, Russell R, Archibald SD, Young JE. Modulation of the antitumor effect of methotrexate by low-dose leucovorin in squamous cell head and neck cancer: a randomized placebo-controlled clinical trial. J Clin Oncol. 1990;8:203–208. doi: 10.1200/JCO.1990.8.2.203. [DOI] [PubMed] [Google Scholar]