

Abstract
Efficient degradation of plant polysaccharides in rumen requires xylanolytic enzymes with a high catalytic capacity. In this study, a full-length xylanase gene (xynA) was retrieved from the sheep rumen. The deduced XynA sequence contains a putative signal peptide, a catalytic motif of glycoside hydrolase family 10 (GH10), and an extra C-terminal proline-rich sequence without a homolog. To determine its function, both mature XynA and its C terminus-truncated mutant, XynA-Tr, were expressed in Escherichia coli. The C-terminal oligopeptide had significant effects on the function and structure of XynA. Compared with XynA-Tr, XynA exhibited improved specific activity (12-fold) and catalytic efficiency (14-fold), a higher temperature optimum (50°C versus 45°C), and broader ranges of temperature and pH optima (pH 5.0 to 7.5 and 40 to 60°C versus pH 5.5 to 6.5 and 40 to 50°C). Moreover, XynA released more xylose than XynA-Tr when using beech wood xylan and wheat arabinoxylan as the substrate. The underlying mechanisms responsible for these changes were analyzed by substrate binding assay, circular dichroism (CD) spectroscopy, isothermal titration calorimetry (ITC), and xylooligosaccharide hydrolysis. XynA had no ability to bind to any of the tested soluble and insoluble polysaccharides. However, it contained more α helices and had a greater affinity and catalytic efficiency toward xylooligosaccharides, which benefited complete substrate degradation. Similar results were obtained when the C-terminal sequence was fused to another GH10 xylanase from sheep rumen. This study reveals an engineering strategy to improve the catalytic performance of enzymes.
INTRODUCTION
Xylan is the most abundant hemicellulose in the world, accounting for 7 to 30% of the dry weight of different plant cell walls (1). It is made up of a xylose backbone linked by β-1,4-glycoside bonds and various glucuronic acid side chains. Complete degradation of xylan requires different enzymes to act synergistically due to its structural heterogeneity. Among them, endo-β-1,4-xylanase (EC 3.2.1.8) is the crucial enzyme that catalyzes the hydrolysis of β-1,4 xylose linkages in the backbone while releasing xylose and short xylooligosaccharides (2). On the basis of the similarity of amino acid sequences, most endo-β-1,4-xylanases are confined to glycosyl hydrolase family 10 (GH10) and GH11 (http://www.cazy.org/) (3). GH10 xylanases have a catalytic domain of a classic (α/β)8 barrel fold, in which the catalytic cleft is located on the surface of the C-terminal side of the central barrel (4, 5). Two conserved glutamic acid residues are located in the open cleft of the catalytic module in order to recognize and interact with the substrate through hydrogen bonds or hydrophobic stacking interactions and act as the nucleophile and acid/base catalyst (3, 6, 7). Xylanases have been attracting extensive attention because of their potential application in various industrial processes, such as textile, pulp and paper, food, animal feed, and biofuel production and biobleaching (8, 9). In recent years, technical, environmental, and economic progress has triggered the exploration of greater microbial resources to obtain more efficient biocatalysts for industrial demands. High catalytic efficiency is a key indicator of favorable xylanases.
Ruminants are known to be a powerful bioreactor of lignocellulose degradation, providing an anoxic environment for many organisms to secrete varieties of enzymes to simultaneously attack the plant cell wall (10). So far, many xylanase genes have been obtained from pure cultures of ruminal microorganisms. However, these cultured bacteria account for only less than 15% of the ruminal microorganisms (11). Moreover, in this particular environment, some ruminal xylanases with special characteristics have been demonstrated, such as the bifunctional xylanase/endoglucanase from yak rumen microorganisms (12), the bifunctional endo-/exo-type cellulase from an anaerobic ruminal bacterium (13), and the cold-active xylanase from goat rumen (14).
We previously studied the expression pattern of GH10 xylanases in sheep rumen during a feeding cycle and found that a few genes were predominant during the entire process (15). Of these, xynA was determined by real-time quantitative PCR to be the most highly transcribed xylanase gene. In this study, we expressed its recombinant protein in Escherichia coli. Analysis of the polypeptide sequence showed that XynA has an extra proline-rich sequence at the C terminus with unknown functions. The objective of this study was to examine the effect of this sequence on the enzyme function and extend its role to other xylanases.
MATERIALS AND METHODS
Strains, plasmids, and chemicals.
E. coli strains Trans I-T1 and BL21(DE3) from TransGen (Beijing, China) were used for plasmid amplification and expression, respectively. The plasmids pGEM-T Easy (Promega, Madison, WI) and pET-22b(+) (Invitrogen, Carlsbad, CA) were used for gene cloning and heterologous gene expression, respectively. LA Taq DNA polymerase, a DNA purification kit, and restriction endonucleases were purchased from TaKaRa (Otsu, Japan), and T4 DNA ligase was obtained from New England BioLabs (Hitchin, United Kingdom). Birch wood xylan, beech wood xylan, soluble/insoluble wheat arabinoxylan, p-nitrophenyl-β-d-xylopyranoside (pNPX), low-viscosity carboxymethyl cellulose sodium (CMC-Na), Avicel PH-101 microcrystalline cellulose, lichenan, barley β-glucan, and xylooligosaccharide standards xylose, xylobiose, xylotriose, xylotetraose, xylopentaose, and xylohexaose were purchased from Sigma (St. Louis, MO). Direct-load prestained protein marker III was purchased from GenStar (Beijing, China). All the other chemicals were of analytical grade and commercially available.
Gene cloning and sequence analysis.
The genomic DNA of sheep rumen contents was extracted using the cetyltrimethylammonium bromide method (16) and purified with a TaKaRa DNA purification kit. On the basis of the partial sequence of xynA (GenBank accession number JX154664) (15), specific primers were designed and used to amplify the 5′ and 3′ flanking regions by thermal asymmetric interlaced (TAIL) PCR with the genomic DNA of sheep rumen contents as the template (17). After sequence assembly, an open reading frame (ORF) was identified with the ORF finder at NCBI (http://www.ncbi.nlm.nih.gov). Comparisons of the sequences with known sequences were conducted with the BLASTN, BLASTX, and BLASTP programs at NCBI. The signal peptide sequence was predicted using the SignalP (version 4.0) server (www.cbs.dtu.dk/services/SignalP/). A multiple-protein-sequence alignment was carried out using the ClustalW program (http://www.ebi.ac.uk/clustalW). The modular structure of the enzyme was predicted by use of SWISS-MODEL online (http://swissmodel.expasy.org) with the xylanase from Geobacillus stearothermophilus as the template (PDB accession number 2Q8X). The molecular mass and isoelectric point of the mature peptide were calculated using Vector NTI (version 10.0) software (Invitrogen).
Mutant construction, protein expression, and purification.
Sequence analysis indicated that XynA had an extra 60 residues at the C terminus. To verify the function of this fragment, we retrieved the nucleotide sequences of mature XynA without the signal peptide-coding sequence and its truncated version (XynA-Tr) without the C-terminal sequence by PCR with primers xynA-expF/xnyA-expR and xynA-expF/xynA-expR-Tr (see Table S1 in the supplemental material), respectively. The PCR fragments were digested by EcoRI and NotI and ligated into the pET-22b(+) vector. The resulting plasmids, pET-xynA and pET-xynA-Tr, respectively, were transformed into E. coli BL21(DE3). After confirmation with restriction digestion and DNA sequencing, the positive transformants harboring pET-xynA or pET-xynA-Tr were used for enzyme production with IPTG (isopropyl-β-d-thiogalactopyranoside) induction (1 mM) at 25°C for 16 h (14). The recombinant proteins were purified to electrophoretic homogeneity by ultrafiltration, ammonium sulfate fractionation (40 to 60% saturation), and elution with 10 to 400 mM imidazole in buffer A (20 mM Tris-HCl, 50 mM NaCl, pH 8.0) at a flow rate of 2 ml s−1 using nickel-affinity chromatography (GE Healthcare, Uppsala, Sweden). Protein fractions were collected and analyzed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE; 12% separation gel and 5% spacer gel). The protein concentration was determined using a protein assay kit (Bio-Rad, Hercules, CA).
Enzymatic activity assay and characterization.
Beech wood xylan was used as the substrate in all the enzymatic reactions except those specifically mentioned. Each reaction system contained 950 μl of 1% (wt/vol) xylan substrate and 50 μl of 2 μM protein sample. Xylanase activity was determined by measuring the release of reducing sugar with the dinitrosalicylic acid (DNS) method (18). One unit (U) of xylanase activity was defined as the amount of enzyme that released 1 μmol of reducing sugar equivalent to xylose per minute under standard conditions (pH 6.0, 37°C, 10 min).
The pH optima for the enzyme activities of recombinant XynA and XynA-Tr were estimated at 37°C in McIlvaine buffer (200 mM sodium phosphate, 100 mM sodium citrate, pH 3.0 to 8.0) for 10 min. To determine the optimal temperature for activity, the assays were performed at temperatures ranging from 10°C to 65°C at the pH optimum determined as described above. For analysis of pH stability, the enzymes were preincubated in McIlvaine buffer (pH 3.0 to 7.0), 200 mM Tris-HCl (pH 7.0 to 9.0), or 200 mM glycine-NaOH (pH 9.0 to 10.0) without substrate at 37°C for 1 h, and residual enzyme activities were measured under standard conditions (pH 6.0, 37°C, 10 min). The thermal stability of the enzymes was determined by measuring the residual enzyme activities after incubation at 45°C, 55°C, or 60°C for various durations.
Determination of substrate specificities and kinetic parameters.
The substrate specificities were determined by measuring the enzyme activity at pH 6.0 and 45°C for XynA or 50°C for Xyn-Tr in McIlvaine buffer containing 1% beech wood xylan, birch wood xylan, soluble/insoluble wheat arabinoxylan, or CMC-Na as the substrate. To determine the xylosidase activity, 2 mM p-nitrophenyl-β-d-xylopyranoside was used as the substrate, as described previously (19). The kinetic parameters were determined in McIlvaine buffer containing 1 to 20 mg ml−1 beech wood xylan with 50 μl of 2 μM protein sample under standard conditions for 5 min. Km and Vmax values were determined on the basis of Lineweaver-Burk plots using GraphPad Prism (version 5) software (GraphPad Software Inc., La Jolla, CA). The experiments were carried out three times, and each experiment included triplicates.
Analysis of hydrolysis products.
The reaction mixture, containing 900 μl of 0.5% (wt/vol) beech wood xylan or soluble wheat arabinoxylan and 100 μl of enzyme sample (10 U of XynA, XynA-Tr, XynB, or an XynB fusion protein [XynB-Fu]), was incubated under optimal conditions for 12 h. The extra enzyme was removed from the reaction system by use of a Nanosep centrifugal 3K device (Pall, New York, NJ). The hydrolysis products of XynA and XynA-Tr against various xylan substrates were determined by high-performance anion-exchange chromatography (HPAEC) on a chromatograph which was equipped with a Dionex CarboPac PA-100 (4- by 250-mm) column (Sunnyvale, CA). The products were eluted by buffer B (100 mM NaOH, 210 mM sodium acetate). Xylose, xylobiose, xylotriose, and xylotetraose were used as standards.
Site-directed mutagenesis of XynA and XynA-Tr.
To avoid a catalysis reaction in the binding assay and titration experiment described below, site-directed mutagenesis was conducted to obtain inactive mutants XynA-E151Q and XynA-Tr-E151Q in which the catalytic residue glutamic acid was replaced by glutamine (E151Q) by overlap PCR with primers XynA-E151QF and XynA-E151QR (see Table S1 in the supplemental material). Protein expression and purification were conducted as described above with IPTG induction at 30°C for 8 h. The recombinant proteins were directly purified by nickel-affinity chromatography and analyzed by SDS-PAGE, as described above. The identities of the protein bands with the expected molecular masses (44.06 kDa for XynA-E151Q and 37.68 kDa for XynA-Tr-E151Q) were verified by nano liquid chromatography–electrospray ionization–collision-induced dissociation–tandem mass spectrometry (nano-LC-ESI-CID-MS/MS). An enzyme assay was conducted to check their xylanase activities.
Assay for binding to insoluble and soluble polysaccharides.
To verify the role of the extra C-terminal oligopeptide in substrate binding, the ability of inactive XynA-E151Q to bind to insoluble and soluble polysaccharides was evaluated by SDS-PAGE as described above and native affinity PAGE (in which no SDS was added), respectively. For insoluble polysaccharides, approximately 20 μg of inactive XynA-E151Q was incubated with 1 to 4% Avicel or insoluble wheat arabinoxylan in 200 mM phosphate buffer (pH 6.0) at 4°C for 1 h with gentle agitation (150 rpm), followed by centrifugation at 12,000 × g for 20 min. Unbound protein in the supernatant was removed, and the precipitate was washed with the same phosphate buffer 5 times to remove residual, unbound protein. The protein fraction that bound to the insoluble polysaccharides, including the target protein, was collected by centrifugation, suspended in 100 μl of phosphate buffer as described above, and boiled at 100°C for 10 min. The recovered protein was subjected to SDS-PAGE analysis as described above. Native PAGE of the soluble polysaccharides, including beech wood xylan, wheat arabinoxylan, CMC-Na, lichenan, and barley β-glucan, was performed at 4°C with a constant voltage of 120 V using 13% acrylamide containing 0.2% (final concentration) each soluble substrate. The protein was visualized by Coomassie blue staining.
CD spectroscopy.
To verify the structural difference between XynA and XynA-Tr, the proteins were monitored by a MOS-450 circular dichroism (CD spectrometer (Bio-Logic, Claix, France) equipped with a TCU-250 Peltier-type temperature control system. Far-UV (190 to 250 nm) and near-UV (250 to 320 nm) CD spectra (20) were measured at 1-nm intervals at room temperature. The proteins (1 mg ml−1) in phosphate-buffered saline (PBS) buffer (1.42 g liter−1 Na2HPO4, 0.8 g liter−1 NaCl, 0.27 g liter−1 KH2PO4, 0.2 g liter−1 KCl, pH 6.0) were subjected to signal measurement in a 5-mm cell. All measurements were repeated three times.
Isothermal titration calorimetry (ITC).
The thermodynamics of inactive XynA-E151Q and XynA-Tr-E151Q were determined by use of an iTC 200 microcalorimeter (GE Healthcare, Waukesha, WI). The proteins and xylooligosaccharide substrates were dialyzed against citric acid-Na2HPO4 buffer (50 mM Na2HPO4, 25 mM citrate, pH 6.0) overnight at 4°C. The protein concentrations were measured three times, and the protein concentration was adjusted to a final concentration of 50 μM. Each substrate (2 μl, 1.0 mM) was injected into the protein cell 20 times with a 4-μl rotating stirrer-syringe. The titration of the ligand with buffer was the control. The calorimetric data were fitted to the binding isotherms by the use of Origin (version 5.0) software (GE Healthcare), and the association constant (ka) was calculated accordingly.
Xylooligosaccharide hydrolysis.
To evaluate the catalytic efficiency of XynA and XynA-Tr toward xylooligosaccharides, each enzyme sample (2 μM) was incubated with 200 μg of xylobiose, xylotriose, xylotetraoside, xylopentaoside, or xylohexaose at each optimal pH and temperature for up to 60 min. Aliquots (100 μl) were collected at each time point, heated at 100°C for 5 min, and filtered through the Nanosep centrifugal 3K device (Pall). The hydrolysis products were analyzed by HPAEC with continuous elution of 100 mM NaOH (5 min) and 0 to 100 mM sodium acetate (15 min). Xylooligosaccharides were used as the standards. The catalytic efficiency values were then determined following the equation described by Ichinose et al. (21).
Effect of the extra C-terminal sequence on another ruminal xylanase.
The coding sequence of the extra C-terminal oligopeptide was fused to another xylanase gene (xynB; GenBank accession number JX154662) from sheep rumen at its C terminus by overlap PCR with primers xynB-F, xynB-A-F, xynB-A-R, and xnyA-expR (see Table S1 in the supplemental material). Expression, purification, and characterization of the fusion protein XynB-Fu were conducted as described above. The wild-type and fused proteins, XynB and XynB-Fu, respectively, were also subjected to analysis of hydrolysis products.
RESULTS
Gene cloning, sequence analysis, and protein expression.
The 1,221-bp full-length xynA was cloned from the genomic DNA of sheep rumen by TAIL PCR. The deduced amino acid sequence exhibited 59.4% identity with xylanase XynB (GenBank accession number AGC54683) of the same sheep rumen and 59% identity with a putative xylanase (GenBank accession number ADX05679) from the uncultured cow rumen microorganism. Alignment of the sequence of the deduced mature XynA with the sequences of 12 closely related ruminal xylanases (see Fig. S1 in the supplemental material) indicated that XynA contained a putative signal peptide (24 residues), a catalytic motif of GH10 (323 residues), and an extra C-terminal oligopeptide (60 residues). Structure modeling with G. stearothermophilus xylanase as the template indicated that without the extra C-terminal sequence, XynA had a structure typical of that of GH10 xylanases and Glu151 and Glu260 corresponded to the catalytic residues. No disulfide bond formed between the four cysteine residues (Cys82/106/113/146). The C-terminal oligopeptide was proline rich, with a proline content of 20%, which was much higher than that of XynA (5.74%). The theoretical molecular masses and the isoelectric points of XynA were 44.06 kDa and 6.45, respectively, and those of its truncated mutant without the C-terminal sequence (XynA-Tr) were 37.68 kDa and 5.88, respectively.
The gene fragments coding for mature XynA and XynA-Tr were amplified from the sheep rumen genomic DNA, ligated into pET-22b(+) to obtain the recombinant plasmids pET-xynA and pET-xynA-Tr, and transformed into E. coli competent cells. The supernatants and the precipitates of positive transformants that harbored the recombinant plasmids were screened for xylanase activities after IPTG induction. Purified recombinant XynA and XynA-Tr each showed a single band at 43 kDa and 37 kDa, respectively, on an SDS-polyacrylamide gel (see Fig. S2 in the supplemental material).
Enzyme properties of XynA and XynA-Tr.
Purified recombinant XynA and its truncated derivative, XynA-Tr, had similar pH optima at 6.0, but XynA retained higher activities than XynA-Tr at pH 5.0 and pH 6.5 to 8.0 (Fig. 1A). The optimal temperature of XynA was 50°C, 5°C higher than that of XynA-Tr (Fig. 1B). Moreover, XynA retained more activity at 55 to 65°C than XynA-Tr. The two enzymes were stable (activities, above 80%) over a broad pH range of 5.0 to 9.0 at 37°C for 1 h (Fig. 1C) and at 55°C and below (Fig. 1D). Compared with XynA, truncated XynA-Tr retained more activity under the physical conditions of the rumen (pH 5.0 to 7.0 and 39°C) (22).
FIG 1.

Characterization of purified recombinant XynA and XynA-Tr. (A) Effect of pH on xylanase activity at 37°C; (B) effect of temperature on xylanase activity at pH 6.0; (C) pH stability at 37°C for 1 h in buffers of pH 3.0 to 10.0; (D) thermostability at 45°C, 55°C, and 60°C.
Substrate specificity and kinetic parameters.
XynA and XynA-Tr exhibited the highest specific activities toward soluble wheat arabinoxylan (35.72 × 10−6 katal mg−1 and 18.83 × 10−6 katal mg−1, respectively) and moderate activity on beech wood xylan (18.92 × 10−6 katal mg−1 and 1.62 × 10−6 katal mg−1, respectively) and birch wood xylan (12.52 × 10−6 katal mg−1 and 3.66 × 10−7 katal mg−1, respectively). No activity was detected when pNPX, CMC-Na, or insoluble wheat arabinoxylan was used as the substrate. The kinetic values of both enzymes are shown in Table 1. XynA had higher values of Vmax, kcat, and hydrolysis efficiency (kcat/Km) than XynA-Tr but lower Km values than XynA-Tr.
TABLE 1.
Kinetic data for the four xylanases with beech wood xylan as the substrate
| Xylanase | Km (mg ml−1) | Vmax (μmol min−1 mg−1) | kcat (s−1) | kcat/Km (ml s−1 mg−1) |
|---|---|---|---|---|
| XynA | 5.40 ± 0.19 | 2,071 ± 30 | 1,521 ± 18 | 282 |
| XynA-Tr | 7.05 ± 1.88 | 222 ± 15 | 139 ± 4 | 19.8 |
| XynB-Fu | 1.71 ± 0.13 | 812 ± 17 | 605 ± 8 | 354 |
| XynB | 4.55 ± 0.43 | 94.4 ± 3.7 | 59.8 ± 2.8 | 13.1 |
Analysis of hydrolysis products.
The hydrolysis products of XynA and XynA-Tr against beech wood xylan and soluble wheat arabinoxylan were analyzed. As shown in Table 2, xylose and xylobiose were the main hydrolysis products of XynA degradation. The hydrolysis product composition of XynA-Tr was more complex, comprising xylose, xylobiose, and xylotriose. The results suggested that XynA degraded the xylan substrate more completely than its truncated derivative.
TABLE 2.
Hydrolysis products of beech wood xylan and soluble wheat arabinoxylan degraded by four xylanasesa
| Hydrolysis product | % hydrolysis producta |
|||||||
|---|---|---|---|---|---|---|---|---|
| Beech wood xylan |
Wheat arabinoxylan |
|||||||
| XynA | XynA-Tr | XynB-Fu | XynB | XynA | XynA-Tr | XynB-Fu | XynB | |
| Xylose | 29.0 | 9.45 | 24.2 | 4.00 | 25.6 | 9.67 | 26.3 | 19.4 |
| Xylobiose | 55.8 | 55.6 | 62.4 | 53.9 | 30.6 | 16.7 | 34.2 | 23.1 |
| Xylotriose | 16.8 | 1.99 | 23.2 | 6.62 | 1.22 | 11.5 | ||
| Xylotetraose | 1.32 | |||||||
| Other xylooligosaccharides | 15.1 | 18.1 | 11.4 | 17.6 | 43.8 | 67.0 | 38.2 | 46.0 |
The sum of the hydrolysis products is defined as 100%, and the amount of each hydrolysis product is presented as a percentage of the total; the detection limit was defined to be 0.01%.
Identification and activity assay of mutant enzymes.
XynA-E151Q and XynA-Tr-E151Q were successfully expressed, purified (see Fig. S2 in the supplemental material), and verified by nano-LC-ESI-CID-MS/MS analysis. The mutant enzymes completely lost the capacity to hydrolyze beech wood xylan on the basis of the results of the DNS method (data not shown).
Abilities to bind to various insoluble and soluble polysaccharides.
As shown in Fig. S3 in the supplemental material, no distance difference was detected between the proteins treated with or without soluble polysaccharides (beech wood xylan, wheat arabinoxylan, CMC-Na, lichenan, and barley β-glucan) in native affinity polyacrylamide gels, and no bound protein was observed in the SDS-polyacrylamide gels when the proteins were treated with the insoluble polysaccharides Avicel and insoluble wheat arabinoxylan. The results indicated that XynA had no capacity to bind to any of the tested soluble and insoluble polysaccharides.
CD and ITC analysis.
The CD spectra in the far-UV and near-UV ranges (190 to 250 nm and 250 to 350 nm, respectively) of XynA-Tr were greatly altered by removal of the extra C-terminal oligopeptide (Fig. 2). Far-UV spectra indicated that the helical and sheet components of XynA-Tr were reduced. After truncation, the contents of aromatic residues (Tyr/Phe/Trp) increased from 11.5% (XynA) to 12.1% (XynA-Tr). This change (0.6%) made a contribution to the increase of the CD spectra in the near-UV region, but it was not as much as that shown in Fig. 2 (>50%). Thus, we assumed that this extra C-terminal oligopeptide had effects on both the secondary and tertiary structures of XynA.
FIG 2.
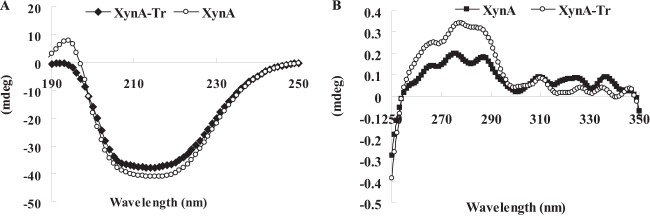
Secondary and tertiary structures of XynA and XynA-Tr verified by CD analysis. (A) Far-UV CD spectra; (B) near-UV CD spectra.
The association constant (ka), catalytic efficiency, and raw binding curves of the two proteins with four xylooligosaccharide substrates are shown in Table 3 and Fig. S4 in the supplemental material. For inactive XynA, the ka value ranged from 3.08 × 104 M−1 to 2.74 × 105 M−1. The ka values of inactive XynA-Tr were much lower, ranging from 4.6 × 102 M−1 to 3.05 × 103 M−1. XynA also showed a greater catalytic efficiency to hydrolyze xylooligosaccharides than its truncated mutant. The results suggested that XynA had a stronger affinity and hydrolysis capacity toward xylooligosaccharide substrates than XynA-Tr.
TABLE 3.
ka and catalytic efficiency (kcat/Km) of XynA and XynA-Tr toward xylooligosaccharides of different lengths
| Hydrolysis product |
ka (M−1 [103]) |
kcat/Km (mM−1 min−1) |
||
|---|---|---|---|---|
| XynA | XynA-Tr | XynA | XynA-Tr | |
| Xylohexaose | 274 ± 21.1 | 1.45 ± 0.13 | 831.4 ± 2.0 | 1.24 ± 0.03 |
| Xylopentaose | 241 ± 14.4 | 3.05 ± 0.08 | 162.2 ± 2.0 | 0.60 ± 0.07 |
| Xylotetraose | 134 ± 36.6 | 1.22 ± 0.03 | 128.2 ± 0.5 | 1.12 ± 0.10 |
| Xylotriose | 30.8 ± 4.1 | 0.46 ± 0.03 | 135.6 ± 1.3 | NDa |
ND, not detected.
Effect of the C-terminal sequences on another xylanase.
The DNA sequence coding for the C-terminal oligopeptide was fused to xynB of the same sheep rumen source at the C terminus. The fused enzyme XynB-Fu was expressed in E. coli, displayed xylanase activity, and was purified to electrophoretic homogeneity, as shown in SDS-polyacrylamide gels (see Fig. S2 in the supplemental material). XynB-Fu showed a pH optimum similar to that of XynB, but the optimal temperature for XynB-Fu increased from 35°C to 40°C (data not shown). Notably, in the presence of the extra C-terminal sequence, XynB-Fu showed a significantly enhanced catalytic efficiency and degraded xylan substrates more completely (Tables 1 and 2).
DISCUSSION
The rumen harbors a complex microbial community with thousands of species which produce enzymes of various functional groups. Because xylan degradation by xylanase is the first step taken to break down the lignocellulose in feedstuff, a xylanase with high catalytic performance is more favorable for feedstuff utilization. In our previous study of xylanase expression in sheep rumen, xynA was found to represent the most predominantly expressed xylanase gene of GH10 (15). We conjecture that XynA has certain favorable characteristics over others in the highly competitive rumen environment. In this study, we cloned the full-length xynA gene, expressed it in E. coli, and determined its enzymatic properties. Compared with two other highly transcribed xylanases, XynB and XynC, of the same rumen source (15), XynA had a similar pH optimum (6.0) but a higher temperature optimum (50°C versus 40°C), a better thermostability at 55°C, and higher catalytic activity (1,135 U mg−1 versus 73.9 and 142.3 U mg−1). The physiological pH and temperature of the rumen are pH 5.0 to 7.0 and 39°C, respectively (22). Although the temperature optimum of XynA is higher than the temperature of the rumen, XynA preserved over 60% of its maximal activity, which is still substantially higher than that of XynB and XynC. Sequence analysis indicated that XynA contains an extra proline-rich sequence at the C terminus that has no homologs in the GenBank database. The effect of this sequence on enzyme function and structure was thus determined in the study.
Besides a catalytic domain, xylanases often exhibit a modular structure composed of a carbohydrate binding module (CBM), a xylan-binding domain, a linker sequence, a thermostabilizing domain, a repeated sequence, and domains whose functions are unknown. These noncatalytic domains have been proven to have effects on the biochemical and biophysical properties of enzymes (2). In addition, terminal sequences also influence enzymes in different ways (23). For example, the C-terminal region of Trichoderma reesei endo-1,4-xylanase changed its half-life (24), deletion of the N- and C-terminal sequences of Aspergillus niger xylanase resulted in a lower temperature optimum and decreased thermostability (25), and disruption of the terminal interaction of a GH10 xylanase resulted in decreased stability (26). The reason might be that removal of these terminal sequences changed the secondary structure contents, aromatic cluster formation, in vivo folding, and Gibbs free energy. In this study, we also identified an extra 60-residue C-terminal sequence that is proline rich. Proline-rich regions occur widely in the N and C termini or between domains of both prokaryotic and eukaryotic proteins (27). These sequences not only function as a linker in many enzymes but also influence enzyme properties. However, these changes are specific; for example, Huang et al. (28) reported that deletion of the C-terminal proline-rich segment of phospholipase A2 changed the substrate specificity and uncoupled calcium and substrate binding, Li et al. (29) indicated that deletion of the glycine/proline-rich linker of XynAS27 changed the pH and temperature ranges but not the catalytic efficiency, and Wen et al. (30) showed that deletion of the C-terminal proline-rich region of a 1,3-1,4-β-d-glucanase improved the enzymatic activity and thermotolerance. In the presence of the C-terminal proline-rich sequence, XynA retained more activity than its truncated mutant, XynA-Tr, over broader pH (pH 5.5 to 6.5 versus pH 5.0 to 7.5) and temperature (40 to 50°C versus 40 to 60°C) ranges. Considering the characteristics of proline, such as a flat hydrophobic surface and a unique amide bond, it may represent a strong hydrogen bond acceptor and may thus play a role in peptide bond formation (31–33). CD analysis indicated that this proline-rich sequence did affect the secondary and tertiary structures of XynA. However, without a homolog to the proline-rich sequence, we failed to predict the structure of XynA.
Catalytic efficiency is a measurement of the enzyme-substrate reaction system. When using beech wood xylan as the substrate, the catalytic efficiency of XynA was 18.2-fold of that of XynA-Tr (Table 1), and XynA released much more xylose than XynA-Tr (29.01% versus 9.45%). When using xylooligosaccharides as the substrate, the catalytic efficiency of XynA improved to 100-fold of that of XynA-Tr (Table 3). The proline-rich region has been reported to interact with substrates at multiple weak binding sites in a rapid and nonspecific way (27). By use of a combination of the Km values (similar to the substrate dissociation constant [Kd] when in the range of 0.4 to 40 μM [34]), the finding of the inability of XynA to bind all tested insoluble and soluble polysaccharides, the finding that that XynA had the capacity to hydrolyze xylooligosaccharides, and the end product compositions obtained by XynA and XynA-Tr hydrolysis, we attempted to understand the function of this terminal sequence in relation to that of the entire enzyme. ITC analysis revealed that this C-terminal proline-rich sequence results in a stronger affinity to all tested xylooligosaccharides, corresponding to the results of xylooligosaccharide hydrolysis. We thus assume that this C-terminal sequence plays a role different from that of CBM, as reported before (35).
Construction of fusion proteins is an important tool in protein engineering to improve natural enzymes. Yang et al. (36) fused an oligopeptide to the N terminus of an α-amylase and improved its catalytic efficiency, thermal stability, and resistance to oxidation. In this study, we fused the proline-rich sequence of XynA to the C terminus of another ruminal xylanase (XynB) of the same source and the same family (15). The fused protein showed an improved catalytic efficiency of 27-fold and more complete hydrolysis products, mainly xylose and xylobiose. The optimal activity of XynB occurred at 40°C and pH 6.0 (15), which are similar to the physiological conditions of the rumen (pH 5.0 to 7.0 and 39°C [22]). Thus, XynB fused with the C-terminal proline-rich sequence could be produced in a high-yield expression system and processed as a feed additive for better feedstuff utilization.
In summary, we identified a C-terminal proline-rich sequence in the ruminal xylanase XynA of GH10. This oligopeptide simultaneously increased the optimal temperature, broadened the pH and temperature ranges, enhanced the catalytic efficiency, and degraded the xylan substrate more completely. Analysis of the mechanisms underlying these changes revealed that this proline-rich sequence not only changed the secondary and tertiary structures but also improved the affinity and hydrolysis capacity of the enzyme toward xylooligosaccharides. Its effectiveness was verified by use of another ruminal xylanase of the same source and same family. Therefore, this method appears to have great potential for the engineering of other microbial enzymes. In future studies, we plan to obtain the crystal structure of XynA to obtain a better mechanistic understanding and to engineer more industrial xylanases for better performance.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Natural Science Foundation for Distinguished Young Scholars of China (31225026), the National Science and Technology Support Program (2011BADB02), the China Modern Agriculture Research System (CARS-42), and the Foundation (no. 2013IM101) of the Key Laboratory of Industrial Fermentation Microbiology of the Ministry of Education and the Tianjin Key Lab of Industrial Microbiology (Tianjin University of Science and Technology).
Footnotes
Published ahead of print 21 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00016-14.
REFERENCES
- 1.Singh S, Madlala AM, Prior BA. 2003. Thermomyces lanuginosus: properties of strains and their hemicellulases. FEMS Microbiol. Rev. 27:3–16. 10.1016/S0168-6445(03)00018-4 [DOI] [PubMed] [Google Scholar]
- 2.Collins T, Gerday C, Feller G. 2005. Xylanases, xylanase families and extremophilic xylanases. FEMS Microbiol. Rev. 29:3–23. 10.1016/j.femsre.2004.06.005 [DOI] [PubMed] [Google Scholar]
- 3.Henrissat B, Davies G. 1997. Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 7:637–644. 10.1016/S0959-440X(97)80072-3 [DOI] [PubMed] [Google Scholar]
- 4.Harris GW, Jenkins JA, Connerton I, Cummings N, Leggio LL, Scott M, Hazlewood GP, Laurie JI, Gilbert HJ, Pickersgill RW. 1994. Structure of the catalytic core of the family F xylanase from Pseudomonas fluorescens and identification of the xylopentaose-binding sites. Structure 2:1107–1116. 10.1016/S0969-2126(94)00112-X [DOI] [PubMed] [Google Scholar]
- 5.Torronen A, Harkki A, Rouvinen J. 1994. Three-dimensional structure of endo-1,4-β-xylanase II from Trichoderma reesei: two conformational states in the active site. EMBO J. 13:2493–2501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Birsan C, Johnson P, Joshi M, MacLeod A, McIntosh L, Monem V, Nitz M, Rose D, Tull D, Wakarchuck W. 1998. Mechanisms of cellulases and xylanases. Biochem. Soc. Trans. 26:156–160 [DOI] [PubMed] [Google Scholar]
- 7.Notenboom V, Birsan C, Nitz M, Rose DR, Warren RAJ, Withers SG. 1998. Insights into transition state stabilization of the β-1,4-glycosidase Cex by covalent intermediate accumulation in active site mutants. Nat. Struct. Mol. Biol. 5:812–818. 10.1038/1852 [DOI] [PubMed] [Google Scholar]
- 8.Menon V, Prakash G, Prabhune A, Rao M. 2010. Biocatalytic approach for the utilization of hemicellulose for ethanol production from agricultural residue using thermostable xylanase and thermotolerant yeast. Bioresour. Technol. 101:5366–5373. 10.1016/j.biortech.2010.01.150 [DOI] [PubMed] [Google Scholar]
- 9.Polizeli ML, Rizzatti AC, Monti R, Terenzi HF, Jorge JA, Amorim DS. 2005. Xylanases from fungi: properties and industrial applications. Appl. Microbiol. Biotechnol. 67:577–591. 10.1007/s00253-005-1904-7 [DOI] [PubMed] [Google Scholar]
- 10.Flint HJ, Bayer EA. 2008. Plant cell wall breakdown by anaerobic microorganisms from the mammalian digestive tract. Ann. N. Y. Acad. Sci. 1125:280–288. 10.1196/annals.1419.022 [DOI] [PubMed] [Google Scholar]
- 11.Krause DO, Denman SE, Mackie RI, Morrison M, Rae AL, Attwood GT, McSweeney CS. 2003. Opportunities to improve fiber degradation in the rumen: microbiology, ecology, and genomics. FEMS Microbiol. Rev. 27:663–693. 10.1016/S0168-6445(03)00072-X [DOI] [PubMed] [Google Scholar]
- 12.Chang L, Ding M, Bao L, Chen Y, Zhou J, Lu H. 2011. Characterization of a bifunctional xylanase/endoglucanase from yak rumen microorganisms. Appl. Microbiol. Biotechnol. 90:1933–1942. 10.1007/s00253-011-3182-x [DOI] [PubMed] [Google Scholar]
- 13.Ko KC, Han Y, Choi JH, Kim GJ, Lee SG, Song JJ. 2011. A novel bifunctional endo-/exo-type cellulase from an anaerobic ruminal bacterium. Appl. Microbiol. Biotechnol. 89:1453–1462. 10.1007/s00253-010-2949-9 [DOI] [PubMed] [Google Scholar]
- 14.Wang G, Luo H, Meng K, Wang Y, Huang H, Shi P, Pan X, Yang P, Diao Q, Zhang H, Yao B. 2011. High genetic diversity and different distributions of glycosyl hydrolase family 10 and 11 xylanases in the goat rumen. PLoS One 6:e16731. 10.1371/journal.pone.0016731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Z, Zhao H, Yang P, Zhao J, Huang H, Xue X, Zhang X, Diao Q, Yao B. 2013. Comparative quantitative analysis of gene expression profiles of glycoside hydrolase family 10 xylanases in the sheep rumen during a feeding cycle. Appl. Environ. Microbiol. 79:1212–1220. 10.1128/AEM.02733-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brady SF. 2007. Construction of soil environmental DNA cosmid libraries and screening for clones that produce biologically active small molecules. Nat. Protoc. 2:1297–1305. 10.1038/nprot.2007.195 [DOI] [PubMed] [Google Scholar]
- 17.Huang H, Wang G, Zhao Y, Shi P, Luo H, Yao B. 2010. Direct and efficient cloning of full-length genes from environmental DNA by RT-qPCR and modified TAIL-PCR. Appl. Microbiol. Biotechnol. 87:1141–1149. 10.1007/s00253-010-2613-4 [DOI] [PubMed] [Google Scholar]
- 18.Miller GL, Blum R, Glennon WE, Burton AL. 1960. Measurement of carboxymethylcellulase activity. Anal. Biochem. 1:127–132. 10.1016/0003-2697(60)90004-X [DOI] [Google Scholar]
- 19.Zhao J, Shi P, Li Z, Yang P, Luo H, Bai Y, Wang Y, Yao B. 2012. Two neutral thermostable cellulases from Phialophora sp. G5 act synergistically in the hydrolysis of filter paper. Bioresour. Technol. 121:404–410. 10.1016/j.biortech.2012.07.027 [DOI] [PubMed] [Google Scholar]
- 20.Roberge M, Lewis RN, Shareck F, Morosoli R, Kluepfel D, Dupont C, McElhaney RN. 2003. Differential scanning calorimetric, circular dichroism, and Fourier transform infrared spectroscopic characterization of the thermal unfolding of xylanase A from Streptomyces lividans. Proteins 50:341–354. 10.1002/prot.10262 [DOI] [PubMed] [Google Scholar]
- 21.Ichinose H, Yoshida M, Kotake T, Kuno A, Igarashi K, Tsumuraya Y, Samejima M, Hirabayashi J, Kobayashi H, Kaneko S. 2005. An exo-β-1,3-galactanase having a novel β-1,3-galactan-binding module from Phanerochaete chrysosporium. J. Biol. Chem. 280:25820–25829. 10.1074/jbc.M501024200 [DOI] [PubMed] [Google Scholar]
- 22.Soest PJ. 1994. Nutritional ecology of the ruminant, 2nd ed. Cornell University Press, Ithaca, NY [Google Scholar]
- 23.Verma D, Satyanarayana T. 2012. Molecular approaches for ameliorating microbial xylanases. Bioresour. Technol. 117:360–367. 10.1016/j.biortech.2012.04.034 [DOI] [PubMed] [Google Scholar]
- 24.Turunen O, Etuaho K, Fenel F, Vehmaanperä J, Wu X, Rouvinen J, Leisola M. 2001. A combination of weakly stabilizing mutations with a disulfide bridge in the alpha-helix region of Trichoderma reesei endo-1,4-β-xylanase II increases the thermal stability through synergism. J. Biotechnol. 88:37–46. 10.1016/S0168-1656(01)00253-X [DOI] [PubMed] [Google Scholar]
- 25.Liu L, Sun X, Yan P, Wang L, Chen H. 2012. Non-structured amino-acid impact on GH11 differs from GH10 xylanase. PLoS One 9:e45762. 10.1371/journal.pone.0045762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhardwaj A, Leelavathi S, Mazumdar-Leighton S, Ghosh A, Ramakumar S, Reddy VS. 2010. The critical role of N- and C-terminal contact in protein stability and folding of a family 10 xylanase under extreme conditions. PLoS One 6:e11347. 10.1371/journal.pone.0011347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williamson MP. 1994. The structure and function of proline-rich regions in proteins. Biochem. J. 297:249–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang B, Yu BZ, Rogers J, Byeon IJL, Sekar K, Chen X, Sundaralingam M, Tsai MD, Jain MK. 1996. Phospholipase A2 engineering. Deletion of the C-terminus segment changes substrate specificity and uncouples calcium and substrate binding at the zwitterionic interface. Biochemistry 35:12164–12174 [DOI] [PubMed] [Google Scholar]
- 29.Li N, Shi P, Yang P, Wang Y, Luo H, Bai Y, Zhou Z, Yao B. 2009. A xylanase with high pH stability from Streptomyces sp. S27 and its carbohydrate-binding module with/without linker-region-truncated versions. Appl. Microbiol. Biotechnol. 83:99–107. 10.1007/s00253-008-1810-x [DOI] [PubMed] [Google Scholar]
- 30.Wen TN, Chen JL, Lee SH, Yang NS, Shyur LF. 2005. A truncated Fibrobacter succinogenes 1,3–1,4-β-d-glucanase with improved enzymatic activity and thermotolerance. Biochemistry 44:9197–9205. 10.1021/bi0500630 [DOI] [PubMed] [Google Scholar]
- 31.Hurley JH, Mason DA, Matthews BW. 1992. Flexible geometry conformational energy maps for the amino acid residue preceding a proline. Biopolymers 32:1443–1446. 10.1002/bip.360321104 [DOI] [PubMed] [Google Scholar]
- 32.MacArthur MW, Thornton JM. 1991. Influence of proline residues on protein conformation. J. Mol. Biol. 218:397–412. 10.1016/0022-2836(91)90721-H [DOI] [PubMed] [Google Scholar]
- 33.Nicholson H, Tronrud D, Becktel W, Matthews B. 1992. Analysis of the effectiveness of proline substitutions and glycine replacements in increasing the stability of phage T4 lysozyme. Biopolymers 32:1431–1441. 10.1002/bip.360321103 [DOI] [PubMed] [Google Scholar]
- 34.Zolotnitsky G, Cogan U, Adir N, Solomon V, Shoham G, Shoham Y. 2004. Mapping glycoside hydrolase substrate subsites by isothermal titration calorimetry. Proc. Natl. Acad. Sci. U. S. A. 101:11275–11280. 10.1073/pnas.0404311101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shoseyov O, Shani Z, Levy I. 2006. Carbohydrate binding modules: biochemical properties and novel applications. Microbiol. Mol. Biol. Rev. 70:283–295. 10.1128/MMBR.00028-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang H, Lu X, Liu L, Li J, Shin HD, Chen RR, Du G, Chen J. 2013. Fusion of an oligopeptide at the N terminus of an alkaline α-amylase from Alkalimonas amylolytica simultaneously improves its catalytic efficiency, thermal stability, and oxidative stability. Appl. Environ. Microbiol. 79:3049–3058. 10.1128/AEM.03785-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.