

Abstract
To develop a stable and marker-free Lactobacillus strain useful for the expression of vaccines, we developed a temperature-sensitive suicide plasmid with expression cassettes containing an HCE promoter, a PgsA anchor, the alpha-toxin gene, and an rrnB T1T2 terminator (PPαT) that uses a 5-fluorouracil (5-FU) counterselectable marker for Lactobacillus casei. Three strains containing the correct PPαT expression cassettes were produced via the selective pressure of 5-FU screening. We confirmed that the upp gene was deleted and that the PPαT expression cassettes were inserted into the upp site of L. casei ATCC 393 by genomic PCR amplification and sequencing. 5-FU resistance in recombinant bacteria could be stably inherited for as long as 40 generations following insertion. However, bacteria containing the integrated DNA grew more slowly than wild-type L. casei. An indirect enzyme-linked immunosorbent assay (ELISA) analysis demonstrated that the alpha-toxin gene was expressed. Also, we visualized expression of the protein on the surface of L. casei cells using laser confocal microscopy. These results taken together demonstrate that these recombinant bacteria should provide a safe tool for effective vaccine production.
INTRODUCTION
Lactic acid bacteria (LAB) have shown significant potential as vaccine delivery vehicles, primarily through the use of plasmids that express bioactive compounds at mucosal surfaces, where they can stimulate appropriate immune responses (1–3). LAB offer several advantages over current systemic vaccination routes. Several strains may act as natural adjuvants, potentially eliminating the use of toxic adjuvants common in systemic vaccines. Strains have also been identified that protect against degradation during passage of the vaccine through the gastrointestinal tract to the mucosal surface, where they may induce both mucosal and systemic immunity (4).
Lactobacillus casei is a lactic acid bacterium, and its effect on immune cells in the gut has been extensively studied (5–9). It has been reported that this probiotic bacterium interacts with gut-associated lymphoid tissue (GALT) and makes contact with immune cells associated with Peyer's patches and the lamina propria of the intestinal mucosa (10). Cells from the innate immune response have been proposed to be the main target of L. casei for the induction of immune stimulation in the gut (9). These observations suggest that probiotic bacteria have the potential for use as vaccine delivery vehicles.
L. casei has been used previously to express plasmid-encoded protective antigens against porcine parvovirus VP2 and Escherichia coli K88 and K99, thus conferring on mice protection against both porcine parvovirus and lethal E. coli challenge (11, 12). Bioactive compounds must be expressed at a high enough level to elicit the desired immune response (3). While the high copy numbers of some plasmids may seem advantageous for antigen expression, plasmid instability and the selective pressure required for plasmid maintenance complicate their use in human clinical applications (13, 14). Integrating genes into the chromosome for expression is expected to eliminate selection requirements and provide genetic stability (4).
The upp gene encodes uracil phosphoribosyltransferase (UPRTase). This enzyme belongs to the pyrimidine salvage pathway and creates UMP from uracil and phosphoribosyl pyrophosphate (15). The toxic antimetabolite 5-fluorouracil (5-FU) is also a substrate of UPRTase and is converted into 5-fluoro-UMP. After conversion, 5-fluoro-UMP acts as a suicide inhibitor of the enzyme thymidylate synthase, which causes cell death. Therefore, microorganisms with active UPRTase are sensitive to 5-FU (16). The upp gene has been identified in L. casei and is not essential for survival (17). In this study, we describe a gene deletion method where the upp gene serves as a counterselection marker. PPαT expression cassettes were created to boost the expression and facilitate antigen expression on the cellular surface. These cassettes contained the HCE constitutive strong promoter of thermostable d-amino acid aminotransferase (d-AAT) from Geobacillus toebii and the PgsA anchor protein from Bacillus subtilis (18), a poly-γ-glutamate (PGA) synthetase complex. PGA is an unusual anionic polypeptide in which glutamate is polymerized via γ-amide linkages. According to a previous report, PgsA functions to stabilize the complex by anchoring it in the cell membrane (19). PgsA is localized to the membrane and is ideally positioned for the cell surface display of heterologous proteins (20). The alpha-toxin gene from Clostridium perfringens serves as an antigen, and the rrnB T1T2 terminator from E. coli was integrated into the upp gene site by site-specific recombination.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Wild-type L. casei ATCC 393 was grown in MRS medium (Sigma), and plasmid-free recombinant L. casei ATCC 393 PPαT Δupp was grown in SDM medium containing 100 μg/ml of 5-FU (Sigma) at 37°C without shaking. Chloramphenicol (Cm) (Sigma) was used at a concentration of 10 μg/ml for L. casei ATCC 393 containing the pGBHCupp-2A2B-PPαT plasmid to create a temperature-sensitive strain. For plasmid propagation, E. coli TG1 was grown in LB medium containing 30 μg/ml of Cm.
pGBHCupp plasmid.
The temperature-sensitive plasmid pGBHCupp was received from Hongyu Cui (Veterinary Laboratory, Northeast Agricultural University) (17) and contained a pWV01 replicon and the upp and chloramphenicol resistance genes. The plasmid pGBWVE1 was received from the Poultry Disease Laboratory, Harbin Veterinary Research Institute, Chinese Academy of Agricultural Sciences, and contained the pWV01 temperature-sensitive replicon. Based on the pGBWVE1 plasmid, upp and Cm resistance genes were inserted into the pGBWVE1 plasmid while keeping the complete replicon element and the multiple-cloning site (MCS) by digestion with restriction enzymes and ligation with DNA ligase. The primers for the upp and Cm resistance genes were as follows (the indicated restriction enzyme sites are underlined): Cm-upper, GGTAAGCTTTATAATGAACTTTAATAAAATTGATTT (HindIII); Cm-lower, TTAACTCGAGTCTCATATTATAAAAGCCAGTCA (XhoI); upp-upper, CGCCATGGGGTTTGCAAACAAATGATTATC (NcoI); and upp-lower, AGAATTCACCTCCTTACTGAAGTGACGTCATATCAT (EcoRI).
Construction of a homologous recombination vector. (i) Construction of the pGBHCupp-2A2B temperature-sensitive vector.
For homologous recombination in L. casei ATCC 393, upstream homologous arm 2A (1,021 bp) and downstream homologous arm 2B (768 bp) were PCR amplified using L. casei ATCC 393 genomic DNA as a template to create an internal 651-bp deletion in the upp gene site. The sense primer for homologous arm 2A was GATCTAGATCAAAGCGGCGCGCCGGCAGCCCAGTTAGTT (BglII), and the antisense primer was TTGGCGCGCCAAGGCTCCTCCTAAACGCATTC (AscI). The sense primer for homologous arm 2B was ACCTTAATTAAATTCTTTGGCATGTGTAAAA (PacI), and the antisense primer was TCCCCGCGGGGAAGGTTGATCGGAAGCA (SacII). The two PCR fragments were gel purified and inserted into the pMD18-T vector (TaKaRa). Correct inserts were identified by sequencing. The pMD18-T-2A plasmid was digested with BglII (TaKaRa) and AscI (NEB). 2A was inserted into the pGBHCupp vector MCS site using the same restriction enzyme digestion, and this construct was named pGBHCupp-2A. The pMD18-T-2B plasmid was digested with SacII (TaKaRa) and PacI (NEB), and 2B was inserted into the pGBHCupp-2A MCS site via the same restriction enzyme digestion and ligated to create the plasmid pGBHCupp-2A2B.
(ii) Construction of the PPαT expression cassettes.
The HCE promoter (218 bp) with the Shine-Dalgarno sequence ----AGGA----, a PgsA anchor (1,140 bp), a multiple-cloning site (MCS), and the rrnB T1T2 terminator (434 bp) were synthesized by Shanghai Biological Engineering Co., Ltd., China, with the restriction enzymes AscI and PacI, and the construct was named pUC57–A1821. An alpha-toxin gene containing the restriction endonuclease sites HpaI and BglII was amplified by PCR using Clostridium perfringens genomic DNA as a template. The sense primer of the alpha-toxin gene was TACGTAATGTTCTGGGATCCTGATACAGA (HpaI), and the antisense primer was GCTCTAGATTTTATATTATAAGTTGAATTTCCTG (BglII). The PCR fragment was gel purified, inserted into the pMD18-T vector (TaKaRa), and identified by sequencing. The alpha-toxin gene was inserted into the A1821 MCS site to create the final construct, pUC57-PPαT.
(iii) Construction of the pGBHCupp-2A2B-PPαT homologous recombination plasmid.
pGBHCupp-2A2B-PPαT was created by digesting the pUC57-PPαT plasmid with AscI and PacI and inserting PPαT into the pGBHCupp-2A2B vector digested with the same restriction endonucleases. This plasmid was both temperature sensitive and chloramphenicol resistant.
Preparation of competent Lactobacillus cells and subsequent transformation.
Preparation of competent Lactobacillus cells and subsequent transformation were performed as previously described with some modifications (21–23). Briefly, a 100-ml culture of L. casei ATCC 393 cells was grown in MRS medium at 37°C for 4.5 h. The cells were placed in an ice bath for 30 min, pelleted at 4°C and 1,717 × g, and then resuspended in 40 ml of ice-cold EPWB buffer (0.6 mmol/liter NaH2PO4 and 0.1 mmol/liter MgCl2 [pH 7.4] with filter sterilization). The cells were pelleted and washed in EPWB buffer two more times, washed in EPB buffer (EPWB plus 0.3 M sucrose), and then finally resuspended in 1 ml EPB buffer, resulting in competent cells. Approximately 500 ng of plasmid DNA (derived from E. coli TG1) was added to a 200-μl aliquot of competent cells on ice and transformed by electroporation in a cold 0.2-cm cuvette at 2.5 kV. Cells recovered in MRS overnight and were then plated on MRS medium containing 5 μg/ml of Cm.
Homologous recombination of L. casei ATCC 393.
For homologous recombination, cultures were serially transferred three times (1% inoculum) and grown to stationary phase with 24 h of incubation in a 42°C water bath to select for single-crossover integrants of the targeting plasmid, thereby rendering the transformants resistant to 5 μg/ml of Cm in MRS solid medium during 36 h of incubation in a 37°C incubator. To obtain double-crossover recombinant bacteria, single colonies were serially grown to stationary phase in antibiotic-free MRS medium three to five times in a 37°C water bath. The cultures were then serially inoculated three times into fresh antibiotic-free MRS medium and grown for 24 h in a 42°C water bath. Clones that had lost the plasmid as a result of the second homologous crossover event were rendered 5-FU resistant, and dilutions of 1,000- to 10,000-fold were selected by plating on semidefined medium with glucose (GSDM) (4, 24) containing 100 μg/ml of 5-FU (25). Colonies were screened for PPαT integration using primers upstream and downstream of the crossover event that corresponded to the number of the upstream gene in each location. The sense primer for screening was 5′-GGCAGCCCAGTTAGTTTTAGTGGTCAGGAATATCATT, and the antisense primer was 5′-AGGTTGATCGGAAGCAGGATTGCGACCGGTGACGCATA. Integrants were confirmed by sequencing. Sequencing primers included SBF-F-I421 (5′-CAAACCGATTGGAATGTTCC), HCE-F1 (5′-GATCTCTCCTTCACAGATTCC), SBF-F-I437 (5′-TCTCAACAGCGCCAACAACC), SBF-F-I512 (5′-CGCGAACTGACGAAAGACTC), SBF-F-I573 (5′-CTAACTCTCAAAAAGGAACAGC), and SBF-F-J021 (5′-GTAGCGCCGATGGTAGTGT).
Growth and hereditary stability of recombinant L. casei strains.
To determine whether the properties of the recombinant strains changed relative to those of wild-type L. casei, growth and hereditary stability were analyzed after the recombinant L. casei strains were screened by PCR and sequencing.
Bacterial growth was measured using the optical density at 600 nm (OD600). Recombinant bacteria were transferred into MRS or SDM medium at a dilution of 1:100 and sampled once every 2 h. The OD600 was determined after a 10-fold dilution.
Three strains containing PPαT chromosomal integrations were analyzed for stability by serially transferring the cultures after 24 h of incubation into SDM medium containing 5-FU at 37°C (1% inoculum; 40 generations). DNA was extracted from the cells, and genomic PCR was used to confirm the presence of a PPαT integrant band in each strain using screened primers for the integration event.
Expression of alpha-toxin protein. (i) Indirect ELISA of alpha-toxin protein.
For expression analysis of the PgsA-toxin fusion protein in recombinant bacteria, integrated bacteria were grown overnight in MRS medium at 37°C. Bacterial cells were collected by centrifugation at 3,000 × g for 15 min. The pellets were washed twice with sterile phosphate-buffered saline (PBS) (pH 7.4) and lysed in a Bead Beater (Biospec, Bartlesville, OK) by vigorous shaking. The cell debris was centrifuged at 3,000 × g for 10 min, and the supernatant was analyzed via indirect enzyme-linked immunosorbent assay (ELISA). ELISAs were performed as follows. Protein extracts (8 μg/ml) were applied to 96-well plates overnight at 4°C. Wild-type L. casei protein was used as a negative-control antigen. Plates were washed three times with PBS plus 0.05% Tween 20 (PBST). After the wells were blocked for 2 h at 37°C with PBS containing 5% skim milk, the polyclonal antibody to the alpha-toxin of Clostridium perfringens was diluted (1:200) in PBS–1% bovine serum albumin (BSA) in six replicates and incubated for 1 h at 37°C. After the plates were washed three times with PBST, horseradish peroxidase-conjugated goat anti-rabbit IgG antibody (Invitrogen) was added to each well (1:2,000) and incubated for an additional 1 h at 37°C. After another round of washing, color development was performed using o-phenylenediamine dihydrochloride as the substrate, and the absorbance was measured at 490 nm (12).
(ii) Expression of the PgsA-toxin fusion protein on the cell surface.
Laser confocal microscopy was used to confirm the expression of alpha-toxin protein on the bacterial surface. Wild-type L. casei ATCC 393 and L. casei ATCC 393 PPαT Δupp were grown overnight in MRS medium at 37°C. One milliliter of culture was collected by centrifugation at 1,700 × g for 10 min, and the pellets were washed twice with PBS (pH 7.4). The polyclonal antibody to alpha-toxin from Clostridium perfringens were diluted (1:200) in PBS, and 200 μl was added and incubated for 1 h at 37°C. After the pellets were washed three times with PBS, fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG antibody (Invitrogen, USA) was added to each microtube (1:1,000) and incubated for an additional 1 h at 37°C. Samples were then washed three times with PBS, dyed with 4′,6′-diamidino-2-phenylindole (DAPI) (Invitrogen, USA) for 30 min at 4°C, washed three times, resuspended in 200 μl PBS, and smeared on a microscope slide. Images were viewed by laser confocal microscopy (model LSM510 META; Zeiss, Germany).
RESULTS
Construction of the pGBHCupp-2A2B-PPαT homologous recombination plasmid.
Homologous arms 2A and 2B and the PPαT expression cassettes were amplified by PCR, digested with the relevant restriction endonucleases, and ligated with T4 DNA ligase as indicated in Fig. 1.
FIG 1.

Schematic diagram of vector construction. 1, PCR products of upstream homologous arm 2A with AscI and BglII, cloned into the pMD18-T vector and named pMD18-T-2A. pMD18-T-2A was digested with AscI and BglII endonucleases and inserted into the pGBHCupp vector using the same endonucleases, and this was named pGBHCupp-2A. 2, pMD18-T-2B, containing downstream homologous arm 2B, was digested with PacI and SacII, and 2B was inserted into pGBHCupp-2A; this construct was named pGBHCupp-2A2B. 3, A1821 was synthesized by Shanghai Biological Engineering Co., Ltd., China, and included the HCE promoter, the PgsA anchor, an MCS, and the T1T2 terminator. This construct was inserted into the pUC57 cloning vector and called pUC57–A1821. The alpha-toxin gene cloned into the pMD18-T vector was digested with HpaI and BglII, inserted into the pUC57–A1821 MCS, and named pUC57-PPαT. 4, pUC57-PPαT was digested with AscI and PacI. PPαT was inserted into the pGBHCupp-2A2B vector using the same endonuclease digestion, and this construct was named pGBHCupp-2A2B-PPαT.
Homologous recombination.
Genomic PCR was performed after recombinant bacteria containing the pGBHCupp-2A2B-PPαT temperature-sensitive plasmid were screened with temperature elevation and exposure to Cm. This analysis identified a 4,456-bp PCR product from the recombinant bacteria that contained a PPαT expression cassette of 2,721 bp, the deleted upp gene of 651 bp, and 2,386 bp from wild-type L. casei (Fig. 2). PCR products were sequenced by Genewiz Biological Technology Company, Ltd., Beijing, China. BLAST results demonstrated that the recombinant PCR product included the HCE promoter, a PgsA anchor, alpha-toxin, and the rrnB T1T2 terminator. The sequence is precisely the same as that of the expression cassette. These results illustrated that the PPαT expression cassettes had integrated successfully into the genome of L. casei.
FIG 2.
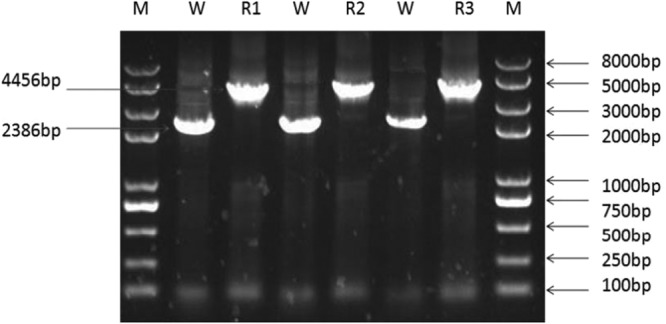
PPαT expression cassettes integrated successfully into the genome of L. casei. Lane M, Trans II DNA marker; lane W, wild-type L. casei genomic PCR product (the spacer is 2,386 bp in size); lanes R1, R2, and R3, 3, 5, and 12 recombinant L. casei (L. casei PPαT Δupp) genomic PCR products (the spacer is 4,456 bp in size).
Recombinant L. casei strains were obtained by screening. Genomic PCR and sequencing were used to determine the stability of PPαT integrations after 40 generations. These results demonstrated that cassettes were stably inherited over several generations (Fig. 3). However, significant changes occurred in the growth patterns of the recombinants relative to those of the wild type, as all recombinant strains grew more slowly, whether they were cultivated in MRS or SDM culture medium (Fig. 4).
FIG 3.
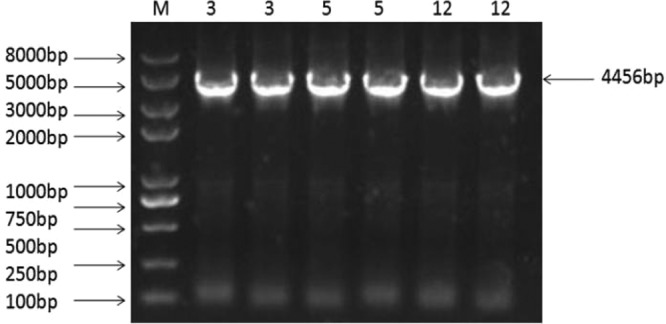
PCR analysis confirms that L. casei PPαT Δupp is genetically stable after 40 generations. Lanes 3, PCR product of L. casei PPαT Δupp 3 genomic DNA after 40 generations; lanes 5, PCR product of L. casei PPαT Δupp 5 genomic DNA after 40 generations; lanes 12, PCR product of L. casei PPαT Δupp 12 genomic DNA after 40 generations. Their spacers are 4,456 bp in size.
FIG 4.
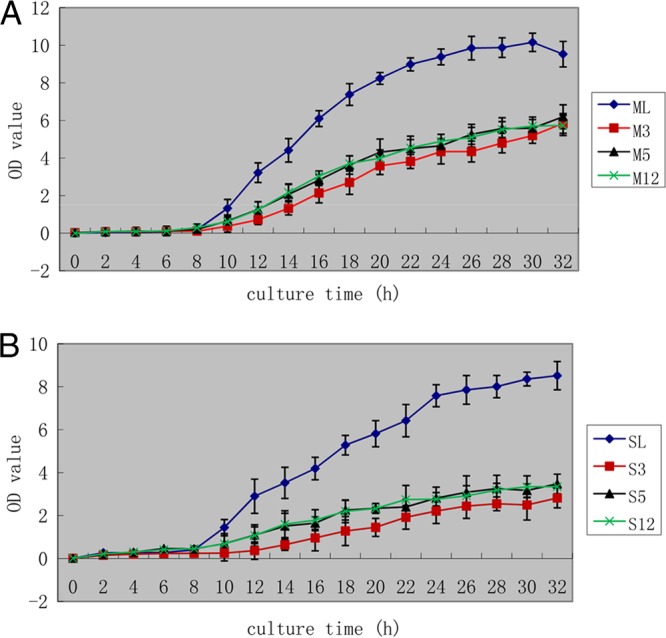
OD600 measurements in MRS medium (A) and SDM medium (B). Bacterial growth was measured using the OD600. Recombinant bacteria were transferred into MRS or SDM medium at a dilution of 1:100 and sampled once every 2 h. The OD600 was determined after a 10-fold dilution. The horizontal axis indicates the culture time in hours; the vertical axis indicates the OD600 value. (A) ML, wild-type L. casei grown in MRS medium; M3, L. casei PPαT Δupp 3 grown in MRS medium; M5, L. casei PPαT Δupp 5 grown in MRS medium; M12, L. casei PPαT Δupp 12 grown in MRS medium. (B) SL, wild-type L. casei grown in SDM medium; S3, L. casei PPαT Δupp 3 grown in SDM medium; S5, L. casei PPαT Δupp 5 grown in SDM medium; S12, L. casei PPαT Δupp 12 grown in SDM medium.
Indirect ELISA.
To analyze the expression of the alpha-toxin gene, an indirect ELISA was used. Assay results were repeated six times and showed that the OD490 for sample 3 was 0.6962 ± 0.1145, that for sample 5 was 0.4935 ± 0.0239, that for sample 12 was 0.5455 ± 0.0318, and that for the negative control was 0.2468 ± 0.0282. The differences between all groups were very significant (P ≤ 0.01). These results suggested that the alpha-toxin protein was expressed and that the protein had biological activity.
Expression of the PgsA-toxin fusion protein on the cell surface.
Laser confocal microscopy was used to determine whether the fusion protein was expressed on the surface of L. casei cells. After cells were treated with FITC-conjugated secondary antibodies and dyed with DAPI, fusion protein was visible on the surface of L. casei cells, as shown in Fig. 5.
FIG 5.
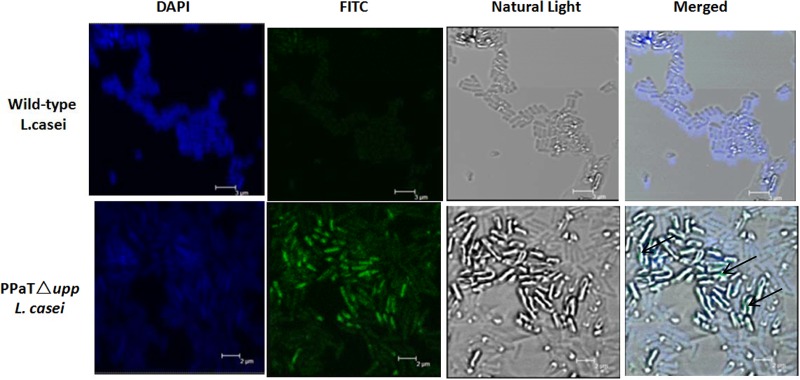
Detection and localization of PgsA-toxin fusion protein in L. casei cells. The PgsA-toxin fusion protein localization in L. casei cells was detected by laser confocal microscopy. All the cells were probed with FITC-conjugated goat anti-rabbit IgG antibody and stained with DAPI. Merged images showed green light (FITC) from the PgsA-toxin fusion protein on the surface of L. casei cells (arrow). Bars, 3 μm for wild-type L. casei and 2 μm for L. casei PPαT Δupp.
DISCUSSION
LAB are widely used as live-vaccine vehicles in mucosal immunization because LAB are safe, exhibit adjuvant properties, and are only weakly immunogenic (23). Integrating genes into the chromosome for expression has the potential to eliminate selection requirements, provide genetic stability (4), and conform to biological safety standards. To integrate sequences into the bacterial chromosome, recombinant plasmids that are suitable for L. casei must be constructed. This plasmid can be eliminated when the homologous arm has been integrated into the L. casei chromosome using various methods. Therefore, we chose the temperature-sensitive, low-copy-number replicon pWV01. At temperatures of 42°C or higher, plasmid replication failed, and the plasmid underwent homologous recombination with the host chromosome. Systems containing two plasmids have been used for chromosomal integration in some bacteria (4, 25). One of the two plasmids must have a defect that prevents replication in strains lacking the repA gene. Therefore, this system requires an E. coli strain containing the repA gene, such as E. coli EC101, for plasmid cloning. A few laboratories possess these E. coli strains, where they can be obtained with some difficultly. Two-plasmid systems also require resistance to more than one antibiotic, such as Cm and erythromycin (Em), and thus these systems require more than one transformation. To simplify this process, we employed a single-plasmid system with the temperature-sensitive plasmid pGBHCupp-2A2B-PPαT. Replication failed when the temperature was elevated above 42°C, and the plasmid underwent homologous recombination with the host chromosome. We performed two temperature changes in this experiment. The first temperature rise deleted the plasmid and produced a single recombinant product that was chloramphenicol resistant. The second temperature rise produced bacteria with two recombination events that were chloramphenicol sensitive and 5-FU resistant. The double-recombinant product was selected on 5-FU-containing SDM plates.
The upp gene encodes a phosphoribosyl transferase (PRTase) (26–31). PRTases recycle free purine or pyrimidine bases by converting them into the corresponding nucleotide monophosphates, thereby sparing the cell the burden of synthesizing these molecules de novo. However, PRTases may also act on base analogs, creating unnatural nucleotides that can be toxic to the cell. Accordingly, PRTase-defective mutants are resistant to the toxic effects of analogs such as 5-FU (32). Kristich et al. developed a method for markerless genetic exchange in Enterococcus faecalis and used it to construct an srtA mutant (32). Goh et al. used the upp gene as a counterselectable marker replacement system for the study of the S-layer protein SlpX of Lactobacillus acidophilus NCFM (25). Braks et al. applied this method to a positive/negative selectable marker system using reverse genetics in Plasmodium (33). In L. casei, the upp gene is nonessential (17) and can serve as a counterselectable marker because the cell becomes 5-FU resistant if the upp gene is deleted. In this study, we took advantage of the deletable nature of the upp gene to create a counterselectable marker, and PPαT expression cassettes were used to replace the upp gene.
In addition, antigens expressed on the surface of bacteria are better recognized by the immune system than those that are intracellular (34). Various anchor genes have been used for this, including those for OprF (a major outer membrane protein of Pseudomonas aeruginosa), FadL (an outer membrane protein involved in long-chain fatty acid transport in Escherichia coli), and PgsA (35–38). PgsA is considered to be a better anchor for LAB. Therefore, we used the PgsA gene product in this study as an anchor for surface display of antigens on LAB. PgsA is a transmembrane protein derived from the poly-γ-glutamic acid synthetase complex (the Pgs-BCA system) of Bacillus subtilis (19, 39). Indeed, the surface display of antigens on the surface of L. casei cells has been confirmed by laser confocal microscopy. Green fluorescence was detected on the surface of L. casei cells when probed with FITC-conjugated goat anti-rabbit IgG antibody, and blue fluorescence was observed in the bodies of cells stained with DAPI.
In an analysis of the properties of the three new recombinant strains, all strains were genetically stable. However, the recombinant strains grew more slowly than wild-type L. casei, in both MRS and SDM media. This may be related to the insertion of the exogenous gene.
The quantity of protein expressed from a genomically integrated gene is less than that from a gene carried by a plasmid, as the genome is present as a single copy and multiple copies of a plasmid are present in the cell. Therefore, we used an ELISA to detect protein expression. The ELISA results demonstrated that the alpha-toxin genes of these three strains were expressed and that the expressed protein had biological activity.
In summary, a single-plasmid system was suitable for the construction of recombinant strains of L. casei. We successfully integrated expression cassettes into the L. casei chromosome and detected expressed protein for the preparation of food-grade vaccines.
ACKNOWLEDGMENT
This work was supported by the National Nature Funds Committee (31272594) of China.
Footnotes
Published ahead of print 21 March 2014
REFERENCES
- 1.Bermúdez-Humarán LG, Cortes-Perez NG, L'Haridon R, Langella P. 2008. Production of biological active murine IFN-gamma by recombinant Lactococcus lactis. FEMS Microbiol. Lett. 280:144–149. 10.1111/j.1574-6968.2007.01038.x [DOI] [PubMed] [Google Scholar]
- 2.Poo H, Pyo HM, Lee TY, Yoon SW, Lee JS, Kim CJ, Sung MH, Lee SH. 2006. Oral administration of human papillomavirus type 16 E7 displayed on Lactobacillus casei induces E7-specific antitumor effects in C57/BL6 mice. Int. J. Cancer 119:1702–1709. 10.1002/ijc.22035 [DOI] [PubMed] [Google Scholar]
- 3.Wells JM, Mercenier A. 2008. Mucosal delivery of therapeutic and prophylactic molecules using lactic acid bacteria. Nat. Rev. Microbiol. 6:349–362. 10.1038/nrmicro1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Douglas GL, Klaenhammer TR. 2011. Directed chromosomal integration and expression of the reporter gene gusA3 in Lactobacillus acidophilus NCFM. Appl. Environ. Microbiol. 77:7365–7371. 10.1128/AEM.06028-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perdigón G, Maldonado Galdeano C, Valdez JC, Medici M. 2002. Interaction of lactic acid bacteria with the gut immune system. Eur. J. Clin. Nutr. 56(Suppl 4):S2. 10.1038/sj.ejcn.1601657 [DOI] [PubMed] [Google Scholar]
- 6.Galdeano CM, Perdigón G. 2004. Role of viability of probiotic strains in their persistence in the gut and in mucosal immune stimulation. J. Appl. Microbiol. 97:673–681. 10.1111/j.1365-2672.2004.02353.x [DOI] [PubMed] [Google Scholar]
- 7.Galdeano CM, Perdigón G. 2006. The probiotic bacterium Lactobacillus casei induces activation of the gut mucosal immune system through innate immunity. Clin. Vaccine Immunol. 13:219–226. 10.1128/CVI.13.2.219-226.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bibas Bonet ME, Chaves S, Mesón O, Perdigon G. 2006. Immunmodulatory and antiinflammatory activity induced by oral adminsitration of a probiotic strain of Lactobacillus casei. Eur. J. Inflam. 4:31–41 [Google Scholar]
- 9.Galdeano CM, de Moreno de LeBlanc A, Vinderola G, Bonet ME, Perdigón G. 2007. Proposed model: mechanisms of immunomodulation induced by probiotic bacteria. Clin. Vaccine Immunol. 14:485–492. 10.1128/CVI.00406-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dogi CA, Galdeano CM, Perdigón G. 2008. Gut immune stimulation by non pathogenic Gram(+) and Gram(−) bacteria. Comparison with a probiotic strain. Cytokine 41:223–231 [DOI] [PubMed] [Google Scholar]
- 11.Wei CH, Liu JK, Hou XL, Yu LY, Lee JS, Kim CJ. 2010. Immunogenicity and protective efficacy of orally or intranasally administered recombinant Lactobacillus casei expressing ETEC K99. Vaccine 28:4113–4118. 319. 10.1016/j.vaccine.2009.05.088 [DOI] [PubMed] [Google Scholar]
- 12.Liu D, Wang X, Ge J, Liu S, Li Y. 2011. Comparison of the immune responses induced by oral immunization of mice with Lactobacillus casei-expressing porcine parvovirus VP2 and VP2 fused to Escherichia coli heat-labile enterotoxin B subunit protein. Comp. mmunol. Microbiol. Infect. Dis. 34:73–81. 10.1016/j.cimid.2010.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Sullivan DJ, Klaenhammer TR. 1993. High- and low-copy-number Lactococcus shuttle cloning vectors with features for clone screening. Gene 137:227–231. 10.1016/0378-1119(93)90011-Q [DOI] [PubMed] [Google Scholar]
- 14.Posno M, Leer RJ, van Luijk N, van Giezen MJ, Heuvelmans PT, Lokman BC, Pouwels PH. 1991. Incompatibility of Lactobacillus vectors with replicons derived from small cryptic Lactobacillus plasmids and segregational instability of the introduced vectors. Appl. Environ. Microbiol. 57:1822–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neuhard J. 1983. Utilization of preformed pyrimidine bases and nucleosides, p 95–148 In Munch-Petersen A. (ed), Metabolism of nucleotides, nucleosides and nucleobases in microorganisms. Academic Press, New York, NY [Google Scholar]
- 16.Graf N, Altenbuchner J. 2011. Development of a method for markerless gene deletion in Pseudomonas putida. Appl. Environ. Microbiol. 77:5549–5552. 10.1128/AEM.05055-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui H. 2011. Construction of nonselection markers genetically modified lactic acid bacteria operating system and pig lactoferrin genic expression cheese in this operating system. Doctoral dissertation. Northeast Agricultural University, Harbin, China [Google Scholar]
- 18.Ashiuchi M, Misono H. 2002. Biochemistry and molecular genetics of poly-gamma-glutamate synthesis. Appl. Microbiol. Biotechnol. 59:9–14. 10.1007/s00253-002-0984-x [DOI] [PubMed] [Google Scholar]
- 19.Ashiuchi M, Nawa C, Kamei T, Song JJ, Hong SP, Sung MH, Soda K, Misono H. 2001. Physiological and biochemical characteristics of poly gamma-glutamate synthetase complex of Bacillus subtilis. Eur. J. Biochem. 268:5321–5328. 10.1046/j.0014-2956.2001.02475.x [DOI] [PubMed] [Google Scholar]
- 20.Narita J, Okano K, Tateno T, Tanino T, Sewaki T, Sung MH, Fukuda H, Kondo A. 2006. Display of active enzymes on the cell surface of Escherichia coli using PgsA anchor protein and their application to bioconversion. Appl. Microbiol. Biotechnol. 70:564–572. 10.1007/s00253-005-0111-x [DOI] [PubMed] [Google Scholar]
- 21.Luchansky JB, Kleeman EG, Raya RR, Klaenhammer TR. 1989. Genetic transfer systems for delivery of plasmid deoxyribonucleic acid to Lactobacillus acidophilus ADH: conjugation, electroporation, and transduction. J. Dairy Sci. 72:1408–1417. 10.3168/jds.S0022-0302(89)79248-1 [DOI] [PubMed] [Google Scholar]
- 22.Walker DC, Aoyama K, Klaenhammer TR. 1996. Electrotransformation of Lactobacillus acidophilus group A1. FEMS Microbiol. Lett. 138:233–237. 10.1111/j.1574-6968.1996.tb08163.x [DOI] [PubMed] [Google Scholar]
- 23.Wei MQ. 1995. An improved method for the transformation of Lactobacillus strains using electroporation. J. Microbiol. Methods 21:97–109. 10.1016/0167-7012(94)00038-9 [DOI] [Google Scholar]
- 24.Kimmel SA, Roberts RF. 1998. Development of a growth medium suitable for exopolysaccharide production by Lactobacillus delbrueckii ssp. bulgaricus RR. Int. J. Food Microbiol. 40:87–92. 10.1016/S0168-1605(98)00023-3 [DOI] [PubMed] [Google Scholar]
- 25.Goh YJ, Azcárate-Peril MA, O'Flaherty S, Durmaz E, Valence F, Jardin J, Lortal S, Klaenhammer TR. 2009. Development and application of a upp-based counterselective gene replacement system for the study of the S-layer protein SlpX of Lactobacillus acidophilus NCFM. Appl. Environ. Microbiol. 75:3093–3105. 10.1128/AEM.02502-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bae T, Kozlowicz B, Dunny GM. 2002. Two targets in pCF10 DNA for PrgX binding: their role in production of Qa and prgX mRNA and in regulation of pheromone-inducible conjugation. J. Mol. Biol. 315:995–1007. 10.1006/jmbi.2001.5294 [DOI] [PubMed] [Google Scholar]
- 27.Fabret C, Ehrlich SD, Noirot P. 2002. A new mutation delivery system for genome-scale approaches in Bacillus subtilis. Mol. Microbiol. 46:25–36. 10.1046/j.1365-2958.2002.03140.x [DOI] [PubMed] [Google Scholar]
- 28.Fukagawa T, Hayward N, Yang J, Azzalin C, Griffin D, Stewart AF, Brown W. 1999. The chicken HPRT gene: a counter selectable marker for the DT40 cell line. Nucleic Acids Res. 27:1966–1969. 10.1093/nar/27.9.1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peck RF, DasSarma S, Krebs MP. 2000. Homologous gene knockout in the archaeon Halobacterium salinarum with ura3 as a counterselectable marker. Mol. Microbiol. 35:667–676. 10.1046/j.1365-2958.2000.01739.x [DOI] [PubMed] [Google Scholar]
- 30.Pritchett MA, Zhang JK, Metcalf WW. 2004. Development of a markerless genetic exchange method for Methanosarcina acetivorans C2A and its use in construction of new genetic tools for methanogenic archaea. Appl. Environ. Microbiol. 70:1425–1433. 10.1128/AEM.70.3.1425-1433.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spring KJ, Mattick JS, Don RH. 1994. Escherichia coli gpt as a positive and negative selectable marker in embryonal stem cells. Biochim. Biophys. Acta 1218:158–162. 10.1016/0167-4781(94)90005-1 [DOI] [PubMed] [Google Scholar]
- 32.Kristich CJ, Manias DA, Dunny GM. 2005. Development of a method for markerless genetic exchange in Enterococcus faecalis and its use in construction of a srtA mutant. Appl. Environ. Microbiol. 71:5837–5849. 10.1128/AEM.71.10.5837-5849.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Braks JA, Franke-Fayard B, Kroeze H, Janse CJ, Waters AP. 2006. Development and application of a positive-negative selectable marker system for use in reverse genetics in Plasmodium. Nucleic Acids Res. 34:e39. 10.1093/nar/gnj033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee JS, Poo H, Han DP, Hong SP, Kim K, Cho MW, Kim E, Sung MH, Kim CJ. 2006. Mucosal immunization with surface-displayed severe acute respiratory syndrome coronavirus spike protein on Lactobacillus casei induces neutralizing antibodies in mice. J. Virol. 80:4079–4087. 10.1128/JVI.80.8.4079-4087.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee SH, Choi JI, Han MJ, Choi JH, Lee SY. 2005. Display of lipase on the cell surface of Escherichia coli using OprF as an anchor and its application to enantioselective resolution in organic solvent. Biotechnol. Bioeng. 90:223–230. 10.1002/bit.20399 [DOI] [PubMed] [Google Scholar]
- 36.Lee SH, Choi JI, Park SJ, Lee SY, Park BC. 2004. Display of bacterial lipase on the Escherichia coli cell surface by using FadL as an anchoring motif and use of the enzyme in enantioselective biocatalysis. Appl. Environ. Microbiol. 70:5074–5080. 10.1128/AEM.70.9.5074-5080.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sung MH, Hong SP, Lee JS, Jung CM, Kim CJ, Soda K, Ashiuchi M. 2003. Surface expression vectors having pgsBCA, the genecoding poly-gamma-glutamate synthetase, and a method for expression of target protein at the surface of microorganism using the vector. International patent WO 03/01436
- 38.Poo H, Song JJ, Hong SP, Choi YH, Yun SW, Kim JH, Lee SC, Lee SG, Sung MH. 2002. Novel high-level constitutive expression system, pHCE vector, for a convenient and cost-effective soluble production of human tumor necrosis factor-α. Biotechnol. Lett. 24:1185–1189. 10.1023/A:1016107230825 [DOI] [Google Scholar]
- 39.Ashiuchi M, Soda K, Misono HA. 1999. poly-gamma-glutamate synthetic system of Bacillus subtilis IFO 3336: gene cloning and biochemical analysis of poly-gamma-glutamate produced by Escherichia coli clone cells. Biochem. Biophys. Res. Commun. 263:6–12. 10.1006/bbrc.1999.1298 [DOI] [PubMed] [Google Scholar]