

Abstract
This study aimed to investigate the salivary microbiota and metabolome of 13 children with celiac disease (CD) under a gluten-free diet (treated celiac disease [T-CD]). The same number of healthy children (HC) was used as controls. The salivary microbiota was analyzed by an integrated approach using culture-dependent and -independent methods. Metabolome analysis was carried out by gas chromatography-mass spectrometry–solid-phase microextraction. Compared to HC, the number of some cultivable bacterial groups (e.g., total anaerobes) significantly (P < 0.05) differed in the saliva samples of the T-CD children. As shown by community-level catabolic profiles, the highest Shannon's diversity and substrate richness were found in HC. Pyrosequencing data showed the highest richness estimator and diversity index values for HC. Levels of Lachnospiraceae, Gemellaceae, and Streptococcus sanguinis were highest for the T-CD children. Streptococcus thermophilus levels were markedly decreased in T-CD children. The saliva of T-CD children showed the largest amount of Bacteroidetes (e.g., Porphyromonas sp., Porphyromonas endodontalis, and Prevotella nanceiensis), together with the smallest amount of Actinobacteria. T-CD children were also characterized by decreased levels of some Actinomyces species, Atopobium species, and Corynebacterium durum. Rothia mucilaginosa was the only Actinobacteria species found at the highest level in T-CD children. As shown by multivariate statistical analyses, the levels of organic volatile compounds markedly differentiated T-CD children. Some compounds (e.g., ethyl-acetate, nonanal, and 2-hexanone) were found to be associated with T-CD children. Correlations (false discovery rate [FDR], <0.05) were found between the relative abundances of bacteria and some volatile organic compounds (VOCs). The findings of this study indicated that CD is associated with oral dysbiosis that could affect the oral metabolome.
INTRODUCTION
Celiac disease (CD) is a chronic immune-mediated enteropathy, which affects the small intestinal mucosa after the ingestion of gluten from wheat, rye, and their cross-related varieties in genetically susceptible individuals (1). CD is one of the most diffuse chronic diseases in Europe, North America, and developing countries (South America, South Africa, and South Asia) (2). Nowadays, the incidence of CD is increasing, and it is estimated to be 0.7 to 2.0% (3). Overall, CD is characterized by steatorrhea and malnutrition coupled with multiple deficiency states. Diverse problems such as dental anomalies, short stature, osteopenic bone disease, lactose intolerance, infertility, and nonspecific abdominal pain are associated with CD (4). Moreover, alterations of the oral ecosystem and of the saliva composition seem to be determined by CD (2, 5, 6). Compared to healthy subjects, the saliva of CD patients contains small amounts of amylase and secretory IgA and IgM (7, 8) and has low buffering capacity, speed of salivary flow, concentration of calcium, and Ca/P ratios (5, 9).
The gluten-free diet (GFD) is effective and safe. At present, the exclusion of gluten-containing products is the only treatment available. Recently, some reports also suggested that the gastrointestinal (GI) microbiota is somewhat affected during CD pathogenesis and treatment with GFD (10, 11). Early microbial infections (12, 13) and imbalances in the composition of the GI microbiota (14–21) were associated with CD. Compared to healthy individuals, GFD lasting 2 years did not completely restore the GI microbiota and, consequently, the metabolome of children with CD (14).
It is doubtless that the oral and GI microbiota play key roles in health and disease (22–28). The human oral cavity is a complex ecosystem populated by ca. 700 bacterial species that reach numbers of 1011 bacteria/g (wet weight) of dental plaque and 108 to 109 CFU/g of saliva (29). Some of these bacterial species are closely associated with the development of oral diseases, mainly dental caries and periodontitis (25, 26). Also, several nonoral diseases, such as bacterial endocarditis (30), heart disease (31), obesity (28), pneumonia (32), atherosclerosis (33), and preterm low birth weight (34), seemed to be somehow related to bacteria from the human oral cavity. Overall, the saliva is considered to be the most suitable tool to obtain information on the microbiota of the oral cavity (35). The composition of saliva includes volatile organic compounds (VOCs) (metabolome) derived from various sources (e.g., serum, blood, microorganisms, and environmental pollution). Metabolites are considered indicators of physiological or pathological states (36). Recently, saliva metabolomics analyses have opened new possibilities for the identification of metabolite biomarkers representative of specific disorders (36, 37).
Previously, few studies dealt with the salivary microbiota of CD patients. It was shown previously that strict adherence to GFD reduces the prevalence of dental caries (38). Compared to healthy individuals, CD children were characterized by a low prevalence of salivary mutans streptococci and lactobacilli (9). On the contrary, no statistically significant differences (P < 0.05) were found for streptococcus and lactobacillus counts between the saliva samples of healthy children (HC) and those of CD children who were subjected to GFD (39). A more in-depth characterization of the oral microbiota of CD patients during GFD is needed, and no previous study combined characterizations of the salivary microbiota and metabolome.
This study aimed to compare the salivary microbiota and metabolomes of CD children subjected to GFD and healthy children.
MATERIALS AND METHODS
Study design.
This study was approved by the Institutional Review Board of the Faculty of Medicine and Surgery of the University of Bari Aldo Moro (Italy). Informed written consent was obtained from parents. Two groups of children (median age, ca. 10 ± 1.4 years) were included in the study: 32 symptom-free CD patients who were treated with GFD for at least 2 years (treated celiac disease [T-CD]) and 37 healthy children (HC) without CD and other known food intolerances, who were referred as the controls. CD diagnosis was based on the positivity of tissue transglutaminase (TTG)-IgA and the antiendomysial antibody (EMA) test in the presence of clinical symptoms and positive histological evidence of villous atrophy with crypt hyperplasia, increases in levels of intraepithelial lymphocytes with a gluten-containing diet, and the disappearance of symptoms with the normalization of a positive serum-specific antibody(ies) with GFD (40). HC were selected after negative results of serological tests for CD were obtained.
Based on the exclusion criteria, 43 children were excluded after the first visit (see Fig. S1 in the supplemental material). The information and characteristics of the recruited children are reported in Table 1.
TABLE 1.
Baseline demographic and clinical characteristics of children
| Characteristic | Value for group |
|
|---|---|---|
| Children with celiac disease (n = 13) | Healthy controls (n = 13) | |
| Mean age (yr) ± SD | 9.7 ± 1.4 | 10.2 ± 1.4 |
| No. of male children/no. of female children | 4/9 | 5/8 |
| Feeding habit | Gluten-free diet | Unrestricted |
| Mean duration of gluten-free diet (yr) ± SD | 4.6 ± 2.2 | |
| Mean BMIa (%) ± SD | 63.5 ± 25.2 | 58.3 ± 31.24 |
| Tooth-brushing habits | 2 times a day | 2 times a day |
| Mean hemoglobin level (g/dl) ± SD | 13.6 ± 0.7 | 13.1 ± 1.2 |
| Mean iron level (ng/ml) ± SD | 76.3 ± 25.1 | 65.2 ± 31.4 |
| Mean ferritin level (ng/ml) ± SD | 33.6 ± 14.5 | 28.8 ± 15.4 |
BMI, body mass index.
Collection of saliva samples.
Unstimulated whole saliva samples were collected in the morning, 2 h after tooth brushing, by direct spitting into a sterile plastic tube in a time span not exceeding 30 min. No intake of food and drink was allowed the morning before sampling. The samples were either immediately subjected to analysis (plate counts and Biolog system) or frozen at −20°C (DNA extraction and metabolome analyses).
Enumeration of cultivable bacteria.
Salivary samples (1 g) were mixed with 9 ml of sterilized physiological solution and homogenized. Counts of viable bacterial cells were carried out as described previously by De Angelis and coworkers (41). The following selective media were used: plate count agar (total facultative aerobes and anaerobes), MRS agar (lactobacilli and enterococci), modified Bifidobacterium agar (bifidobacteria) (Becton, Dickinson, France SA, Le Pont de Claix, France), glucose-M17 medium (lactococci and streptococci), Baird Parker plus egg yolk tellurite emulsion (staphylococci and micrococci), Wilkins-Chalgren anaerobe agar (total anaerobes), Wilkins-Chalgren anaerobe agar plus G-N selective supplements and defibrinated sheep blood (Bacteroides, Porphyromonas, and Prevotella), violet red bile agar (enterobacteria), and Slanetz-Bartley medium (enterococci). Except for modified Bifidobacterium agar, all media were purchased from Oxoid Ltd. (Hampshire, England).
Community-level catabolic profiles (CLCPs).
Carbon source utilization patterns of the salivary microbiota were assessed by using Biolog 96-well Eco microplates (Biolog, Inc., Hayward, CA) (42). Microplates contained 31 different carbon sources (carbohydrates, carboxylic acids, polymers, amino acids, amines, and miscellaneous substrates) in triplicate. Ten grams of saliva was homogenized with 90 ml of a sterile sodium chloride (0.9% [wt/vol]) solution (Classic Blender) and centrifuged at 12,500 × g for 15 min at 4°C. The pellet was washed with 50 mM Tris-HCl (pH 7.0) and then washed with a sterile sodium chloride solution and centrifuged at 12,500 × g for 15 min at 4°C. The cellular suspension was diluted (1:10) into the sterile sodium chloride solution and subsequently dispensed (150 μl) into each of the 96 wells of the Biolog Eco microplates. The microplates were incubated at 30°C in the dark, and color development was measured at 590 nm every 24 h with a microplate reader (Biolog Microstation). Three indices were determined (43–45). Shannon's diversity (H′), indicating the substrate utilization pattern, was calculated as H′ = −Σ pi ln(pi), where pi is the ratio of the activity of a particular substrate to the sums of activities of all substrates at 120 h. Substrate richness (S), measuring the number of different substrates used, was calculated as the number of wells with a corrected absorbance of >0.25. Substrate evenness (E) was defined as the equitability of activities across all utilized substrates and was calculated as E = H′/log S.
16S rRNA gene amplicon library preparation and sequencing.
Total DNA extraction was carried out on the pellet of 2 ml of saliva by using a Biostic Bacteremia DNA isolation kit (Mo Bio Laboratories, Inc., Carlsbad, CA).
Microbial diversity was studied by pyrosequencing of the amplified V1–V3 region of the 16S rRNA gene. A fragment of 520 bp was amplified by using primers and PCR conditions described previously (46). 454 adaptors were included in the forward primer, followed by a 10-bp sample-specific multiplex identifier (MID). After agarose gel electrophoresis was performed, PCR products were purified twice by using an Agencourt AMPure kit (Beckman Coulter, Milan, Italy) and quantified by using the QuantiFluor system (Promega, Milan, Italy), and an equimolar pool was obtained prior to further processing. Duplicate PCR products were pooled for each sample. The amplicon pool was used for pyrosequencing on a GS Junior platform (454 Life Sciences, Roche, Italy) according to the manufacturer's instructions and using titanium chemistry.
Bioinformatics and data analysis.
Raw reads were first filtered according to the 454 processing pipeline. Sequences were then analyzed by using QIIME 1.7.0 software (47). Raw reads were demultiplexed and further filtered through the split_library.py script of QIIME. In order to guarantee a higher level of accuracy, the reads were excluded from the analysis if they had an average quality score of <25, if there were ambiguous base calls, if there were primer mismatches, and if they were <300 bp. Sequences that passed the quality filter were denoised (48), and singletons were excluded. Operational taxonomic units (OTUs) defined by 97% similarity were picked by using the uclust method (49), and the representative sequences, chosen as the most abundant in each cluster, were submitted to the RDPII classifier (50) to obtain the taxonomy assignment and the relative abundance of each OTU by using the Greengenes 16S rRNA gene database (51). Alpha- and beta-diversities were evaluated by QIIME, as recently described (52). Adonis and Anosim statistical tests were performed with the compare_category.py script of QIIME, in order to verify if there were differences between the two types of individuals. For categorical variables, analysis of variance (ANOVA) and G tests were carried out with the otu_category_significance.py script of QIIME in order to test whether the presence/abundance of any OTUs was significantly associated with a specific subject type or the other variables, while Spearman correlations were computed between OTUs and metabolite concentrations.
Gas chromatography-mass spectrometry–solid-phase microextraction analysis of salivary volatile compounds.
After preconditioning according to the manufacturer's instructions, a Carboxen-polydimethylsiloxane (CAR-PDMS) (85-μm) fiber and a manual solid-phase microextraction (SPME) holder (Supelco, Inc., Bellefonte, PA, USA) were used. Before headspace sampling was performed, the fiber was exposed to a gas chromatography (GC) inlet for 5 min for thermal desorption at 250°C. Three grams of salivary sample was placed into 10-ml glass vials and added with 10 μl of 4-methyl-2-pentanol (final concentration of 33 mg/liter) as the internal standard. Samples were then equilibrated for 10 min at 45°C. The SPME fiber was exposed to each sample for 40 min. Both equilibration and absorption phases were carried out with stirring. The fiber was then inserted into the injection port of the gas chromatograph for 10 min of sample desorption. GC-mass spectrometry (MS) analyses were carried out with an Agilent 7890A gas chromatograph (Agilent Technologies, Palo Alto, CA) coupled to an Agilent 5975C mass selective detector operating in the electron impact mode (ionization voltage, 70 eV). A Varian CP7773 Wax 52 CB capillary column (length, 50 m; inside diameter, 0.32 mm) (Agilent Technologies) was used. The temperature program was 40°C for 1 min, followed by an increase to 65°C, at a rate of 4.5°C/min; an increase to 230°C, at a rate of 10°C/min; and then 230°C for 17 min. The injector, interface, and ion source temperatures were 250°C, 250°C, and 230°C, respectively. The mass-to-charge ratio interval was 30 to 350 Da at a rate of 2.9 scans per s. Injection was carried out in the splitless mode, and helium (flow rate, 1 ml/min) was used as the carrier gas. Molecules were identified based on comparisons of their retention times with those of pure compounds (Sigma-Aldrich, Milan, Italy). Identities were confirmed by searching mass spectra in the available databases (NIST, version 2005, and Wiley, version 1996). All the GC-MS raw files were converted to the netCDF format via a Chemstation system (Agilent Technologies) and subsequently processed with the XCMS toolbox (http://metlin.scripps.edu/download/). XCMS software allows automatic and simultaneous retention time alignment, matched filtration, peak detection, and peak matching. The resulting table, containing information such as peak indices (retention time-m/z pair) and normalized peak areas, was exported into R (http://www.r-project.org/) for subsequent statistical or multivariate analyses. Quantitative data for the compounds identified were obtained by the interpolation of the relative areas versus the internal standard area. GC-MS-SPME data were organized into a matrix and analyzed by canonical discriminant analysis of principal coordinates (CAP) (41).
Statistical analysis.
Culture-dependent and metabolome data were obtained in at least triplicates. ANOVA was carried out on transformed data, followed by separation of means with Tukey's honestly significant difference (HSD) test, using Statistica for Windows statistical software (Statistica 6.0 for Windows 1998; StatSoft). Significantly different groups (P < 0.05) by Tukey's test are indicated in Table 2. CAP analysis was also carried out for GC-MS-SPME data. The hypothesis of nonsignificant differences in the multivariate location within groups was tested by using the trace statistic based on 9,999 permutations (53). The correlation between the concentration of metabolites and the number of predominant bacterial cells and genera was examined by linear regression analysis.
TABLE 2.
Cultivable bacteria in saliva samples from children with celiac disease who were treated with a gluten-free diet for at least 2 years and from healthy childrenb
| Microbial group | Median no. of cultivable cells, log CFU/ml (range) |
P value | |
|---|---|---|---|
| T-CD children | HC | ||
| Total aerobic bacteria | 7.29 (6.22–8.02) | 7.55 (6.92–7.89) | 0.057 |
| Total anaerobes | 7.62a (6.00–8.36) | 7.81a (7.00–8.93) | 0.029 |
| Enterococcus and Lactobacillus | 7.23 (5.85–8.51) | 7.42 (6.06–8.02) | 0.27 |
| Lactococcus and Streptococcus | 7.02 (6.00–7.90) | 7.05 (6.14–7.88) | 0.156 |
| Staphylococcus and Micrococcus | 5.70 (5.10–6.13) | 5.50 (4.87–7.18) | 0.178 |
| Bacteroides, Porphyromonas, and Prevotella | 6.10 (5.09–7.01) | 6.07 (5.02–6.90) | 0.074 |
| Enterobacteriaceae | 3.66a (1.50–4.59) | 1.00a (0–5.59) | 0.042 |
| Bifidobacterium | 7.32 (6.04–8.43) | 7.18 (6.90–8.03) | 0.285 |
Significantly different groups (P < 0.05) by Tukey's test.
Data are the means of three independent experiments (n = 3) for each child.
Nucleotide sequence accession number.
The 16S rRNA sequences produced in this study are available at the Sequence Read Archive of the NCBI (SRP035361).
RESULTS
Enumeration of cultivable bacteria.
Selective media were used to enumerate cultivable bacteria (Table 2). Compared to HC, the number of total anaerobes was significantly (P < 0.05) decreased in the saliva of T-CD children. The other significant (P < 0.05) difference concerned the number of presumptive enterobacteria. No significant (P > 0.05) differences were found between the T-CD and HC groups for the other microbial groups.
Community-level catabolic profiles.
The substrate utilization pattern (H′ index) and substrate richness (S index) values were calculated (Fig. 1). Compared to HC, the H′ and S indices were significantly (P < 0.05) decreased in the saliva of T-CD children. The E index, which measures the statistical significance (equitability) of the H′ and S index values, confirmed the above-described significant (P < 0.05) differences.
FIG 1.
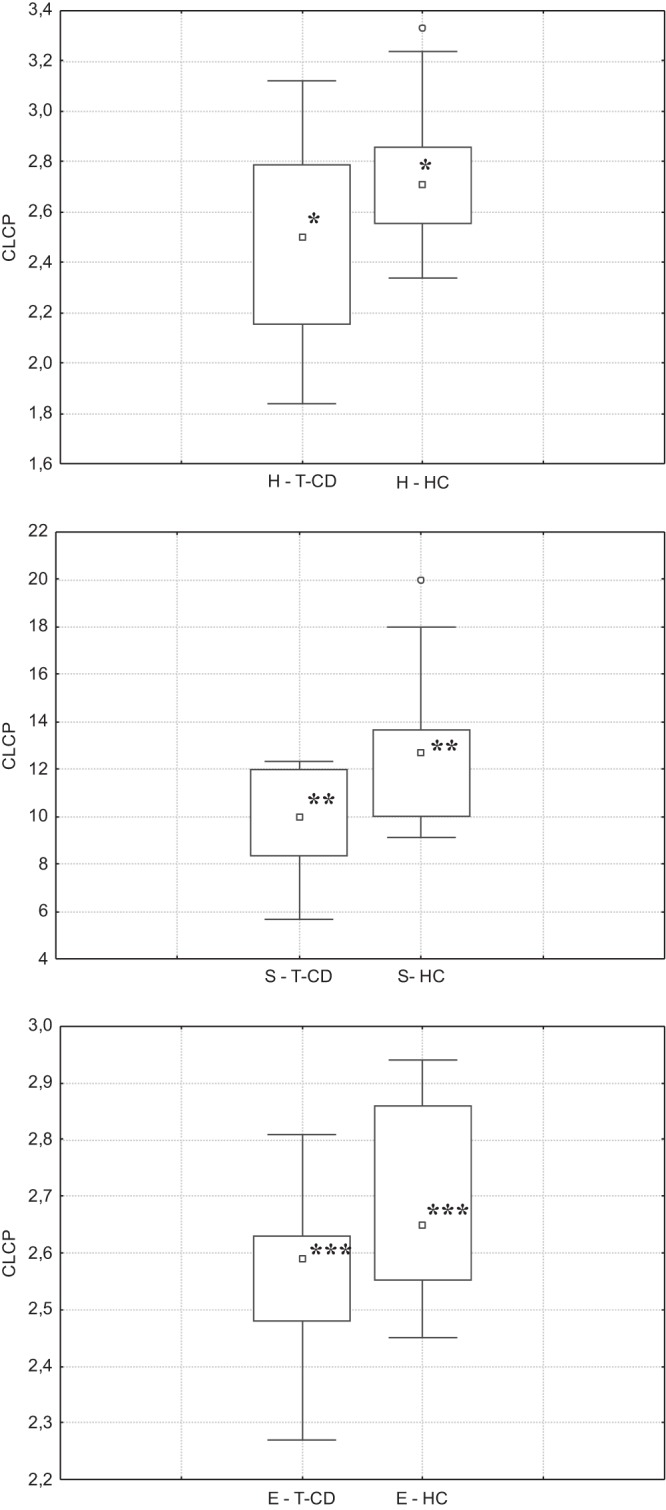
CLCP indices (utilization pattern substrate [H′], substrate richness [S], and equitability [E]) of the salivary microbiota of children with celiac disease who were treated with a gluten-free diet for at least 2 years (T-CD children) and healthy children (HC). Data are the means of three independent experiments (n = 3). The center line of each box represents the median, and the top and bottom of the box represent the 75th and 25th percentiles of the data, respectively. The top and bottom of the error bars represent the 5th and 95th percentiles of the data, respectively. The circles in each box plot extend to the outliers of the data.
Richness and diversity of the salivary microbiota based on 16S rRNA gene sequencing data analysis.
A total of 235,481 raw sequences was obtained and analyzed; 179,276 reads passed the filters applied through the QIIME split_library.py script, with an average value of 7,171 reads/sample and an average length of 499 bp calculated after primer removal. Good's estimated sample coverage (median value of 98%; P > 0.05) indicated that satisfactory coverage was reached for all samples analyzed. The average number of species (OTUs) identified in the salivary samples of T-CD children (median value of 319) significantly (P = 0.005) differed from that in the salivary samples from HC (median value of 393). Overall, six phyla (Firmicutes, Bacteroidetes, Proteobacteria, Fusobacteria, Actinobacteria, and Tenericutes) and two candidate divisions (TM7 and SR1) were identified (Fig. 2; see also Table S1 in the supplemental material). Compared to HC, the saliva of T-CD children differed only in the levels of Bacteroidetes (40 versus 33%; P = 0.001) and Actinobacteria (5 versus 7%; P = 0.023) (Fig. 2). The bacterial diversity among the microbial communities of T-CD children and HC was also estimated by using the richness estimator (Chao1) and diversity (Shannon) indices (Fig. 3). Chao1 and Shannon diversity index values were lower (P > 0.05) for the saliva of T-CD children than for the saliva of HC. The difference in the community structure was further confirmed by using three phylogeny-based beta-diversity measures. The salivary microbiota of HC and T-CD children were clearly differentiated based on bacterial-lineage-specific principal-coordinate analysis with a weighted UniFrac distance matrix (Fig. 4). In addition, both Adonis and Anosim statistical tests indicated a significant influence of subject type (HC versus T-CD children) on microbial diversity.
FIG 2.

Relative abundance (percent) of total bacteria, which were found at the phylum level in the saliva of T-CD children and healthy children.
FIG 3.
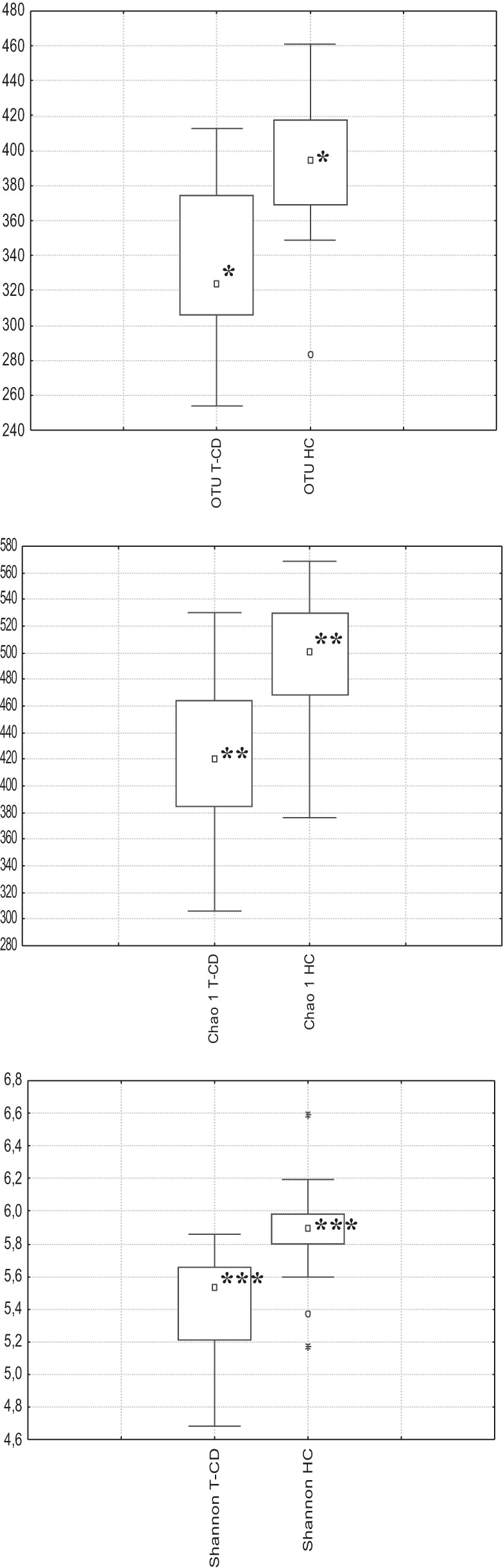
Average number of species (OTUs), richness (Chao1), and diversity (Shannon index) values for the saliva of T-CD children and HC. The center line of each box represents the median, and the top and bottom of the box represent the 75th and 25th percentiles of the data, respectively. The top and bottom of the error bars represent the 5th and 95th percentiles of the data, respectively. The circles and asterisks in each box plot extend to the outliers and extremes of the data, respectively.
FIG 4.

Principal-coordinate analysis based on weighted UniFrac analysis of all 16S rRNA gene sequences found in the saliva of T-CD children and HC.
Distinctive salivary microbiome associated with CD.
The differences (P < 0.05) in the relative abundances of OTUs associated with the saliva of T-CD children or HC are shown in Fig. 5. The abundances of some OTUs belonging to the Firmicutes phylum significantly differed between T-CD children and HC (P < 0.05). Streptococcus sanguinis, Gemella species, and Lachnospiraceae were more abundant in the saliva of T-CD children, while levels of some lactic acid bacteria (e.g., Streptococcus thermophilus) were highest in HC. Granulicatella adiacens, Mogibacterium sp., Selenomonas sp., and Veillonella parvula were also positively associated (P < 0.05) with HC. Within the phylum Bacteroidetes, Porphyromonas sp., Porphyromonas endodontalis, and Prevotella nanceiensis showed the highest abundances in the saliva of T-CD children. Actinomyces oris, Atopobium sp., and Corynebacterium durum were less abundant Actinobacteria in the saliva of HC. The candidate division SR1 was more abundant in saliva of T-CD children (P < 0.05).
FIG 5.

Relative proportions (percent) of predominant bacteria, showing significant (P < 0.05) differences between the saliva samples of T-CD children and those of HC.
Volatile organic compound profiling of saliva.
Overall, the content of various metabolites largely varied within the same group (T-CD children or HC), and the total median values of some volatile organic compounds (VOCs) differed significantly (P < 0.05) between T-CD children and HC. GC-MS-SPME data were analyzed by canonical discriminant analysis of principal coordinates (CAP). Compounds with negative values were those significantly (P < 0.05) associated with saliva of T-CD children, while those on the positive axis were significantly (P < 0.05) associated with saliva of HC (Fig. 6A). The median values for alcohols and phenols [e.g., 2-ethyl-1-hexanol, 4-(1,1,3,3-tetramethylbutyl)-phenol, and ethyl alcohol] were significantly (P < 0.05) higher in HC (Fig. 6A; see also Table S2 in the supplemental material). Within aldehydes, nonanal was found at the highest level in T-CD children, and octanal was found at the highest level in HC. The levels of butanoic acid 2-methyloctyl ester and acetic acid ethyl ester were highest in HC and T-CD children, respectively. Hydrocarbons were the largest group of VOCs. Overall, hydrocarbons (e.g., 1-octadecene) were found at the highest level in HC. On the other hand, halogenated and aromatic hydrocarbons (e.g., 1-chlorodecane and trichloromethane) were significantly associated with T-CD children. Carbone disulfide was also associated with T-CD children. With few exceptions, ketones, terpenes, and thiophenes were found at the highest levels in the saliva of HC. According to the main VOC composition, the two groups of children were separated by CAP (Fig. 6B).
FIG 6.

CAP loading coefficient plot (A) and score plot (B) of the volatile organic compounds found in saliva of T-CD children and HC. n.d., not defined.
Correlation between microbiome and metabolome data.
Spearman's correlation analysis run with QIIME allowed us to find some correlations between the relative abundances of salivary bacteria and metabolites (data not shown). A positive correlation (false discovery rate [FDR], <0.05) was found between the abundances of P. endodontalis and Prevotella sp. and the levels of nonanal and 1-chlorodecane. C. durum was positively correlated with 1,2,3-trimethylbenzene, 2,6-dimethyl-4-heptanone, 4-methyl-2-hexanone, and 4-methyl-3-penten-2-one, and G. adiacens, Atopobium sp., and Bacilli levels were correlated (FDR, <0.05) with the levels of 1(3H)-isobenzofuranone (γ-lactone) and 1-octadecene. Also, γ-lactone and 1-chlorodecane were correlated with V. parvula, S. thermophilus, and the candidate division SR1.
DISCUSSION
According to the World Health Organization, the oral human microbiota plays a crucial role in the health or disease status of the human host (54–56). Previously, variations of the composition of the oral microbiota were described for CD patients, but the trend and the importance were not defined (9, 39).
To the best of our knowledge, this study represents the largest effort to characterize the diversity, population structure, and metabolome of the oral microbiota of CD children on a GFD.
Some cultivable microbial populations differed between the saliva of T-CD children and the saliva of HC. In agreement with this study, higher levels of total anaerobic bacteria were found in the fecal samples of HC than in the fecal samples of T-CD children (14). According to culture-dependent data, the community-level catabolic profiles (CLCPs) also varied between T-CD children and HC. As previously shown for other ecosystems (45, 57, 58), CLCPs successfully described the global metabolic activities of the salivary microbiota. The substrate with the lowest Shannon's index (H′ index) was found in the saliva of T-CD children. The capacity for the use of multiple substrates (e.g., carbohydrates, carboxylic acids, polymers, amino acids, amines, and miscellaneous compounds) was usually linked to the microbial diversity of a certain microbial ecosystem. As shown by the Shannon-Weaver index, CLCP analysis showed the highest level of microbial diversity in HC.
Analysis of pyrosequencing data showed that bacterial groups from the saliva of HC were similar to those found in previous studies (55, 56). In agreement with the CLCPs, sequencing data showed the highest richness estimator (Chao1) and diversity index (Shannon) values for the saliva of HC. The gut and oral microbiota may have common structures (29), and the bacterial species richness of both these ecosystems usually decreases during inflammatory bowel disease (IBD) (59–61). It was hypothesized that high microbial richness and diversity values, which characterized the healthy microbiota, may have a protective effect on humans (59). The composition of the main bacterial phyla differed between the salivary microbiota of T-CD children and that of HC. Compared to HC, Lachnospiraceae, Gemellaceae (genus Gemella), and S. sanguinis were most abundant in the saliva of T-CD children. On the contrary, the abundance of S. thermophilus (the main Streptococcus species) was markedly decreased in T-CD children. S. sanguinis was found to be associated with endocarditis and other distant-site infections (30). Streptococcus is the main genus of the healthy oral microbiota (25, 59, 62). Similar results were found for the saliva of individuals affected by IBD (59). Moreover, duodenal and fecal samples of CD patients (both at diagnosis and during GFD) were associated with a decrease in the level of Streptococcaceae (63). Other Firmicutes (e.g., V. parvula) also associated with oral health (64) were found at the highest levels in the saliva of HC. The saliva of T-CD children harbored the highest levels of Bacteroidetes (e.g., Porphyromonas sp., P. endodontalis, and Prevotella nanceiensis) and the lowest levels of Actinobacteria. The fecal and duodenal microbiota of CD patients were characterized by the largest numbers of Gram-negative bacteria (Bacteroidetes and Enterobacteria) and the smallest numbers of Gram-positive bacteria such as Firmicutes (e.g., lactic acid bacteria) and Actinobacteria (e.g., Bifidobacterium) (14, 20, 21). Some Porphyromonas species (e.g., P. endodontalis) were previously associated with periodontal diseases (65). Within the phylum Actinobacteria, the saliva of T-CD children was characterized by decreased levels of some Actinomyces species (including A. oris), Atopobium sp., and C. durum. Actinomyces spp. are predominant Gram-positive members of the human oral commensal microbiota and are known as the initial colonizers of tooth surfaces (66). Rothia mucilaginosa was the only Actinobacteria species found at the highest level in saliva of T-CD children. Rothia species are components of the oral microbiota and were also identified in duodenal biopsy specimens (67). Although some infections by R. mucilaginosa have been described, this species is considered a harmless colonizer of the oral cavity (68). Recently, it was hypothesized that some species of the Rothia genus, including R. mucilaginosa, are involved in gluten degradation (69, 70).
Overall, the human-associated microbiota interacts directly with the host by means of metabolic products (71, 72). This study combined characterizations of the salivary microbiota and the related metabolome. Analyses of the saliva metabolome have been successfully used in several fields of physiology, diagnostics, functional genomics, pharmacology, toxicology, and nutrition (36, 73). CAP of the GC-MS–SPME metabolic profiles allowed the identification of VOCs, which changed in the saliva of T-CD children. Many of the VOCs described in this study corresponded to those previously reported for the saliva of healthy individuals (37, 74). These compounds may have various origins, as there are many possible routes of entry into the salivary flow, including environmental exposure through inhalation of air and/or water vapor through the lungs, ingestion through the mouth, food intake, and transdermal absorption through the skin. Moreover, microbial metabolic activities at the level of the oral cavity may also affect the synthesis of VOCs (37, 74). First, this study showed some correlations between the microbiome and metabolome of children with CD. It was hypothesized that the salivary microbiota affects the development of the gut microbiota (59). According to this hypothesis, high levels of nonanal, 4-methyl-2-hexanone (2-hexanone), and ethyl-acetate (acetic acid ethyl ester) were found in both saliva (this study) and fecal (14, 75) samples of T-CD children.
Some microbial indices (e.g., ratios of some Firmicutes and Actinobacteria to Bacteroidetes) and the levels of some metabolites (e.g., nonanal, ethyl-acetate, and 2-exanone) are signatures of CD patients. GFD lasting at least 2 years did not completely restore the salivary microbiota and, consequently, the salivary metabolome of T-CD children. Similar data were also found for duodenal (63) and fecal (14, 75) samples. Further screenings with a wider sample size could provide informative sources for discovering CD-specific noninvasive salivary biomarkers. The limitation of this study was related to the small number of T-CD children analyzed. Further studies dealing with different severities of CD may be useful to highlight the correlations between the severity of CD and the salivary microbiome. Different GFDs could be analyzed to determine the effects of specific foods on the salivary microbiota of T-CD children. In addition, dietary implementation with probiotics could be regarded as an alternative strategy to correct oral dysbiosis.
Supplementary Material
Footnotes
Published ahead of print 21 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00362-14.
REFERENCES
- 1.Tye-Din J, Anderson R. 2008. Immunopathogenesis of celiac disease. Curr. Gastroenterol. Rep. 10:458–465. 10.1007/s11894-008-0085-9 [DOI] [PubMed] [Google Scholar]
- 2.Mina S, Riga C, Azcurra AI, Brunotto M. 2012. Oral ecosystem alterations in celiac children: a follow-up study. Arch. Oral Biol. 57:154–160. 10.1016/j.archoralbio.2011.08.017 [DOI] [PubMed] [Google Scholar]
- 3.Lamireau T, Clouzeau H. 2013. Epidemiology of celiac disease. Pathol. Biol. (Paris) 61:e1–e4. 10.1016/j.patbio.2011.03.005 [DOI] [PubMed] [Google Scholar]
- 4.Murray JA. 1999. The widening spectrum of celiac disease. Am. J. Clin. Nutr. 69:354–365 [DOI] [PubMed] [Google Scholar]
- 5.Mina SS, Azcurra AI, Dorronsoro S, Brunotto MN. 2008. Alterations of the oral ecosystem in children with celiac disease. Acta Odontol. Latinoam. 21:121–126 http://www.actaodontologicalat.com/archivo/v21n2/fulltext/articulo2.pdf [PubMed] [Google Scholar]
- 6.Toscano V, Conti FG, Anastasi E, Mariani P, Tiberti C, Poggi M, Montuori M, Monti S, Laureti S, Cipolletta E, Gemme G, Caiola S, Di Mario U, Bonamico M. 2000. Importance of gluten in the induction of endocrine autoantibodies and organ dysfunction in adolescent celiac patients. Am. J. Gastroenterol. 95:1742–1748. 10.1111/j.1572-0241.2000.02187.x [DOI] [PubMed] [Google Scholar]
- 7.Lenander-Lumikari M, Ihalin R, Lähteenoja H. 2000. Changes in whole saliva in patients with coeliac disease. Arch. Oral Biol. 45:347–354. 10.1016/S0003-9969(00)00008-X [DOI] [PubMed] [Google Scholar]
- 8.Samaşca G, Iancu M, Farcău D, Butnariu A, Pop T, Pîrvan A, Andreica M, Miu N, Cristea V, Dejica D. 2011. IgA anti-tissue transglutaminase antibodies, first line in the diagnosis of celiac disease. Clin. Lab. 57:695–701 [PubMed] [Google Scholar]
- 9.Acar S, Yetkiner AA, Ersin N, Oncag O, Aydogdu S, Arikan C. 2012. Oral findings and salivary parameters in children with celiac disease: a preliminary study. Med. Princ. Pract. 21:129–133. 10.1159/000331794 [DOI] [PubMed] [Google Scholar]
- 10.Sanz Y. 2009. Novel perspectives in celiac disease therapy. Mini Rev. Med. Chem. 9:359–367. 10.2174/1389557510909030359 [DOI] [PubMed] [Google Scholar]
- 11.Niewinski MM. 2008. Advances in celiac disease and gluten-free diet. J. Am. Diet. Assoc. 108:661–672. 10.1016/j.jada.2008.01.011 [DOI] [PubMed] [Google Scholar]
- 12.Stene LC, Honeyman MC, Hoffenberg EJ, Haas JE, Sokol RJ, Emery L, Taki I, Norris JM, Erlich HA, Eisenbarth GS, Rewers M. 2006. Rotavirus infection frequency and risk of celiac disease autoimmunity in early childhood: a longitudinal study. Am. J. Gastroenterol. 101:2333–2340. 10.1111/j.1572-0241.2006.00741.x [DOI] [PubMed] [Google Scholar]
- 13.Walters JR, Bamford KB, Ghosh S. 2008. Coeliac disease and the risk of infections. Gut 57:1034–1035. 10.1136/gut.2008.151571 [DOI] [PubMed] [Google Scholar]
- 14.Di Cagno R, De Angelis M, De Pasquale I, Ndagijimana M, Vernocchi P, Ricciuti P, Gagliardi F, Laghi L, Crecchio C, Guerzoni ME, Gobbetti M, Francavilla R. 2011. Duodenal and faecal microbiota of celiac children: molecular, phenotype and metabolome characterization. BMC Microbiol. 11:219. 10.1186/1471-2180-11-219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sánchez E, Nadal I, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y. 2008. Reduced diversity and increased virulence-gene carriage in intestinal enterobacteria of coeliac children. BMC Gastroenterol. 8:50. 10.1186/1471-230X-8-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sánchez E, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y. 2010. Intestinal Bacteroides species associated with coeliac disease. J. Clin. Pathol. 63:1105–1111. 10.1136/jcp.2010.076950 [DOI] [PubMed] [Google Scholar]
- 17.Sánchez E, Ribes-Koninckx C, Calabuig M, Sanz Y. 2012. Intestinal Staphylococcus spp. and virulent features associated with coeliac disease. J. Clin. Pathol. 65:830–834. 10.1136/jclinpath-2012-200759 [DOI] [PubMed] [Google Scholar]
- 18.Sanz Y, Sanchez E, Marzotto M, Calabuig M, Torriani S, Dellaglio F. 2007. Differences in faecal bacterial communities in coeliac and healthy children as detected by PCR and denaturing gradient gel electrophoresis. FEMS Immunol. Med. Microbiol. 51:562–568. 10.1111/j.1574-695X.2007.00337.x [DOI] [PubMed] [Google Scholar]
- 19.Schippa S, Iebba V, Barbato M, Di Nardo G, Totino V, Checchi MP, Longhi C, Maiella G, Cucchiara S, Conte MP. 2010. A distinctive ‘microbial signature' in celiac pediatric patients. BMC Microbiol. 10:175. 10.1186/1471-2180-10-175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Collado MC, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y. 2009. Specific duodenal and faecal bacterial groups associated with pediatric coeliac disease. J. Clin. Pathol. 62:264–269. 10.1136/jcp.2008.061366 [DOI] [PubMed] [Google Scholar]
- 21.Nadal I, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y. 2007. Imbalance in the composition of the duodenal microbiota of children with coeliac disease. J. Med. Microbiol. 56:1669–1674. 10.1099/jmm.0.47410-0 [DOI] [PubMed] [Google Scholar]
- 22.Petrof EO, Claud EC, Gloor GB, Allen-Vercoe E. 2013. Microbial ecosystems therapeutics: a new paradigm in medicine? Benef. Microbes 4:53–61. 10.3920/BM2012.0039 [DOI] [PubMed] [Google Scholar]
- 23.Blaser MJ. 2010. Harnessing the power of the human microbiome. Proc. Natl. Acad. Sci. U. S. A. 107:6125–6126. 10.1073/pnas.1002112107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sekirov I, Russel SL, Antunes LCM, Finlay BB. 2010. Gut microbiota in health and disease. Physiol. Rev. 90:859–904. 10.1152/physrev.00045.2009 [DOI] [PubMed] [Google Scholar]
- 25.Bik ME, Long CD, Armitage GC, Loomer P, Emerson J, Mongodin EF, Nelson KE, Gill SR, Fraser-Liggett CM, Relman DA. 2010. Bacterial diversity in the oral cavity of ten healthy individuals. ISME J. 4:962–974. 10.1038/ismej.2010.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ling Z, Kong J, Jia P, Wei C, Wang Y, Pan Z, Huang W, Li L, Chen H, Xiang C. 2010. Analysis of oral microbiota in children with dental caries by PCR-DGGE and barcoded pyrosequencing. Microb. Ecol. 60:677–690. 10.1007/s00248-010-9712-8 [DOI] [PubMed] [Google Scholar]
- 27.Francavilla R, Calasso M, Calace L, Siragusa S, Ndagijimana M, Vernocchi P, Brunetti L, Mancino G, Tedeschi G, Guerzoni E, Indrio F, Laghi L, Miniello V, Gobbetti M, De Angelis M. 2012. Effect of lactose on gut microbiota and metabolome of infants with cow's milk allergy. Pediatr. Allergy Immunol. 23:420–427. 10.1111/j.1399-3038.2012.01286.x [DOI] [PubMed] [Google Scholar]
- 28.Piombino P, Genevose A, Esposito S, Moio L, Cutolo PP, Chambery A, Severino V, Moneta E, Smith DP, Owens SM, Gilbert JA, Ercolini D. 2014. Saliva from obese individuals suppresses the release of aroma compounds from wine. PLoS One 9:e85611. 10.1371/journal.pone.0085611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maukonen J, Matto J, Suihko ML, Saarela M. 2008. Intra-individual diversity and similarity of salivary and faecal microbiota. J. Med. Microbiol. 57:1560–1568. 10.1099/jmm.0.47352-0 [DOI] [PubMed] [Google Scholar]
- 30.Lockhart PB, Durak DT. 1999. Oral microflora as a cause of endocarditis and other distant site infections. Infect. Dis. Clin. N. Am. 13:833–850. 10.1016/S0891-5520(05)70111-2 [DOI] [PubMed] [Google Scholar]
- 31.Beck JD, Eke P, Heiss G, Manadios P, Couper D, Lin D, Moss K, Elter J, Offenbacher S. 2005. Periodontal disease and coronary heart disease: a reappraisal of the exposure. Circulation 112:19–24. 10.1161/CIRCULATIONAHA.104.511998 [DOI] [PubMed] [Google Scholar]
- 32.Paju S, Scannapeico FA. 2007. Oral biofilm, periodontitis, and pulmonary infections. Oral Dis. 13:508–512. 10.1111/j.1601-0825.2007.01410a.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koren O, Spor A, Felin J, Fak F, Stombaugh J, Tremaroli V, Behre CJ, Knight R, Fagerberg B, Ley RE, Backhed F. 2011. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc. Natl. Acad. Sci. U. S. A. 108:4592–4598. 10.1073/pnas.1011383107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boggess KA, Beck JD, Murtha AP, Moss K, Offenbacher S. 2006. Maternal periodontal disease in early pregnancy and risk for a small-for-gestational-age infant. Am. J. Obstet. Gynecol. 194:1316–1322. 10.1016/j.ajog.2005.11.059 [DOI] [PubMed] [Google Scholar]
- 35.Takeshita T, Nakano Y, Kumagai T, Yasui M, Kamio N, Shibata Y, Shiota S, Yamashita Y. 2009. The ecological proportion of indigenous bacterial populations in saliva is correlated with oral health status. ISME J. 3:65–78. 10.1038/ismej.2008.91 [DOI] [PubMed] [Google Scholar]
- 36.Zhang A, Sun H, Wang X. 2012. Saliva metabolomics opens door to biomarker discovery, disease diagnosis, and treatment. Appl. Biochem. Biotechnol. 168:1718–1727. 10.1007/s12010-012-9891-5 [DOI] [PubMed] [Google Scholar]
- 37.AL-Kateb H, de Lacy Costello B, Ratcliffe N. 2013. An investigation of volatile organic compounds from the saliva of healthy individuals using headspace-trap/GC-MS. J. Breath Res. 7:036004. 10.1088/1752-7155/7/3/036004 [DOI] [PubMed] [Google Scholar]
- 38.Avsar A, Kalayci AG. 2008. The presence and distribution of dental enamel defects and caries in children with celiac disease. Turk. J. Pediatr. 50:45–50 http://www.turkishjournalpediatrics.org/?fullTextId=467&lang=eng [PubMed] [Google Scholar]
- 39.Shteyer E, Berson T, Lachmanovitz O, Hidas A, Wilschanski M, Menachem M, Shachar E, Shapira J, Steinberg D, Moskovitz M. 2013. Oral health status and salivary properties in relation to gluten free diet in children with celiac disease. J. Pediatr. Gastroenterol. Nutr. 57:49–52. 10.1097/MPG.0b013e31828b3705 [DOI] [PubMed] [Google Scholar]
- 40.Husby S, Koletzko S, Korponay-Szabó IR, Mearin ML, Phillips A, Shamir R, Troncone R, Giersiepen K, Branski D, Catassi C, Lelgeman M, Mäki M, Ribes-Koninckx C, Ventura A, Zimmer KP, ESPGHAN Working Group on Coeliac Disease Diagnosis, ESPGHAN Gastroenterology Committee, European Society for Pediatric Gastroenterology, Hepatology, and Nutrition 2012. European Society for Pediatric Gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease. J. Pediatr. Gastroenterol. Nutr. 54:136–160. 10.1097/MPG.0b013e31821a23d0 [DOI] [PubMed] [Google Scholar]
- 41.De Angelis M, Piccolo M, Vannini L, Siragusa S, De Giacomo A, Serrazzanetti DI, Cristofori F, Guerzoni ME, Gobbetti M, Francavilla R. 2013. Fecal microbiota and metabolome of children with autism and pervasive developmental disorder not otherwise specified. PLoS One 8:e76993. 10.1371/journal.pone.0076993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crecchio C, Gelsomino A, Ambrosoli R, Minati JL, Ruggiero P. 2004. Functional and molecular responses of soil microbial communities under differing soil management practices. Soil Biol. Biochem. 36:1873–1883. 10.1016/j.soilbio.2004.05.008 [DOI] [Google Scholar]
- 43.Shannon CE. 1948. A mathematical theory of communication. Bell Syst. Tech. J. 27:379–423. 10.1002/j.1538-7305.1948.tb01338.x [DOI] [Google Scholar]
- 44.Shannon CE. 1948. A mathematical theory of communication. Bell Labs Tech. J. 27:623–656. 10.1002/j.1538-7305.1948.tb00917.x [DOI] [Google Scholar]
- 45.Zak JC, Willing MR, Moorhead DL, Wildman HG. 1994. Functional diversity of microbial communities: a quantitative approach. Soil Biol. Biochem. 26:1101–1108. 10.1016/0038-0717(94)90131-7 [DOI] [Google Scholar]
- 46.Ercolini D, De Filippis F, La Storia A, Iacono M. 2012. “Remake” by high throughput sequencing of the microbiota involved in the production of water buffalo mozzarella cheese. Appl. Environ. Microbiol. 78:8142–8145. 10.1128/AEM.02218-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reeder J, Knight R. 2010. Rapidly denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Nat. Methods 7:668–669. 10.1038/nmeth0910-668b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- 50.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naïve Bayesan classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73:5261–5267. 10.1128/AEM.00062-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McDonald D, Price MN, Goodrich J, Nawrocki EP, De Santis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. 2012. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6:610–618. 10.1038/ismej.2011.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.De Filippis F, La Storia A, Villani F, Ercolini D. 2013. Exploring the sources of beefsteaks contamination by culture-independent high-throughput sequencing. PLoS One 8:e70222. 10.1371/journal.pone.0070222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ndagijimana M, Laghi L, Vitali B, Placucci G, Brigidi P, Guerzoni ME. 2009. Effect of a synbiotic food consumption on human gut metabolic profiles evaluated by 1H nuclear magnetic resonance spectroscopy. Int. J. Food Microbiol. 134:147–153. 10.1016/j.ijfoodmicro.2009.04.016 [DOI] [PubMed] [Google Scholar]
- 54.Petersen PE. 2003. The world oral health report 2003: Continuous improvement of oral health in the 21st century—the approach of the WHO Global Oral Health Programme. Community Dent. Oral Epidemiol. 31:3–23. 10.1046/j..2003.com122.x [DOI] [PubMed] [Google Scholar]
- 55.Ling Z, Xia L, Wang Y, Li L, Luo Y, Yuan L, Nelson KE, Xiang C. 2013. Pyrosequencing analysis of the human microbiota of healthy Chinese undergraduates. BMG Genomics 14:390. 10.1186/1471-2164-14-390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ling Z, Liu X, Wang Y, Li L, Xiang C. 2013. Pyrosequencing analysis of the salivary microbiota of healthy Chinese children and adults. Microb. Ecol. 65:487–495. 10.1007/s00248-012-0123-x [DOI] [PubMed] [Google Scholar]
- 57.Siragusa S, Di Cagno R, Ercolini E, Minervini F, Gobbetti M, De Angelis M. 2009. Taxonomic structure and monitoring of the dominant population of lactic acid bacteria during wheat flour sourdough type I propagation using Lactobacillus sanfranciscensis starters. Appl. Environ. Microbiol. 75:1099–1109. 10.1128/AEM.01524-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Minervini F, De Angelis M, Di Cagno R, Pinto D, Siragusa S, Rizzello CG, Gobbetti M. 2010. Robustness of Lactobacillus plantarum starters during daily propagation of wheat flour sourdough type I. Food Microbiol. 27:897–908. 10.1016/j.fm.2010.05.021 [DOI] [PubMed] [Google Scholar]
- 59.Said SH, Susa W, Nakagome S, Chinen H, Oshima K, Kim S, Kimura R, Iraha A, Fujita J, Mano S, Morita H, Dohi T, Oota H, Hattori M. 2014. Dysbiosis of salivary microbiota in inflammatory bowel disease and its association with oral immunological biomarkers. DNA Res. 21:15–25. 10.1093/dnares/dst037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, Roca J, Dore J. 2006. Reduced diversity of faecal microbiota in Crohn's disease revealed by a metagenomic approach. Gut 55:205–211. 10.1136/gut.2005.073817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, Grangette C, Vasquez N, Pochart P, Trugnan G, Thomas G, Blottière HM, Doré J, Marteau P, Seksik P, Langella P. 2008. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. U. S. A. 105:16731–16736. 10.1073/pnas.0804812105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kang J-G, Kim SH, Ahn T-Y. 2006. Bacterial diversity in the human saliva from different ages. J. Microbiol. 44:572–576 http://www.msk.or.kr/jsp/view_old_journalD.jsp?paperSeq=2438 [PubMed] [Google Scholar]
- 63.Sánchez E, Donat E, Ribes-Koninckx C, Fernández-Murga ML, Sanz Y. 2013. Duodenal-mucosal bacteria associated with celiac disease in children. Appl. Environ. Microbiol. 79:5472–5479. 10.1128/AEM.00869-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kumar PS, Griffen AL, Moeschberger ML, Leys EJ. 2005. Identification of candidate periodontal pathogens and beneficial species by quantitative 16S clonal analysis. J. Clin. Microbiol. 43:3944–3955. 10.1128/JCM.43.8.3944-3955.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wade WG. 2013. The oral microbiome in health and disease. Pharmacol. Res. 69:137–143. 10.1016/j.phrs.2012.11.006 [DOI] [PubMed] [Google Scholar]
- 66.Kolenbrander PE, Egland PG, Diaz PI, Palmer RJ., Jr 2005. Genome-genome interactions: bacterial communities in initial dental plaque. Trends Microbiol. 13:11–15. 10.1016/j.tim.2004.11.005 [DOI] [PubMed] [Google Scholar]
- 67.Ou G, Hedberg M, Horstedt P, Baranov V, Forsberg G, Drobni M, Sandström O, Wai SN, Johansson I, Hammarström ML, Hernell O, Hammarström S. 2009. Proximal small intestinal microbiota and identification of rod-shaped bacteria associated with childhood celiac disease. Am. J. Gastroenterol. 104:3058–3067. 10.1038/ajg.2009.524 [DOI] [PubMed] [Google Scholar]
- 68.Yamane K, Nambu T, Yamanaka T, Mashimo C, Sugimori C, Leung KP, Fukushima H. 2010. Complete genome sequence of Rothia mucilaginosa DY-18: a clinical isolate with dense meshworklike structures from a persistent apical periodontitis lesion. Sequencing 2010:457236 http://www.hindawi.com/archive/2010/457236/ [Google Scholar]
- 69.Zamakhchari M, Wei G, Dewhirst F, Lee J, Schuppan D, Oppenheim FG, Helmerhorst EJ. 2011. Identification of Rothia bacteria as gluten-degrading natural colonizers of the upper gastro-intestinal tract. PLoS One 6:e24455. 10.1371/journal.pone.0024455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fernandez-Feo M, Ewi G, Blumenjranz G, Dewhirst FE, Schuppan D, Oppenheim FG, Helmerhorst EF. 2013. The cultivable human oral gluten-degrading microbiome and its potential implications in celiac disease and gluten sensitivity. Clin. Microbiol. Infect. 19:e386–e394. 10.1111/1469-0691.12249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Human Microbiome Consortium. 2012. Structure, function and diversity of the human microbiome in an adult reference population. Nature 486:207–214. 10.1038/nature11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Want EJ, Nordstrom A, Morita H, Siuzdak G. 2007. From exogenous to endogenous: the inevitable imprint of mass spectrometry in metabolomics. J. Proteome Res. 6:459–468. 10.1021/pr060505+ [DOI] [PubMed] [Google Scholar]
- 73.Winder CL, Cornmell R, Schuler S, Jarvis RM, Stephens GM, Goodacre R. 2011. Metabolic fingerprinting as a tool to monitor whole-cell biotransformations. Anal. Bioanal. Chem. 399:387–401. 10.1007/s00216-010-4342-z [DOI] [PubMed] [Google Scholar]
- 74.Kusano M, Mendez E, Furton KG. 2013. Comparison of the volatile organic compounds from different biological specimens for profiling potential. J. Forensic Sci. 58:29–39. 10.1111/j.1556-4029.2012.02215.x [DOI] [PubMed] [Google Scholar]
- 75.Di Cagno R, Rizzello CG, Gagliano F, Ricciuti P, Ndagijimana M, Francavilla R, Guerzoni ME, Crecchio C, Gobbetti M, De Angelis M. 2009. Different fecal microbiotas and volatile organic compounds in treated and untreated children with celiac disease. Appl. Environ. Microbiol. 75:3963–3971. 10.1128/AEM.02793-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.