

Abstract
The spore-forming bacterium Bacillus licheniformis is a common contaminant of milk and milk products. Strains of this species isolated from dairy products can be differentiated into three major groups, namely, G, F1, and F2, using random amplification of polymorphic DNA (RAPD) analysis; however, little is known about the genomic differences between these groups and the identity of the fragments that make up their RAPD profiles. In this work we obtained high-quality draft genomes of representative strains from each of the three RAPD groups (designated strain G-1, strain F1-1, and strain F2-1) and compared them to each other and to B. licheniformis ATCC 14580 and Bacillus subtilis 168. Whole-genome comparison and multilocus sequence typing revealed that strain G-1 contains significant sequence variability and belongs to a lineage distinct from the group F strains. Strain G-1 was found to contain genes coding for a type I restriction modification system, urease production, and bacitracin synthesis, as well as the 8-kbp plasmid pFL7, and these genes were not present in strains F1-1 and F2-1. In agreement with this, all isolates of group G, but no group F isolates, were found to possess urease activity and antimicrobial activity against Micrococcus. Identification of RAPD band sequences revealed that differences in the RAPD profiles were due to differences in gene lengths, 3′ ends of predicted primer binding sites, or gene presence or absence. This work provides a greater understanding of the phylogenetic and phenotypic differences observed within the B. licheniformis species.
INTRODUCTION
Spores of Bacillus licheniformis can cause spoilage or specification compliance issues in dairy products, and therefore further understanding of their features, ecology, and phylogeny is needed. It has been found to be the second most common thermophilic spore former after Anoxybacillus flavithermus in milk powder from 18 different countries (1) and has been reported to be the most common aerobic spore former in Australian raw milk, with isolates representing 69% (2) and 67% (3) of the total spore-forming bacteria analyzed.
Molecular methods for identification and genotyping of B. licheniformis isolates have been developed (4–6). B. licheniformis isolates from dairy products have been grouped broadly into random amplification of polymorphic DNA (RAPD) groups G, F1, and F2 (6). Sequence-based genotyping has also been conducted using rpoB and gyrA sequencing (7) and bacitracin synthetase gene sequences (8). These genotyping methods have assigned the isolates to only two or three groups or clusters. Multilocus sequence typing (MLST) of B. licheniformis has identified 27 different sequence types among 53 isolates, suggesting greater heterogeneity than previously observed (9). In agreement with this, we also found significant heterogeneity of dairy B. licheniformis isolates using multilocus variable-number tandem repeat analysis in our previous study, where 19 genotypes were observed among 52 isolates analyzed (5). A more in-depth understanding of different genotypes can be obtained through comparative genomics of representative strains. To date, the genomes of several B. licheniformis strains have been published and include the B. licheniformis strain ATCC 14580 (DSM 13) (10, 11), strain 10-1-A, strain 5-2-D (12), strain WX-02 (13), strain CGMCC 3963 (14), and strain 9945A (15). These are industrial B. licheniformis strains used for producing microbial enzymes and chemicals. However, no genome sequences of B. licheniformis isolated from food are available in any public database.
To investigate the diversity among the dairy strains of B. licheniformis, we carried out whole-genome sequencing of representative strains of the three major RAPD groups, G, F1, and F2. These strains were named according to their RAPD groups (G-1, F1-1, and F2-1). This paper describes the assembly and the annotation of the genomes of these strains and compares their draft genomes to each other, to B. subtilis 168, and to B. licheniformis ATCC 14580. Using the draft genomes, we also identified the DNA sequences of fragments of several RAPD profile bands that are used to differentiate groups G, F1, and F2 and provide an explanation as to why different RAPD profiles are generated.
MATERIALS AND METHODS
Bacterial strains and genomic DNA extraction.
Whole-genome sequencing was carried out on B. licheniformis strains G-1 (RAPD group G), F1-1 (RAPD group F1), and F2-1 (RAPD group F2) which had been isolated from milk powder and genotyped previously (5). Genomic DNA was extracted as described previously (16), and the purity and concentration were determined by agarose gel electrophoresis and by using a NanoDrop instrument (Thermo Scientific). Additional group G isolates (n = 2), group F1 isolates (n = 9), and group F2 isolates (n = 9) of B. licheniformis obtained from milk powder samples and genotyped in our previous study (5) were also used in this study. Gene and genome comparisons with other organisms were carried out, and their genomes were accessed from the following GenBank accession numbers: B. subtilis strain 168 (AL009126.3) (17) and B. licheniformis strains ATCC 14580 (NC_006322.1) (11), 10-1-A (AJLV01000001 to AJLV01000031), 5-2-D (AJLW01000001 to AJLW01000046) (12), WX-02 (AHIF01000001 to AHIF01000003) (13), and CGMCC 3963 (AMWQ01000001 to AMWQ01000152) (14).
Genome sequencing, assembly, annotation, and gene comparison.
Whole-genome shotgun sequencing was performed using the Illumina HiSeq2000 platform at Macrogen (South Korea) to generate raw 101-bp paired reads with an insert size of 200 bp. Different software was used for assembling contigs, and the assembly with the best quality was chosen and annotated. Paired end reads for each strain generated during sequencing, which were in separate FASTAQ files, were brought together into a common file using Geneious, version 5.6.4 (18). Reads were then extracted from Geneious in various multiples of 5 million (5, 10, and 15 million and so on) and exported to individual files. Each of these files was input into VAGUE (Velvet assembler graphics user environment), version 1.0.3, that uses Velvet, version 1.2.07 (19). For each file, different runs were performed using an auto-coverage cutoff option along with the VAGUE-estimated k-mer value and other self-selected k-mer values around this estimated value. The k-mer was estimated by inputting 4,200,000 bp as the estimated genome size for all strains, which is near to the size of the reference genome of strain ATCC 14580. All assemblies performed were evaluated on the basis of N50, maximum contig size, and contig number. For each strain, the assembly having the highest N50 value among all runs was selected. Wherever the assemblies had the same or very near N50 values, the one having the larger maximum contig size and lower contig number was chosen. After assembly, any contamination of extraneous sequence or contigs was removed using BLAST. The draft genomes of strains G-1, F1-1, and F2-1 were then annotated with rapid annotation using subsystem technology (RAST), version 4 (20). The outcomes of genome annotations for these strains by RAST were compared using options in the server itself. Differences in genes or gene functions of the three strains shown by RAST were verified using TBLASTN. Differences in genes as agreed by both the RAST server and TBLASTN programs are reported along with information about their location in the genomes, their presence in B. subtilis 168 and B. licheniformis ATCC 14580, and the highest scoring hits in the NCBI database using the regions of these genes and functions as queries for BLASTP. The regions which were absent in one strain by RAST and TBLASTN were also checked for their presence or absence by mapping their raw reads from the strain which did not contain the regions to the contigs containing the target regions in other strains where the gene was present using Geneious, version 5.6.4 (18).
Whole-genome comparison.
We used Mauve, version 2.3.1 (21), to compare FASTA sequences of the draft genomes of strains G-1, F2-1, and F1-1 with the complete genome sequence of strain ATCC 14580. First, the contigs of strains G-1, F2-1, and F1-1 were individually reordered relative to the genome of strain ATCC 14580. Then the reordered draft genomes were aligned together with strain ATCC 14580 as a reference. Aligning of reordered genomes helped in viewing the regions (local colinear blocks, or LCBs) of draft genomes which were presumably homologous and internally free from genomic rearrangements relative to the parts of the complete genome of strain ATCC 14580.
MLST.
Multilocus sequence typing (MLST) based on internal sequences of adk, ccpA, recF, rpoB, spo0A, and sucC genes (9) was carried out on strains G-1, F1-1, F2-1, 10-1-A, 5-2-D, WX-02, ATCC 14580, and B. subtilis 168. Sequences were concatenated using a Perl script, and phylogenetic trees were constructed using MEGA5 software by the neighbor-joining method with branch lengths estimated by the maximum composite likelihood method (22). Branch quality was assessed by the bootstrap test using 500 replicates. The MLST sequence types for strains G-1, F1-1, and F2-1 were identified using the MLST database of B. licheniformis (9), and a minimum spanning tree diagram using PHYLOViZ software (23) was constructed using data from this database and from the three strains to show their phylogenetic positions.
Gene homology study.
The complete genome sequence of B. subtilis 168 published previously (17) was used as the main reference to find the gene homologs in the assembled contigs of three B. licheniformis strains, G-1, F1-1, and F2-1. The DNA and protein sequences for the genes were obtained from SubtiList (24). At first, the protein sequences from B. subtilis 168 were used to find homologs in the contigs of strains G-1, F2-1, and F1-1 using TBLASTN. Both BLOSUM62 and BLOSUM45 matrices were used with TBLASTN, with other algorithm parameters remaining as the default. If any difference was seen in the result using two matrices, results obtained using BLOSUM62 were preferred to those obtained using BLOSUM45 as the former program is the NCBI default for finding protein homologs. Alternatively, if no homologs were found using the BLOSUM62 matrix, then those found (if any) using BLOSUM45 are reported. If none of these matrices identified a significant homology with a cutoff score of 80 or more and an E value of less than 10−6 (lesser scores and E values less than 10−4 were used for smaller genes), then BLASTN with discontiguous MegaBLAST was used to find homologs. If the homologs could not be found using the above approaches, then genes from the genome of B. licheniformis ATCC 14580 were searched against strains G-1, F2-1, and F1-1 using the matrices and BLAST programs as described above. For the genes of B. subtilis 168 which were not found in strain ATCC 14580, we used the Web tool Gene Synonym Finder (http://www.bioinformatics.org/textknowledge/synonym.php) to ensure that these genes were absent. Finally, the presence or absence of genes was concluded after the raw reads of the strain showing the absence of genes were compared to the draft genome of that strain showing the presence of genes. For the identification of prophages in the strains, the Web-based Phage search tool (PHAST) was used (25).
Organization of homology hits.
The results of a homology search were organized in tables, with each table containing all genes of a particular functional category described previously for B. subtilis strain 168 (17, 24). Only some of the functional categories were considered for our study. We have recorded the contigs containing the regions of homology, the coordinates of the regions, and the percent identity of amino acids (or nucleotides, wherever needed). We have also highlighted in the tables those regions which were obtained using discontiguous MegaBLAST and those regions which covered 95% or less of query sequences.
Antibiotic assays.
Erythromycin resistance of 23 isolates of B. licheniformis was determined using a disc diffusion assay similar to that described earlier (26). For each isolate, 100 μl of overnight culture grown aerobically (without shaking) in Luria Broth at 37°C was mixed with 5 ml of soft Luria agar (0.5%) and then overlaid on 15 ml of Luria agar (1.5%) base. To 8-mm-diameter discs (Advantec, Dublin, CA), 50 μl of erythromycin (50 μg/ml) was added, left for 20 min for antibiotic diffusion, and then incubated overnight at 37°C. The diameters of inhibition zones were measured after incubation, and two biological replicates were carried out on separate days.
The production of antimicrobial compounds by the 23 B. licheniformis isolates against Micrococcus luteus ATCC 10240 was investigated. Supernatants were collected from the B. licheniformis strains grown overnight in Luria broth at 37°C without shaking and filter sterilized using 0.2-μm-pore-size filters (Advantec, Tokyo). M. luteus ATCC 10240 was cultured overnight at 30°C in nutrient agar (without shaking), and 100 μl of the culture was mixed with 5 ml of soft Luria agar (0.5%) and overlaid onto 15 ml of Luria agar (1.5%). The filtered supernatants (50 μl) were added to 8-mm discs that were placed onto agar plates. After 20 min the plates were incubated overnight at 30°C. The diameter of the inhibition zone was measured after incubation, and two biological replicates were carried out on separate days.
Determination of the presence or absence of bacB.
Primers (bacB-F, TCGGCGGACACTCGTTAAAA; bacB-R, GTCTGTTCCAACTCCTCCCG) were designed from within the bacB gene, and PCR was carried out on the 23 B. licheniformis isolates using Platinum SYBR green Supermix UDG (Invitrogen) in Rotorgene Q (Qiagen) with the following steps: 95°C for 2 min, followed by 40 cycles of 95°C for 20s, 55°C for 30s, and 72°C for 60s, with a final step at 72°C for 4 min.
RAPD analysis and cloning of RAPD fragments.
RAPD analysis was carried out as described previously using the primer OPR-13 (5′-GGACGACAAG-3′) (6) on strains G-1, F1-1, and F2-1. The PCR products were separated using a 1.5% agarose gel at 80 V for 3 h, followed by staining with SERVA DNA stain G (SERVA Electrophoresis). RAPD fragments were excised from the agarose gel and purified using a PCR product purification kit (Roche) and then cloned into the pGEM-T vector (Promega). The plasmids were purified from Escherichia coli JM109 using a QIAprep Spin Miniprep kit (Qiagen), and the inserts were sequenced using Sanger sequencing with primers FUP (5′-GTAAAACGACGGCCAGTG-3′) and RUP (5′-CAGGAAACAGCTATGAC-3′) at the Australian Genome Research Facility (AGRF), Brisbane, Australia. The sequences of fragments were compared using BLASTN to filter out vector sequence and were also compared with the three sequenced genomes described here.
Nucleotide sequence accession numbers.
This whole-genome shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession numbers AZSK00000000, AZSL00000000, and AZSM00000000 for strains G-1, F1-1, and F2-1, respectively. The versions described in this paper are versions AZSK01000000, AZSL01000000, and AZSM01000000.
RESULTS AND DISCUSSION
Genome sequencing outputs, assembly, and annotation.
Next-generation sequencing using the Illumina HiSeq2000 platform was carried out for B. licheniformis strains G-1, F1-1, and F2-1. The statistics of the output data are shown in Table S0 in the supplemental material. The total number of paired reads obtained for each strain was more than 65 million. The Q20 and Q30 values, which represent inferred base call accuracies of 99% and 99.9% (27, 28), were assigned to more than 93% and 85% of bases, respectively. On the other hand, ambiguous N bases were present at the rate of 1 out of 100. These values indicate that the probability of an incorrect base call was minimal for the raw sequences, and hence the raw reads were considered appropriate to be used for de novo assemblies.
The statistics for the de novo genome assemblies of three strains are shown in Table 1. For each strain, variation occurred with respect to the effective number of reads input into VAGUE and k-mer size for achieving high-quality assembly. Table 1 represents the highest quality assembly results achieved following several attempts performed using different combinations of read number and k-mer size, and the assemblies corresponding to these results were used for further analysis. Some of the contigs were scaffolded in all three strains so that ambiguous N residues connecting smaller contigs could be seen intermittently in the assembled contigs. The numbers of reads used were chosen for calculating the overall genome coverage for all three strains as shown in Table 1, while the fold coverage for individual contigs for strains G-1, F1-1, and F2-1 ranged from 121- to 4,103-fold, 170-fold to 2,112-fold, and 59-fold to 802-fold, respectively. Comparisons of the values of N50, number of contigs, and maximum contig size for these strains to other published draft genomes indicate that good-quality assemblies have been achieved (19, 29). The total number of nucleotides of the assembled genomes of the three strains was near to the genome size of the completely sequenced strain ATCC 14580, which is 4.22 Mb (10). The G+C contents of all three strains are close to those of other B. licheniformis strains, which are between 45.6 and 46.2% (10, 12, 13).
TABLE 1.
Statistics for the de novo genome assemblies of B. licheniformis strains G-1, F1-1, and F2-1 using VAGUE
| Statistic | Strain G-1 | Strain F1-1 | Strain F2-1 |
|---|---|---|---|
| No. of input reads | 60,000,000 | 55,000,000 | 20,000,000 |
| No. of reads used | 53,905,237 | 49,623,177 | 14,524,929 |
| k-mer size used | 85 | 81 | 77 |
| N50 of contig size (bp) | 504,613 | 661,289 | 317,026 |
| Total no. of nucleotides in assembled genomes | 4,430,231 | 4,112,350 | 4,240,430 |
| No. of contigs | 37 | 30 | 49 |
| GC content (%) | 45.7 | 46.3 | 46.1 |
| Contig size (bp) | |||
| Shortest | 508 | 540 | 566 |
| Median | 27,774 | 55,777 | 9,175 |
| Mean | 119,736 | 137,078 | 86,539 |
| Maximum | 1,169,746 | 1,425,480 | 665,380 |
| Calculated genome coveragea | 1,289× | 1,187× | 347× |
Calculated based on the genome size of B. licheniformis ATCC 14580 (NC_006322.1).
Comparison of three B. licheniformis strain draft genomes using RAST.
RAST was used for annotation of draft genomes into protein-encoding genes (PEGs), RNA-encoding genes (REGs), and hypothetical PEGs. Similar to that in B. licheniformis ATCC 14580 (10), a large proportion of the genome of each strain (approximately one-fourth) is made up of hypothetical PEGs.
Comparison of gene presence or absence among strains was determined by RAST, TBLASTN, and raw-read mapping, and the results are presented in Table S1 in the supplemental material. Regions encoding type I restriction modification systems (subunits M, R, and S) are present in strain G-1 but not in strains F1-1 and F2-1. In the previous study of strain ATCC 14580, two loci each containing genes encoding HsdS, HsdM, and HsdR subunits were found in its genome. These are supposed to be type I restriction modification systems and were hypothesized to cause reduced transformation efficiency (10). However, the type I restriction modification system found in strain G-1 in this study is homologous to, but has weak identity with, HsdS, HsdM, and HsdR from strain ATCC 14580. Similarly, the genes encoding spore germination protein (GerHB/GerIB), potassium-transporting ATPases, urease accessory proteins (UreD, UreE, UreF, and UreG), and urease subunit alpha, beta, and gamma enzymes were found only in strain G-1, not in strain F1-1, F2-1, or ATCC 14580 (10). We confirmed experimentally that all members of group G (three isolates) showed urease production, and all members of groups F1 and F2 (10 each) showed no such activity in Stuart's urea broth (data not shown), thereby agreeing with the prediction based on genome analysis. A urease production test can therefore potentially be used to differentiate RAPD groups G and F, but a higher number of group G isolates would be required to confirm this.
Whole-genome comparison.
The pairwise alignment of the draft genomes of strains G-1, F2-1, and F1-1 with the complete genome of ATCC 14580 using Mauve is shown in Fig. 1. Each locally colinear block (LCB) contains a colored similarity profile of the local sequence, with the height of the colored profile corresponding to the average degree of sequence conservation in that region. Areas that are white, such as those seen frequently in the genome of strain G-1, suggest that this genome contains a significant amount of sequence variability compared with the other three genomes shown in Fig. 1.
FIG 1.
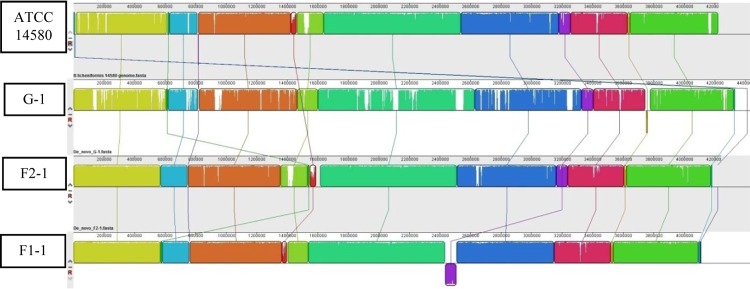
Whole-genome comparison of B. licheniformis strains ATCC 14580, G-1, F2-1, and F1-1 using Mauve, version 2.3.1. The pairwise alignment of draft genomes is shown. The similarly colored blocks are presumably homologous (and internally free of rearrangements) among genomes. White areas within blocks indicate sequences which were not aligned to other genomes and represent nonhomologous regions.
MLST analysis.
An MLST scheme for B. licheniformis based on six housekeeping genes (adk, ccpA, recF, rpoB, spo0A, and sucC) has recently been developed (9), and sequences of these genes have been deposited in the MLST database (http://www.pubmlst.org). MEGA5 (22) was used to analyze the phylogeny of B. licheniformis G-1, F1-1, and F2-1 based on the internal fragment sequences of these housekeeping genes. An unrooted phylogenetic tree showed that strains F1-1 and F2-1 are evolutionarily more closely related to each other and to other strains of B. licheniformis in the NCBI genome database than strain G-1 (Fig. 2). When B. subtilis 168 is introduced in the tree as an outgroup, close clustering of all B. licheniformis strains could be seen. When sequences of the housekeeping genes of strains G-1, F1-1, and F2-1 were compared with those of the sequence types within the B. licheniformis MLST database, they were found to be the sequence types 15, 25, and 26, respectively (Fig. 3). When this was illustrated by constructing a minimum spanning tree using PHYLOViZ, sequence types 25 and 26 fell in one cluster, and sequence type 15 fell in another cluster, which is in agreement with our previous study (5).
FIG 2.

Evolutionary relationship between B. licheniformis strains using the MLST database. An unrooted phylogenetic tree of B. licheniformis strains was based on concatenated sequences of six housekeeping genes in the absence (top) and presence (bottom) of B. subtilis 168 using the neighbor-joining method. It can be inferred that the lineage of strain G-1 separated from the lineage of other strains earlier, but, as expected, it is still very close to other B. licheniformis strains when a B. subtilis 168 outgroup is included.
FIG 3.
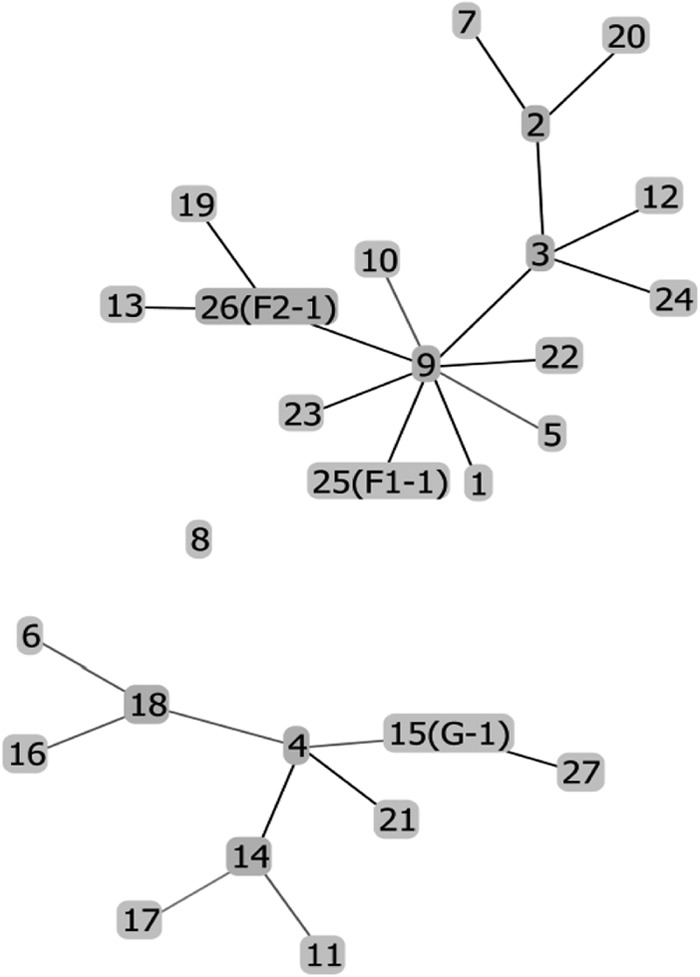
Minimum spanning tree showing the position of the nodes representing the 27 sequence types of B. licheniformis and location of strains G-1, F1-1, and F2-1. This tree was constructed using PHYLOViZ with the sequences of six housekeeping genes (adk, ccpA, recF, rpoB, spo0A, and sucC) from all of the 27 sequence types of B. licheniformis present in the MLST database (http://www.pubmlst.org) and those from strains G-1, F1-1, and F2-1. The tree shows that strains G-1, F1-1, and F2-1 are different sequence types individually and that groups F and G fall in different clusters.
Homology search results.
In the sections below, the presence or absence of genes in different functional categories of relevance to growth, competition, and survival in the environment or in dairy processing plants are investigated, and several related phenotypic characteristics are tested.
Mobility and chemotaxis.
A total of 55 mobility and chemotaxis genes in B. subtilis 168 have been listed previously (17). Sequences of these genes and the proteins encoded by them can be derived from the Web server SubtiList by searching for the functional category “mobility and chemotaxis” (24). The same process was used to search for genes in other functional categories in this study. All of these genes or the proteins encoded by them have regions of significant homology in the contigs of strains G-1, F1-1, and F2-1 (see Tables S2A and S2B in the supplemental material).
Protein secretion.
Twenty-six protein secretion genes are present in B. subtilis 168 according to the SubtiList server (24). The search for homology for these genes in the contigs of our three B. licheniformis strains showed homologs for all of them, with some exceptions (see Tables S3A and S3B in the supplemental material). The homolog of the lytA gene, whose function in B. subtilis is to assist in the secretion of autolysin LytC (24), was not found in strain G-1. This is in contrast to findings in strains F1-1 and F2-1. The tatAC gene involved in the twin-arginine export pathway was absent in B. licheniformis ATCC 14580 and therefore could not be used to find homologs in our strains.
Metabolism of phosphate and sulfur.
Nine genes were shown to be involved in the metabolism of phosphate in B. subtilis 168 previously (17), and one extra gene involved in this process has been mentioned in SubtiList (24). Homologs for all of these genes are present in strains G-1, F1-1, and F2-1 (see Table S4 in the supplemental material). Eight genes have been identified as being responsible for sulfur metabolism in B. subtilis 168 (17) and in the SubtiList server (24). Homologs for all of these genes are present in strains G-1, F1-1, and F2-1 (see Table S5).
Transformation and competence.
In B. subtilis 168, 25 genes have been listed under the functional category of transformation and competence (17, 24). All of these genes were detected in our three strains (see Tables S6A and S6B in the supplemental material). In B. licheniformis ATCC 14580, comP is interrupted by the insertion sequence element IS3Bli1 (10). Interestingly, comP in strains G-1, F1-1, and F2-1 was not interrupted by IS3Bli1. However, IS3Bli1 was present in the genomes of strains F1-1 and F2-1 although it was absent from the genome of strain G-1. ComP has been shown to be essential for competence (30); however, in B. licheniformis ATCC 14580 it was shown that the introduction of an intact comP gene did not improve transformation efficiency (31). The presence of comS in B. licheniformis strains including the noncompetent strain ATCC 14580 and the naturally competent strain 9945A has been shown (31). The sequence of ComS differs between these strains, and this has been assumed to be the important factor for different transformation efficiencies in these strains. The C-terminal extension sequence is present in the ComS of strain ATCC 14580 but not in that of strain 9945A. Additionally, the potential core sequence in ComS for MecA binding differs in these strains. Among our strains, strains F1-1 and F2-1 have ComS sequences more similar to the sequence of strain ATCC 14580, and strain G-1 has an intermediate ComS sequence in which the MecA binding sequence is similar to that of strain 9945A and the C-terminal extension is present. Study of the competency of strain G-1 in the future can add more clarity to the difference in transformation efficiencies among B. licheniformis strains.
Sporulation and germination.
Out of the 164 sporulation genes in SubtiList (24), two genes were found in strains F1-1 and F2-1 but not in strain G-1 (see Tables S7A and S7B in the supplemental material). These genes were phrG, which is a regulator of the phosphatase rapG, and cotW, the product of which is an insoluble spore coat protein (24). Most genes of phr and sps operons were not found in any of the three strains. Among other genes, 31 genes could be compared only with B. subtilis 168 genes, in which case no homology was seen, but they could not be obtained from B. licheniformis ATCC 14580 for comparison. Previous work has shown that many genes of the sps operon were absent in strain ATCC 14580 as well (10). The function of the sps operon is to synthesize polysaccharides in the spore coat (24, 32). Strains G-1, F1-1, and F2-1 were found to produce spores at equivalent frequencies when plated onto nutrient agar with 0.2% starch (data not shown).
Twenty-six genes are listed on the SubtiList server (24) as being involved in spore germination of B. subtilis 168 (see Tables S8A and S8B in the supplemental material). Homologs for all 26 genes except yfkT were found in strains G-1, F1-1, and F2-1.
Biofilm-related genes.
Biofilm-related genes in B. subtilis 168 include those in the epsA-O operon responsible for enzymes producing exopolysaccharide constituents of biofilms and the yqxM-sipW-tasA operon responsible for the production of biofilm proteins. Both of these operons are repressed by the product of sinR, which in turn can be derepressed by the product of sinI (33). Orthologs for the epsA-O operon, the yqxM-sipW-tasA operon, and the genes sinR and sinI, queried after extraction from the genome of B. licheniformis ATCC 14580, are present in strains G-1, F1-1, and F2-1 (see Table S9 in the supplemental material).
Adaptation to atypical conditions.
Out of 81 genes listed for adaptation of B. subtilis to atypical conditions in SubtiList (24), 6 genes were absent in strains G-1, F1-1, and F2-1: rsbP, which positively regulates sigma B activity during energy stress (34); yocM, which codes for a low-molecular-weight heat shock protein (24); and ynzF, yetI, yokG, and ykxI (see Tables S10A and S10B). This suggests that stress responses in B. licheniformis will in large part be similar to those of B. subtilis; however, experimental characterization is needed to further examine this area.
Antibiotic/surfactin and siderophore production.
There are 35 genes studied in B. subtilis 168 which are related to antibiotic production (17, 24). When the homologs for these genes were searched in strains G-1, F1-1, and F2-1, only a few genes were found (see Tables S11A and S11B in the supplemental material). Strains F1-1, F2-1, and G-1 all contained pnbA, the pps operon involved in plipastatin synthesis (24) and the sfp and srfA operons involved in surfactin biosynthesis (36). In contrast, it was previously reported that strain ATCC 14580 did not contain the necessary genes for surfactin or plipastatin synthesis (10), suggesting that large genes are readily lost by industrial strains of B. licheniformis. Lichenysin is a lipopeptide surfactant which can be produced by B. licheniformis. The lichenysin biosynthesis operon in B. licheniformis contains the genes lichenysin synthetase A (licA), lichenysin synthetase B (licB), lichenysin synthetase C (licC), and thioesterase (licTE) genes (37), all of which are present in strains G-1, F1-1, and F2-1. B. licheniformis has been shown to produce a lantibiotic called lichenicidin (38). The homologous regions for all genes of the gene cluster responsible for the production of this lantibiotic were seen in strain F1-1 and strain F2-1 but not in strain G-1. Similar to B. licheniformis ATCC 14580, strains G-1, F1-1, and F2-1 contain a siderophore biosynthesis gene cluster, dhbABCEF, suggesting that all three groups have iron-scavenging ability.
B. licheniformis strains have been grouped into two clusters based on the presence or absence of the bacitracin synthetase genes cluster (8). Among our sequenced isolates, this operon is present only in strain G-1 and not in strains F1-1 and F2-1. These genes are also absent from all other reference B. licheniformis strains cited in this study. Amplification of a fragment of the bacB from isolates of B. licheniformis showed that the threshold cycle (CT) values for group G isolates were equal to or less than 21 with a fluorescence threshold setting of 0.05, and there was no amplification of group F1 and F2 isolates, which revealed that all group G isolates (n = 3) contained the gene while group F1 or F2 isolates (n = 20) did not possess the gene. Antibiotic production from strains of the three groups was also tested, and results showed that there was activity against the target M. luteus similar to that described previously (8). Zones of inhibition from the supernatants of all group G isolates but not group F1 and F2 isolates were observed, suggesting that the activity is probably due to bacitracin (Fig. 4). We also tested the cell-free supernatant from active group G isolates against group F1 and F2 isolates and vice versa using a disc diffusion assay; however, no zone of inhibition was seen (data not shown), indicating that the isolates do not produce antimicrobials which are active against different B. licheniformis groups.
FIG 4.

Group G isolates, but not group F1 or F2 isolates, possess antimicrobial activity against M. luteus. Using a disk diffusion assay, the zone of inhibition of M. luteus ATCC 10240 generated from 50 μl of supernatant of B. licheniformis is expressed as the total diameter of the clearing zone minus the diameter of the filter paper disk (8 mm).
A previous study had shown that all B. licheniformis strains that possess the bacitracin synthetase genes were erythromycin resistant while most of the B. licheniformis strains that did not contain the bacitracin synthetase genes were erythromycin sensitive (8). Similarly, in our study, all three isolates belonging to group G which have bacitracin synthetase genes were erythromycin resistant; however, some group F1 and F2 isolates which did not have the bacitracin synthetase genes were also erythromycin resistant (Fig. 5). This suggests that erythromycin resistance is independent of bacitracin production. Bacitracin and lichenicidin production, therefore, may be useful for differentiating group G strains from group F1 and F2 strains.
FIG 5.

Variable resistance to erythromycin of different groups of B. licheniformis isolates. Using a disk diffusion assay, the erythromycin sensitivity of different B. licheniformis isolates is expressed as the total diameter of the clearing zone minus the diameter of the filter paper disk (8 mm).
Detoxification.
B. subtilis 168 has 89 reported genes for detoxification (17, 24). Most of these genes had regions of homology in strains G-1, F1-1, and F2-1 (see Tables S12A and S12B in the supplemental material). However, among the detoxification genes, tetL, tmrB, ycbR, yetM, yfnC, yokD, ytnJ, yvdP, yxeK, and yyaR did not show any significant homology with any region in strains G-1, F1-1, and F2-1. These genes are responsible for the proteins related to the resistance of tetracycline, tunicamycin, fosmidomycin, fosfomycin, and others (24, 39, 40). Other genes, namely, nap, ybfK, ybfO, and ydhU, had regions of homology only in contigs from strain G-1. The gene ybfO is similar to that for erythromycin esterase, which is responsible for erythromycin resistance in bacteria.
Selected carbohydrate degradation and metabolism genes.
Several carbohydrate degradation and metabolism genes described previously (17) were searched for in strains G-1, F1-1, and F2-1. Genes for α-amylase (amyE) and chitosanase (csn) were not found in strains G-1, F1-1, and F2-1, while those for lichenan degradation (bglS) and fructose 1,6-bisphosphatase (fbp) were found only in strain G-1 (see Table S13 in the supplemental material).
Prophages and plasmids.
Results obtained using PHAST (25) showed that there are phages or phage-related regions in all three strains. These are shown in Table S14 in the supplemental material, which reveals coordinates within contigs where they are present. The completeness of prophages is based upon the score during alignment with the sequences in the database used by PHAST. Out of the three categories, intact prophage is the most complete, and incomplete phage is the least complete. The most closely related phages are also shown, and these have the highest number of similar proteins to those in the B. licheniformis genomes (25). Overall, an intact phage was found in strains F1-1 and F2-1 but not in strain G-1.
Some contigs of strain G-1 either completely or partially showed homology or similarity with plasmids from other strains or species revealed by BLASTX or discontiguous MegaBLAST, respectively. The complete sequence of contig 7 of strain G-1 showed significant homology with a 7,848-bp pFL7 cryptic plasmid (NCBI reference sequence NC_005308.1) of B. licheniformis strain FL7 isolated from pasture land soil (41) and includes the Rep protein required for initiating plasmid replication. There is also a region coding the Rap protein, which has the putative function of delaying sporulation, and traR, required for horizontal transfer during bacterial conjugation. This contig has a G+C content of 43.4%, which is lower than that of the genome of G-1 (45.7%). On the other hand, a large middle part with coordinates of 5041 to 36233 of contig 3 contains various stretches of DNA which show significant similarity but intermittent gaps with different regions of the p19 plasmid of B. subtilis strain 19 (GenBank accession number FJ434456.1). This contig has a much lower G+C content of 35.62%. Contigs of strains F1-1 and F2-1 show no plasmid-related sequences.
Repeat regions.
The CRISPR (clustered regularly interspaced short palindromic repeats) finder program (42) was used to screen probable CRISPR regions of these strains. This program indicates two CRISPR-like regions in strain G-1 in contig 30, one ranging from coordinate 373966 to 374054 and another ranging from 1068213 to 1068311 and each having a repeat of length 24 bp, and one CRISPR region in strain F2-1, ranging from coordinate 603093 to 603199 in contig 35 and having a repeat length of 26 bp. No CRISPR region was detected in strain F1-1.
Analysis of DNA sequences of fragments in RAPD bands.
Genotyping of dairy spore formers, including B. licheniformis, using RAPD has been carried out by numerous researchers (5, 6, 35); however, the identity of the fragments present in the RAPD profiles that are used for differentiating strains is not known. From the three sequenced strains we attempted first to identify and then to explain why certain fragments are seen in the RAPD profile of one group and not in others. Several RAPD bands generated from strains G-1, F1-1, and F2-1 were excised from an agarose gel, cloned into a plasmid vector, and sequenced. The fragments that could be successfully cloned and sequenced are labeled in Fig. 6.
FIG 6.
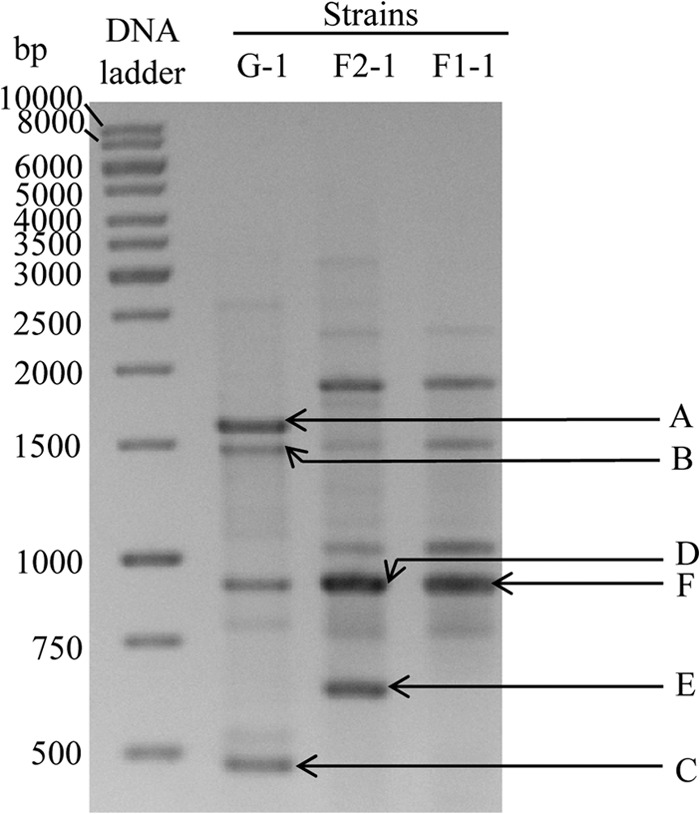
RAPD profiles of B. licheniformis strains G-1, F2-1, and F1-1. The RAPD bands A to F were successfully cloned and sequenced.
Table 2 shows the identities of the fragments present in RAPD bands A to F in relation to the assembled genome of strains G-1, F2-1, and F1-1 using BLAST. The sequences corresponding to RAPD OPR-13 primer (5′-GGACGACAAG-3′) binding sites were obtained from the genome sequences of these three strains and are presented. Differences from the OPR-13 primer sequence are shown in boldface. It was found that most binding sites contained mismatches. All cloned and sequenced fragments from the bands were of expected size, corresponding to the position in the gel. The differences in the RAPD profiles between groups were found to be likely due to different gene lengths (fragment B), differences in the 3′ end of the predicted primer binding sites (fragments A and C), or simply the presence or absence of a region (fragment E). The lengths of the sequences of fragment B from strains F1-1 and F2-1 are slightly greater than the length in strain G-1, as expected; however, they are similar regions (Fig. 6 and Table 2). Sequences of fragments A and C were present in all three strains; however, only group G generated fragments in the RAPD analysis (Table 2 and Fig. 6). Closer inspection revealed mismatches near the 3′ terminal ends of the predicted OPR-13 primer binding site for fragments A and C in strain F1-1 and F2-1 (Table 2), which likely led to inefficient primer annealing and subsequent amplification. The sequence of fragment E, which was present in strain F2-1 only, showed no identity with the genome of strain G-1 or strain F1-1 (Table 2). This fragment shows homology with a gene encoding a hypothetical protein of B. licheniformis WX-02 that has homology to an ABC-type exporter (probably of bacteriocin/lantibiotic) and peptidase domains. The fragments sequenced from bands D and F which appear to be of equal sizes in the gel differ by 9 bp and are actually different DNA fragments. Both of these fragments are present in all three strains, which indicates that they both are involved in the formation of thick bands D and F and likely the similarly sized band of strain G-1 (Fig. 6).
TABLE 2.
Contigs, coordinates, and terminal sequences corresponding to the OPR-13 primer of the RAPD fragments of strains G-1, F1-1, and F2-1
| Fragment and strain | Gene product(s) | Contig no. | Coordinates (nt) | Sequence corresponding to OPR-13a | Length (bp) | Band presenceb |
|---|---|---|---|---|---|---|
| A | Amido-phospho-ribosyl transferase PurF, phospho-ribosyl-formyl glycinamidine cyclo-ligase PurM | |||||
| Strain G-1 | 12 | 46579–48192 | 5′-GCACGACAAG-3′ | 1,614 | Y | |
| 5′-AGACGACAAG-3′ | ||||||
| Strain F1-1 | 30 | 88368–89980 | 5′-GCACGACGAG-3′ | 1,614 | N | |
| 5′-AGACGACGAG-3′ | ||||||
| Strain F2-1 | 29 | 76720–78332 | 5′-GCACGACGAG-3′ | 1,614 | N | |
| 5′-AGACGACGAG-3′ | ||||||
| B | Transmembrane protein YrbG, putative integral inner membrane protein YrzE | |||||
| Strain G-1 | 30 | 353213–354689 | 5′-TGCCGACAAG-3′ | 1,477 | Y | |
| 5′-AAACGACAAG-3′ | ||||||
| Strain F1-1 | 1 | 1223031–1224525 | 5′-TGCCGACAAG-3′ | 1,503 | Y | |
| 5′-AAACGACAAG-3′ | ||||||
| Strain F2-1 | 35 | 105908–107402 | 5′-TGCCGACAAG-3′ | 1,504 | Y | |
| 5′-AAACGACAAG-3′ | ||||||
| C | Arabinose metabolism transcriptional repressor protein | |||||
| Strain G-1 | 4 | 1276–1751 | 5′-CGACGACAAG-3′ | 476 | Y | |
| 5′-GGACGACAAG-3′ | ||||||
| Strain F1-1 | 15 | 1297–1773 | 5′-CGACGACCAA-3′ | 478 | N | |
| 5′-GGACGACAAG-3′ | ||||||
| Strain F2-1 | 13 | 1293–1769 | 5′-CGACGACCAA-3′ | 478 | N | |
| 5′-GGACGACAAG-3′ | ||||||
| D | Rod-shape-determining protein RodA | |||||
| Strain G-1 | 15 | 347230–348138 | 5′-ATACGACAAG-3′ | 910 | Y | |
| 5′-GCACGACAAG-3′ | ||||||
| Strain F1-1 | 5 | 27148–28056 | 5′-ATACGACAAG-3′ | 909 | Y | |
| 5′-GCACGACAAG-3′ | ||||||
| Strain F2-1 | 6 | 62068–62976 | 5′-GCACGACAAG-3′ | 909 | Y | |
| 5′-ATACGACAAG-3′ | ||||||
| E | Homologous, with a region encoding a hypothetical protein of B. licheniformis WX-02 that has homology to an ABC-type exporter (of probably bacteriocin/lantibiotic) and peptidase domains | |||||
| Strain G-1 | N | |||||
| Strain F1-1 | N | |||||
| Strain F2-1 | 35 | 237929–238561 | 5′-GGAAGACAAG-3′ | 633 | Y | |
| 5′-AGACGACAAG-3′ | ||||||
| F | Phospholipid biosynthesis protein PlsX, malonyl coenzyme A-acyl carrier protein transacylase | |||||
| Strain G-1 | 31 | 223684–224583 | 5′-GGACGCCAAG-3′ | 900 | Y | |
| 5′-AAACGACAAG-3′ | ||||||
| Strain F1-1 | 1 | 129878–130777 | 5′-GGACGCCAAG-3′ | 900 | Y | |
| 5′-AAACGACAAG-3′ | ||||||
| Strain F2-1 | 27 | 129870–130769 | 5′-GGACGCCAAG-3′ | 900 | Y | |
| 5′-AAACGACAAG-3′ |
Bases in boldface are the differences in the regions corresponding to the OPR-13 primer (5′-GGACGACAAG-3′) compared to the genome sequence. Note that although there are mismatches in the regions corresponding to the OPR-13 primer in most of the terminal ends of these sequences, there are additional mismatches in the fragments A and C at the 3′ end of one or both these regions in strains F1-1 and F2-1 compared to the sequence in strain G-1. This is probably the reason that these bands were seen in the RAPD gel only in the case of strain G-1.
Presence (Y) or absence (N) of the bands in the RAPD gel.
Conclusions.
This study has used a comparative genomics approach to further understand the genetic heterogeneity of representatives of the most common B. licheniformis genotypes isolated from dairy foods. Group G B. licheniformis is clearly distinct from group F1 and group F2 B. licheniformis. Guided by genome variation, several phenotypic tests were carried out (e.g., urease production and bacitracin production) which yielded concordant results with strains of the same group. Erythromycin resistance, however, was found to be variable within strains of the same genotype group (i.e., groups F1 and F2). Therefore, from the data presented here, new phenotypic tests may be developed to differentiate isolates of B. licheniformis. Also, more simplified genotyping tests may be developed as the basis of the RAPD banding profiles has been identified. Through a better understanding of the genetic potential of B. licheniformis and application of genotyping and phenotyping methods, the identification of contamination sources and persistent strains can be achieved, in particular for raw milk and milk powder foods where B. licheniformis is common. This will assist with the ultimate aim of improving food quality and safety.
Supplementary Material
ACKNOWLEDGMENTS
This study was funded by The Geoffrey Gardiner Dairy Foundation and Dairy Innovation Australia Limited. Rajat Dhakal is an Australian Government Endeavor postgraduate award holder.
We thank the dairy processing plants in Victoria that supplied samples for this study and Ian B. Powell and Christopher J. Pillidge for arranging sample transport from Victoria to our laboratory.
Footnotes
Published ahead of print 21 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00065-14.
REFERENCES
- 1.Ruckert A, Ronimus RS, Morgan HW. 2004. A RAPD-based survey of thermophilic bacilli in milk powders from different countries. Int. J. Food Microbiol. 96:263–272. 10.1016/j.ijfoodmicro.2004.03.020 [DOI] [PubMed] [Google Scholar]
- 2.Williams DJ. 1958. Bacillus spores in raw milk in Victoria and New South Wales. Aust. J. Dairy Technol. 13:3–10 [Google Scholar]
- 3.Cook GM, Sandeman RM. 2000. Sources and characterisation of spore-forming bacteria in raw milk. Aust. J. Dairy Technol 55:119–126 [Google Scholar]
- 4.Chauhan K, Dhakal R, Seale RB, Deeth HC, Pillidge CJ, Powell IB, Craven H, Turner MS. 2013. Rapid identification of dairy mesophilic and thermophilic sporeforming bacteria using DNA high resolution melt analysis of variable 16S rDNA regions. Int. J. Food Microbiol. 165:175–183. 10.1016/j.ijfoodmicro.2013.05.007 [DOI] [PubMed] [Google Scholar]
- 5.Dhakal R, Chauhan K, Seale RB, Deeth HC, Pillidge CJ, Powell IB, Craven H, Turner MS. 2013. Genotyping of dairy Bacillus licheniformis isolates by high resolution melt analysis of multiple variable number tandem repeat loci. Food Microbiol. 34:344–351. 10.1016/j.fm.2013.01.006 [DOI] [PubMed] [Google Scholar]
- 6.Ronimus RS, Parker LE, Turner N, Poudel S, Ruckert A, Morgan HW. 2003. A RAPD-based comparison of thermophilic bacilli from milk powders. Int. J. Food Microbiol. 85:45–61. 10.1016/S0168-1605(02)00480-4 [DOI] [PubMed] [Google Scholar]
- 7.De Clerck E, Vos P. 2004. Genotypic diversity among Bacillus licheniformis strains from various sources. FEMS Microbiol. Lett. 231:91–98. 10.1016/S0378-1097(03)00935-2 [DOI] [PubMed] [Google Scholar]
- 8.Ishihara H, Takoh M, Nishibayashi R, Sato A. 2002. Distribution and variation of bacitracin synthetase gene sequences in laboratory stock strains of Bacillus licheniformis. Curr. Microbiol. 45:18–23. 10.1007/s00284-001-0041-5 [DOI] [PubMed] [Google Scholar]
- 9.Madslien E, Olsen J, Granum P, Blatny J. 2012. Genotyping of B. licheniformis based on a novel multi-locus sequence typing (MLST) scheme. BMC Microbiol. 12:230. 10.1186/1471-2180-12-230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rey M, Ramaiya P, Nelson B, Brody-Karpin S, Zaretsky E, Tang M, de Leon A, Xiang H, Gusti V, Clausen I. 2004. Complete genome sequence of the industrial bacterium Bacillus licheniformis and comparisons with closely related Bacillus species. Genome Biol. 5:R77. 10.1186/gb-2004-5-10-r77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Veith B, Herzberg C, Steckel S, Feesche J, Maurer K, Ehrenreich P, Baumer S, Henne A, Liesegang H, Merkl R, Ehrenreich A, Gottschalk G. 2004. The complete genome sequence of Bacillus licheniformis DSM13, an organism with great industrial potential. J. Mol. Microbiol. Biotechnol. 7:204–211. 10.1159/000079829 [DOI] [PubMed] [Google Scholar]
- 12.Li L, Su F, Wang Y, Zhang L, Liu C, Li J, Ma C, Xu P. 2012. Genome sequences of two thermophilic Bacillus licheniformis strains, efficient producers of platform chemical 2, 3-butanediol. J. Bacteriol. 194:4133–4134. 10.1128/JB.00768-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yangtse W, Zhou Y, Lei Y, Qiu Y, Wei X, Ji Z, Qi G, Yong Y, Chen L, Chen S. 2012. Genome sequence of Bacillus licheniformis WX-02. J. Bacteriol. 194:3561–3562. 10.1128/JB.00572-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu Q, Peng S, Yu Y, Li Y, Xu Y. 2013. Genome sequence of Bacillus licheniformis CGMCC3963, a stress-resistant strain isolated in a Chinese traditional solid-state liquor-making process. Genome Announc. 1:e00060-12. 10.1128/genomeA.00060-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rachinger M, Volland S, Meinhardt F, Daniel R, Liesegang H. 2013. First insights into the completely annotated genome sequence of Bacillus licheniformis strain 9945A. Genome Announc. 1:e00525-13. 10.1128/genomeA.00525-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prasad P, Turner MS. 2011. What bacteria are living in my food? An open-ended practical series involving identification of unknown foodborne bacteria using molecular techniques. Biochem. Mol. Biol. Educ. 39:384–390. 10.1002/bmb.20532 [DOI] [PubMed] [Google Scholar]
- 17.Kunst F, Ogasawara N, Moszer I, Albertini A, Alloni G, Azevedo V, Bertero M, Bessieres P, Bolotin A, Borchert S. 1997. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 390:249–256. 10.1038/36786 [DOI] [PubMed] [Google Scholar]
- 18.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. 10.1093/bioinformatics/bts199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829. 10.1101/gr.074492.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M. 2008. The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. 10.1186/1471-2164-9-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. 10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Francisco AP, Vaz C, Monteiro PT, Melo-Cristino J, Ramirez M, Carriço JA. 2012. PHYLOViZ: phylogenetic inference and data visualization for sequence based typing methods. BMC Bioinformatics 13:87. 10.1186/1471-2105-13-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moszer I, Jones LM, Moreira S, Fabry C, Danchin A. 2002. SubtiList: the reference database for the Bacillus subtilis genome. Nucleic Acids Res. 30:62–65. 10.1093/nar/30.1.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. 2011. PHAST: a fast phage search tool. Nucleic Acids Res. 39:W347–W352. 10.1093/nar/gkr485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo Y, Helmann JD. 2012. Analysis of the role of Bacillus subtilis σM in β-lactam resistance reveals an essential role for c-di-AMP in peptidoglycan homeostasis. Mol. Microbiol. 83:623–639. 10.1111/j.1365-2958.2011.07953.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ewing B, Green P. 1998. Base-calling of automated sequencer traces using Phred. II. Error probabilities. Genome Res. 8:186–194 [PubMed] [Google Scholar]
- 28.Ewing B, Hillier L, Wendl MC, Green P. 1998. Base-calling of automated sequencer traces using Phred. I. Accuracy assessment. Genome Res. 8:175–185 [DOI] [PubMed] [Google Scholar]
- 29.Marcelletti S, Ferrante P, Petriccione M, Firrao G, Scortichini M. 2011. Pseudomonas syringae pv. actinidiae draft genomes comparison reveal strain-specific features involved in adaptation and virulence to Actinidia species. PLoS One 6:e27297. 10.1371/journal.pone.0027297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Piazza F, Tortosa P, Dubnau D. 1999. Mutational analysis and membrane topology of ComP, a quorum-sensing histidine kinase of Bacillus subtilis controlling competence development. J. Bacteriol. 181:4540–4548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoffmann K, Wollherr A, Larsen M, Rachinger M, Liesegang H, Ehrenreich A, Meinhardt F. 2010. Facilitation of direct conditional knockout of essential genes in Bacillus licheniformis DSM13 by comparative genetic analysis and manipulation of genetic competence. Appl. Environ. Microbiol. 76:5046–5057. 10.1128/AEM.00660-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Charnock SJ, Davies GJ. 1999. Structure of the nucleotide-diphospho-sugar transferase, SpsA from Bacillus subtilis, in native and nucleotide-complexed forms. Biochemistry 38:6380–6385. 10.1021/bi990270y [DOI] [PubMed] [Google Scholar]
- 33.Chai Y, Chu F, Kolter R, Losick R. 2008. Bistability and biofilm formation in Bacillus subtilis. Mol. Microbiol. 67:254–263. 10.1111/j.1365-2958.2007.06040.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vijay K, Brody MS, Fredlund E, Price CW. 2000. A PP2C phosphatase containing a PAS domain is required to convey signals of energy stress to the σB transcription factor of Bacillus subtilis. Mol. Microbiol. 35:180–188. 10.1046/j.1365-2958.2000.01697.x [DOI] [PubMed] [Google Scholar]
- 35.Reginensi SM, González MJ, Olivera JA, Sosa M, Juliano P, Bermúdez J. 2011. RAPD-based screening for spore-forming bacterial populations in Uruguayan commercial powdered milk. Int. J. Food Microbiol. 148:36–41. 10.1016/j.ijfoodmicro.2011.04.020 [DOI] [PubMed] [Google Scholar]
- 36.Peypoux F, Bonmatin J, Wallach J. 1999. Recent trends in the biochemistry of surfactin. Appl. Microbiol. Biotechnol. 51:553–563. 10.1007/s002530051432 [DOI] [PubMed] [Google Scholar]
- 37.Konz D, Doekel S, Marahiel MA. 1999. Molecular and biochemical characterization of the protein template controlling biosynthesis of the lipopeptide lichenysin. J. Bacteriol. 181:133–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shenkarev ZO, Finkina EI, Nurmukhamedova EK, Balandin SV, Mineev KS, Nadezhdin KD, Yakimenko ZA, Tagaev AA, Temirov YV, Arseniev AS. 2010. Isolation, structure elucidation, and synergistic antibacterial activity of a novel two-component lantibiotic lichenicidin from Bacillus licheniformis VK21. Biochemistry 49:6462–6472. 10.1021/bi100871b [DOI] [PubMed] [Google Scholar]
- 39.Bechhofer DH, Stasinopoulos SJ. 1998. tetA(L) mutants of a tetracycline-sensitive strain of Bacillus subtilis with the polynucleotide phosphorylase gene deleted. J. Bacteriol. 180:3470–3473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stasinopoulos SJ, Farr GA, Bechhofer DH. 1998. Bacillus subtilis tetA(L) gene expression: evidence for regulation by translational reinitiation. Mol. Microbiol. 30:923–932. 10.1046/j.1365-2958.1998.01119.x [DOI] [PubMed] [Google Scholar]
- 41.Parini C, Guglielmetti S, Mora D, Ricci G. 2004. Complete sequence and structural organization of pFL5 and pFL7, two cryptic plasmids from Bacillus licheniformis. Plasmid 51:192–202. 10.1016/j.plasmid.2004.02.001 [DOI] [PubMed] [Google Scholar]
- 42.Grissa I, Vergnaud G, Pourcel C. 2007. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35:W52–W57. 10.1093/nar/gkm360 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.