

Abstract
F-specific (F+) RNA phages are recommended as indicators of fecal contamination and the presence of enteric viruses and as viral surrogates to elucidate the resistance of viruses to adverse conditions or to assess the effectiveness of inactivating processes. Reverse transcription (RT)-PCR methods have been used to detect, quantify, or identify subgroups of F+ RNA phages. However, these methods may overestimate the infectivity of F+ RNA phages in test samples, since the presence of both infectious and inactivated phages (or naked RNA) can lead to positive RT-PCR signals. In this study, we evaluated the ability of an enzyme treatment (ET) with proteinase K and RNase A prior to RNA extraction, followed by RT-PCR, to differentiate infectious and inactivated F+ RNA phages. The results indicated that ET RT-PCR reduced, but did not completely eliminate, false-positive signals encountered with RT-PCR alone. The two-step ET RT-PCR, in which the enzymes were added sequentially, was more effective at reducing false-positive signals than the one-step ET RT-PCR, which involved addition of both enzymes together. Despite its inability to completely eliminate false-positive signals, ET RT-PCR gave more reliable information on the infectivity of F+ RNA phages. Thus, the method is better than RT-PCR alone for detecting F+ RNA phages as indicators to assess the risk of fecal contamination by enteric pathogens or to evaluate the effectiveness of virus-inactivating processes.
INTRODUCTION
F-specific (F+) RNA phages are positive-sense, single-stranded RNA phages belonging to the family Leviviridae (1). These phages have been widely used as indicators to predict or track the source of fecal contamination, since they are a constituent of human and/or animal feces. F+ RNA phages are also used as viral surrogates to elucidate the resistance of viruses to adverse conditions and to assess the effectiveness of virus-inactivating processes because of their similarity in structure, size, and biochemical properties to enteric viruses (2–5).
Reverse transcription (RT)-PCR has been widely used for the detection of F+ RNA phages from various environmental samples (6–8). Based on the differences in their genome sequences, RT-PCR has been successfully utilized for the identification of subgroups of F+ RNA phages. RT-PCR methods have also been successfully used to quantify F+ RNA phages by the use of SYBR green dye or specific TaqMan probes (6, 9). However, since loss of infectivity may not result in the loss of the target sequence of the genome, RT-PCR methods are inadequate to quantify infectious F+ RNA phages because of the production of false-positive signals caused by the presence of inactivated phage particles or even naked RNA (10). Hot et al. (11) detected enterovirus genomes in 60 of 68 tested samples, but infectious enteroviruses were isolated in only two of the same samples. Previous studies have demonstrated that the levels of F+ RNA phages were clearly correlated with the levels of norovirus, and the inactivation pattern of F+ RNA phages is similar to the inactivation of enteric viruses (12, 13). Due to the fact that the risk of fecal contamination to human health is caused by the presence of infectious viral or bacterial pathogens and that neither inactivated pathogens nor their naked genomes are able to cause disease, the presence of inactivated F+ RNA phages is incapable of predicting the infectiousness of pathogens potentially present. Thus, RT-PCR methods that are capable of differentiating infectious and inactivated F+ RNA phages will give more reliable information on the risk posed by fecal contamination to human health.
The limitation of PCR-based methods in detecting infectious viruses has been recognized by the scientific community, and studies have been performed to evaluate the usefulness of PCR-based methods to quantify the infectivity of viruses (10, 14). Fittipaldi et al. (15) reported a real-time PCR method to discriminate the infectious phage T4 by the use of propidium monoazide. Their method was able to differentiate phage particles inactivated by a heat treatment at 110°C, but not at 85°C, from infectious phage. Shieh et al. (16) evaluated an integrated cell culture RT-PCR to detect naturally occurring enteroviruses in water and demonstrated that direct RT-PCR could lead to more than 90-fold overestimation of naturally occurring infectious viruses in water.
The genome of F+ RNA phages is enclosed in a capsid mainly composed of 180 copies of coat protein, which has a protective effect on the integrity of the viral genome. Inactivated and infectious F+ RNA phages may differ in the resistance of their protein capsids to proteolysis (17, 18). Denaturation of coat protein may always occur when F+ RNA phages are inactivated, since the capsid acts as the outer shell of the viral particle and protects the genome, even though theoretically any damage to the genome or receptor sites on the capsid of F+ RNA phages may lead to the loss of infectivity. Denaturation of protein exposes more available sites to proteases and thus renders it more susceptible to proteolysis. Following degradation of the coat protein, the RNA genome should be more susceptible to RNase degradation than RNA of infectious phages, in which the RNA genome is surrounded by the intact protein capsid (17). Thus, treatment of F+ RNA phages with protease and RNase before RNA extraction, followed by quantitative RT-PCR, should detect mainly infectious phages, since naked RNA or RNA of inactivated phage particles is more readily degraded.
MATERIALS AND METHODS
F+ RNA phages and the host bacterium.
Phages MS2, GA, Qβ, and SP, belonging to subgroups I, II, III, and IV, respectively, were used as references for F+ RNA phages, with Escherichia coli Famp (ATCC 700891) used as the host bacterium. Both the phages and the host bacterium were kindly provided by Lawrence Goodridge, McGill University. E. coli Famp was grown in tryptic soy broth (TSB) (BD, Mississauga, Canada) containing 15 μg ml−1 ampicillin/streptomycin at 37°C, and bacterial cells in the exponential growth phase were used for phage propagation and enumeration. Phage propagation and enumeration were performed by the double-agar-layer (DAL) method using tryptic soy agar (TSA) (BD, Mississauga, Canada) plates containing 15 μg ml−1 ampicillin/streptomycin (19), and dilution was performed with phosphate-buffered saline (PBS) when necessary to ensure that plaques on the plates were enumerable. Phage titers were expressed as PFU ml−1, and only plates with 30 to 300 PFU were used for enumeration.
Susceptibility of infectious F+ RNA phages to RNase.
The susceptibility of infectious F+ RNA phages to RNase was tested with phage MS2. E. coli Famp (200 μl; ∼8 log10 CFU ml−1), soft TSA (TSB with 0.5% agar; 15 ml, prewarmed at 55°C), and RNase A (Thermo Scientific, Logan, UT) were mixed and dispersed into a 15-cm-diameter petri dish (Fisher Scientific, Toronto, Canada) to prepare host cell-seeded plates containing RNase A (3 or 15 μg ml−1). Aliquots (10 μl) of phage suspensions of various titers were dispersed onto the RNase A-supplemented host cell-seeded plates, which were then dried at room temperature for 20 min and incubated at 37°C overnight. A negative-control experiment was also performed by dispersing phage suspensions onto a host cell-seeded soft-TSA plate in the absence of RNase A.
Alternatively, phage MS2 (200 μl; ∼7 log10 PFU ml−1) was treated with 2 μl of RNase A (10 μg μl−1) at 37°C for 0, 10, 20, or 30 min, followed by the addition of 2 μl of RNaseOut Recombinant RNase Inhibitor (40 U μl−1; Life Technologies Inc., Burlington, Canada) to inactivate RNase A. The treated phage suspension was 10-fold serially diluted with PBS, and 10 μl of each of the dilutions was spotted onto a host cell-seeded soft-TSA plate and incubated at 37°C overnight. Untreated phage suspension (at the same concentration) was used as a negative control and considered the 0-min treatment. This was done to determine the susceptibility of free infectious F+ RNA phages (phages in suspension, not attached to host cells) to RNase.
The susceptibility of infectious F+ RNA phages to the treatment with proteinase K and RNase A was also tested in a similar way. Phage MS2 was treated with Engyodontium album proteinase K (333 U ml−1; Life Technologies, Burlington, Canada) and 2 μl of RNase A at 37°C for 10, 20, or 30 min. After that, 2 μl of RNase inhibitor was added to inactivate the RNase A. The infectivity of the treated phage suspension was tested by plaque assay as described above. A phage suspension that was not treated with proteinase K or RNase A was included as a negative control and considered the 0-min treatment. Proteinase K was dissolved in PBS and freshly prepared for each experiment.
Primers used to detect F+ RNA phages.
The primers used for the detection of F+ RNA phages were selected from previous studies (9, 20) or designed based on the alignment of complete or partial sequences of F+ RNA phages (MS2, fr, M12, R17, GA, BZ13, KU1, Qβ, M11, MX1, TW18SP, and NL95) and synthesized by the Laboratory Service Division, University of Guelph, Guelph, Canada. The sequences of the primers used for F+ RNA phage detection are shown in Table 1. Subgroup-specific primers were used in order to detect all F+ RNA phages potentially present in environmental samples. Each of the chosen primer sets was designed to be specific for the corresponding subgroup of F+ RNA phages with the capacity to detect as many strains within the subgroup as possible. The specificity of the primers was tested by aligning the primers with sequences of F+ RNA phages available in GenBank, National Center for Biology Information (NCBI), and also tested by RT-PCR using the Qiagen one-step RT-PCR kit (Qiagen, Toronto, Canada) according to the manufacturer's instructions in a Mastercycler (Eppendorf, Hamburg, Germany), with phages MS2, GA, Qβ, and SP as reference phages for subgroups I, II, III, and IV, respectively. The RT-PCR products were sized electrophoretically on 2% agarose gels stained with ethidium bromide (2 μg liter−1) and visualized under UV light with a Gel Doc system (Bio-Rad, Mississauga, Canada).
TABLE 1.
Primers used for the detection of F+ RNA phages
| Primer | Sequence (5′–3′) | Tmb | Product length (bp) |
|---|---|---|---|
| FRNA-I-Ra | CTTGTTCAGCGAACTTCTTRTA | 53 | 142 |
| FRAN-I-Fa | CAAACCAGCATCCGTAGCC | 57 | |
| FRNA-II-R | GAAAACAAACCGTTGCCG | 53 | 183 |
| FRAN-II-F | GGTTCAAGTTGCGGGATG | 55 | |
| FRNA-III-Ra | TTACGATTGCGAGAAGGCTG | 57 | 115 |
| FRAN-III-Fa | CCGCGTGGGGTAAATCC | 57 | |
| FRNA-IV-R | TTCCAGCCRGGCTCGAT | 57 | 183 |
| FRAN-IV-F | CGTGGAAGCATGCCTGT | 57 |
Partial inactivation of F+ RNA phages.
Suspensions of F+ RNA phages at initial concentrations of approximately 6 to 7 log10 PFU ml−1 were used for partial inactivation. The choice of working phage titers was intended to avoid complete inactivation of the phages in these suspensions. The methods used for inactivation were heat treatment, UV irradiation, and innate inactivation at room temperature. Partially inactivated phage suspensions were divided into four subsamples for phage enumeration by the plaque method, quantitative RT-PCR, and enzyme treatment (ET) RT-PCR (both one step and two step, as described below), all of which were performed immediately after the inactivation treatment.
Heat treatment was performed at 55°C to avoid complete destruction of the phage coat protein. For all four phages, a working phage suspension was 10-fold diluted in PBS, which was prewarmed at 55°C in a water bath, and incubated at the same temperature for 40 min. Preliminary studies indicated that a loss of approximately 1 to 2 log10 PFU ml−1 was achieved by this treatment. The phage suspension was immediately cooled on ice after heat treatment. The phage titer of the working suspension was determined by plaque assay prior to heat treatment.
UV irradiation was performed by the use of an FB-TIV-88A Transilluminator (Fisher Scientific, Toronto, Canada) with a wavelength of 312 nm. The phage suspension (7 ml) was dispersed in a 10-cm-diameter petri dish (Fisher Scientific, Toronto, Canada) and exposed to UV irradiation for 20 min. The UV intensity was 0.19 mW cm−2, as measured with a PMA2100 Radiometer (Solar Light, Glenside, PA, USA). Preliminary studies indicated that a loss of approximately 1.5 to 3.0 log10 PFU ml−1 was achieved by this treatment. Aliquots of the phage suspensions were assayed by the plaque method prior to UV irradiation to determine the initial concentrations of infectious phages.
Innate inactivation of F+ RNA phages following incubation at room temperature was determined using phage suspensions in PBS and stored at 25°C ± 1°C for 14 days. Preliminary studies indicated that a loss of approximately 0.5 to 1.5 log10 PFU ml−1 was achieved during this period. The initial phage titers of the working suspensions were determined by plaque assay.
Enzyme treatment.
Enzyme treatment with proteinase K and RNase A was performed in order to selectively degrade the RNA of inactivated F+ RNA phages. Both one-step and two-step enzyme treatments were tested.
For the one-step treatment, the partially inactivated phage suspension (200 μl) was mixed with proteinase K (final concentration, 333 U ml−1) and RNase A (10 μg μl−1; 2 μl) and incubated at 37°C in a water bath for 30 min, followed by the addition of 2 μl of RNase inhibitor (40 U μl−1) to inactivate the RNase A.
For the two-step ET, partially inactivated phage suspensions were first treated with proteinase K at 37°C for 30 min, followed by the addition of phenylmethylsulfonyl fluoride (PMSF) (Sigma, Oakville, Canada) at a final concentration of 5 mmol liter−1 to inactivate the proteinase K. RNase A was added, and the suspension was incubated at 37°C for a further 30 min, followed by the addition of 2 μl of RNase inhibitor to inactivate the RNase A. The PMSF solution (1 mol liter−1) was prepared in ethanol (Sigma, Oakville, Canada) because of its limited water solubility.
RNA extraction.
Extraction of RNA from phage suspensions was performed using the Total RNA Purification Kit (Norgen, Thorold, Canada), according to the manufacturer's instructions. Briefly, the process included lysing the bacteriophage chemically in a microtube with the lysis solution provided to release RNA from the phage particles, and the contents of the tube was loaded onto a small chromatographic column. After washing three times with the washing solution provided, the RNA was eluted with elution buffer. The extracted phage RNA was stored at −20°C and assayed by real-time RT-PCR within 2 weeks.
Standard curves.
Freshly prepared phage suspensions were used to construct standard curves in order to quantify F+ RNA phages by real-time RT-PCR as described below. The phage suspensions were 10-fold serially diluted with PBS, and appropriate dilutions were assayed by plaque assay to determine phage titers (PFU ml−1). At the same time, RNA was extracted from aliquots (200 μl) of these dilutions. The resulting RNA was assayed by a SYBR green real-time RT-PCR assay, and the CT values (cycle threshold; the number of cycles in a real-time PCR process needed for the fluorescent signal to exceed threshold) were recorded. Experiments were performed in triplicate. The CT values were plotted against the corresponding starting phage titers (logarithmic value) to obtain a standard curve. Standard curves for each of the F+ RNA phages were established individually.
Quantitative RT-PCR.
One-step real-time RT-PCR, with or without ET, was performed using the QuantiFast SYBR green RT-PCR Kit (Qiagen, Toronto, Canada) to quantify F+ RNA phages. The reaction mixture (25 μl) contained 12.5 μl of master mix, 0.25 μl of RT mix, 1 μl each of reverse and forward primers (10 pmol μl−1), 2 μl of RNA template, and 8.25 μl of RNase-free water. The master mix contained HotStarTaq Plus DNA polymerase, QuantiFast SYBR green RT-PCR buffer, deoxynucleoside triphosphate (dNTP) mixture, and fluorescent dyes. The reaction mixtures were prepared in MicroAmp Fast 8-tube strips (Invitrogen, Burlington, Canada), and all the reagents were kept on ice to avoid temperature abuse. The real-time RT-PCR was conducted in a ViiA 7 real-time PCR system (Invitrogen, Burlington, Canada), and the thermal-cycling conditions were as follows: reverse transcription at 50°C for 10 min, activation of HotStarTap Plus DNA polymerase at 95°C for 5 min, followed by 40 cycles of denaturation at 95°C for 10 s and combined annealing/extension at 56°C for 30 s. Finally, a melting curve analysis was performed, from 56 to 95°C, to confirm the specificity of the amplicon.
Detection of naturally occurring F+ RNA phages.
The abilities of the enzyme treatments in combination with RT-PCR to quantify infectious F+ RNA phages were tested with raw sewage and primary treated wastewater, which were collected from the wastewater treatment plant of Guelph, Canada. Raw sewage and primary treated wastewater were filtered through a 0.45-μm membrane filter (Fisher Scientific, Toronto, Canada), and phage titers in the filtrate were tested by plaque assay and RT-PCR with or without a two-step ET prior to RNA extraction. The results obtained by these methods were compared to verify the suitability of ET RT-PCR to quantify infectious F+ RNA phages naturally occurring in environmental samples.
Statistical analysis.
Statistical analysis was performed using SPASS 16.0 (IBM, New York, USA). A significant difference was recorded when the P value was <0.05.
RESULTS
Susceptibility of F+ RNA phages to RNase.
The titer of the phage MS2 working stock was ∼7 log10 PFU ml−1, and this was 10-fold serially diluted with PBS to produce phage suspensions of various concentrations. With RNase A supplemented in host cell-seeded plates, clear zones were not observed on the bacterial lawn where phage suspensions were dispensed, indicating that F+ RNA phages were unable to infect host cells in the presence of RNase A, in contrast to medium not containing RNase A. The presence of 3 μg ml−1 RNase A was able to prevent at least ∼7 log10 PFU ml−1 F+ RNA phages from infecting host cells.
When a phage MS2 suspension was treated with RNase A, which was subsequently inactivated by RNase inhibitor, clear zones were not observed on the bacterial lawn where undiluted phage suspensions were dispersed, which indicated the absence of host cell infection by F+ RNA phages. However, when the treated phage suspensions were diluted with PBS and aliquots (10 μl) of these dilutions were dispensed on the same plates, clear zones were formed on the bacterial lawn where 10−1, 10−2, 10−3, 10−4, or 10−5 dilutions of the treated phage suspensions were located. These clear zones on the bacterial lawns indicated the infectivity of phage MS2 after such treatment and thus the inability of RNase A to inactivate free F+ RNA phages.
When phage MS2 that was treated with proteinase K and RNase A was spotted on a host cell-seeded plate, clear zones were observed on the bacterial lawn where these treated phage suspensions (or dilutions) were dispensed. As observed for the negative control, when phage suspensions were treated with the enzymes for 10, 20, or 30 min, isolated plaques were formed on the bacterial lawn where the 10−6 dilution of the treated phage suspension was dispensed, indicating that little decrease in phage infectivity was achieved after such treatment.
Specificity of primers.
The primer sets FRNA-I, FRNA-II, and FRNA-IV target a conserved region in the replicase gene of phages representative of subgroups I, II, and IV, respectively, while primer set FRNA-III targets a conserved region in the coat protein gene of phages belonging to subgroup III. The target region was found in, but not limited to, phages MS2 and fr when aligning FRNA-I with sequences of subgroup I phages; phages GA, BZ13, and KU1 when aligning FRNA-II with sequences of subgroup II; Qβ, M11, MX1, and TW18 when aligning FRNA-III with sequences of subgroup III; and phages SP and NL95 when aligning FRNA-IV with sequences of subgroup IV. When testing the specificity of primers by RT-PCR, products were obtained only when the primers were used to amplify phages belonging to the corresponding subgroups. The primer set FRNA-I detected phage MS2, primer set FRNA-II detected phage GA, primer set FRNA-III detected Qβ, and primer set FRNA-IV detected phage SP. Cross-reactivity of the selected primer sets among the different subgroups of F+ RNA phages was not observed. The melting curve profiles of the four F+ RNA phages also confirmed the specificity of these primers. Even though the RT-PCR products of primer sets FRNA-II and FRNA-IV are the same length and indistinguishable electrophoretically, different melting points were observed between phages GA and SP (86.2 and 83.7°C, respectively).
Standard curves.
Four 10-fold dilution series, one for each F+ RNA phage, were independently prepared and assayed by real-time RT-PCR to establish standard curves. For each F+ RNA phage, a linear relationship was observed when the average CT values were plotted against the starting phage titers. The squared correlation coefficient (r2) for phages MS2, GA, Qβ, and SP were 0.999, 0.990, 0.989, and 0.989, respectively. The PCR efficiency (E) was calculated according to the following equation: E = 10−1/S − 1, where S is the slope of the standard curve (21). The slopes of the standard curves for phages MS2, GA, Qβ, and GA were −3.89, −4.36, −3.65, and −3.87, representing PCR efficiencies of 81%, 70%, 88%, and 81%, respectively.
Quantification of infectious F+ RNA phages.
Partially inactivated phage suspensions were used to evaluate the ability of the ET RT-PCR to quantify infectious F+ RNA phages. Before partial inactivation, the titers of phage suspensions containing MS2, GA, Qβ, and SP were 6.6, 6.8, 6.2, and 6.3 log10 PFU ml−1, respectively. After treatment with UV irradiation, heat at 55°C, or innate inactivation, only a portion of the phage populations in these suspensions was inactivated, and the residual phage titers were in the range of 3.00 to 5.67 log10 PFU ml−1 when determined by plaque assay (Table 2).
TABLE 2.
Phage titers detected in partially inactivated phage suspensions by plaque assay, RT-PCR, one-step ET RT-PCR, and two-step ET RT-PCR
| Phage | Initial titer (log10 PFU ml−1) | Treatment | Phage titer after partial inactivation (log10 PFU ml−1) |
|||
|---|---|---|---|---|---|---|
| RT-PCR | ET RT-PCR |
Plaque assay | ||||
| One step | Two step | |||||
| MS2 | 6.6 | UVa | 6.47 ± 0.25 | 5.57 ± 0.31 | 4.27 ± 0.45 | 3.70 ± 0.20 |
| Room tempb | 6.67 ± 0.25 | 6.17 ± 0.21 | 5.57 ± 0.12 | 5.10 ± 0.20 | ||
| 55°Cc | 6.60 ± 0.36 | 6.03 ± 0.15 | 5.27 ± 0.06 | 4.87 ± 0.21 | ||
| GA | 6.8 | UV | 6.80 ± 0.10 | 5.93 ± 0.15 | 4.97 ± 0.15 | 4.53 ± 0.21 |
| Room temp | 6.87 ± 0.15 | 6.37 ± 0.06 | 5.97 ± 0.12 | 5.67 ± 0.12 | ||
| 55°C | 6.80 ± 0.26 | 5.97 ± 0.23 | 5.10 ± 0.26 | 5.07 ± 0.25 | ||
| Qβ | 6.2 | UV | 6.30 ± 0.20 | 5.50 ± 0.26 | 4.13 ± 0.21 | 3.77 ± 0.15 |
| Room temp | 6.35 ± 0.21 | 5.93 ± 0.06 | 5.23 ± 0.15 | 4.63 ± 0.15 | ||
| 55°C | 6.27 ± 0.15 | 5.70 ± 0.17 | 4.70 ± 0.36 | 4.23 ± 0.15 | ||
| SP | 6.3 | UV | 6.43 ± 0.15 | 5.53 ± 0.31 | 3.57 ± 0.23 | 3.00 ± 0.26 |
| Room temp | 6.33 ± 0.21 | 5.73 ± 0.15 | 5.33 ± 0.15 | 4.77 ± 0.15 | ||
| 55°C | 6.17 ± 0.06 | 5.30 ± 0.17 | 3.53 ± 0.32 | 3.20 ± 0.26 | ||
UV exposure of 228 mJ cm−2.
Room temp, 25 ± 1°C for 14 days.
Heated at 55°C for 40 min.
Immediately after exposure to the inactivation process, phage suspensions were assayed by RT-PCR, one-step ET RT-PCR, two-step ET RT-PCR, and plaque assay. Phage titers detected by the RT-PCR methods (both with and without ET) were estimated using the RT-PCR standard curves. Despite the reduction of 1.20 to 3.43 log10 PFU ml−1 in phage titers (determined by plaque assay) after treatment with UV irradiation, heat treatment at 55°C, or innate inactivation, the reductions in phage titers of these partially inactivated suspensions were insignificant (P > 0.05) when detected by quantitative RT-PCR alone. However, when the one-step ET was performed prior to RNA extraction, significant (P < 0.05) reductions in phage titers were detected by RT-PCR compared with those determined by RT-PCR alone (Table 2). The phage titers in the partially inactivated suspensions obtained by the one-step ET RT-PCR were 0.70 to 2.53 log10 PFU ml−1 higher than those obtained by the plaque assay. This implied that the one-step ET RT-PCR decreased, but did not eliminate, false-positive signals.
The two-step ET RT-PCR was more effective at reducing the incidence of false-positive signals than the one-step RT-PCR. However, as with the one-step ET RT-PCR, it did not totally eliminate false-positive signals. A reduction of 0.90 to 2.86 log10 PFU ml−1 in phage titers was detected by the two-step ET RT-PCR compared with that obtained by RT-PCR alone (Table 2). Compared with results obtained by the plaque assay, phage titers in the partially inactivated suspensions were 0.03 to 0.60 log10 PFU ml−1 higher when detected by the two-step ET RT-PCR.
Phage titers in the partially inactivated suspensions obtained by RT-PCR, one-step ET RT-PCR, and two-step ET RT-PCR plotted against phage titers determined by the plaque assay are shown in Fig. 1. The regression equation (y = Ax + B, where y is the phage titer obtained by RT-PCR with or without enzyme treatment, x is the phage titer determined by the plaque assay, A is the slope of the regression line, and B is a constant) was obtained by performing linear regression. Perfect agreement between the plaque assay and the PCR-based assay would be achieved if A was equal to 1 and B was equal to 0. The values of A were 0.20, 0.35, and 0.94, and the values of B were 5.63, 4.27, and 0.71, respectively, for RT-PCR, one-step ET RT-PCR, and two-step ET RT-PCR. These data indicate that the two-step ET RT-PCR assay more accurately predicts the number of infectious phages present in a sample than the other two assays.
FIG 1.
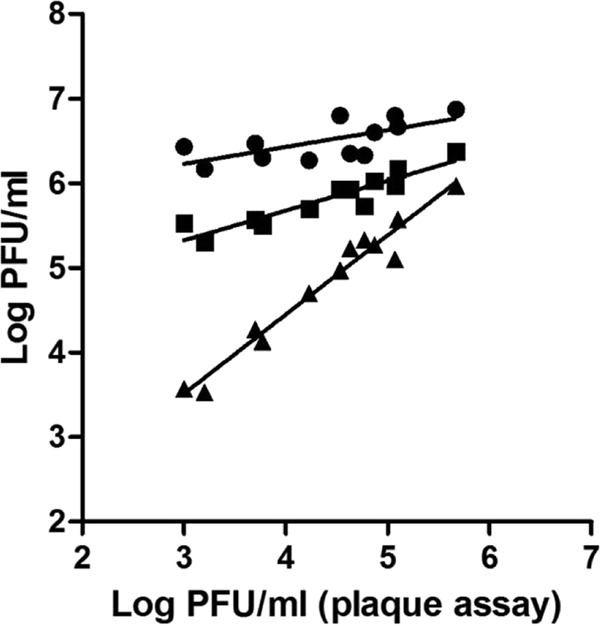
Phage titers in partially inactivated suspensions determined by different methods. Partial inactivation was achieved by treatment with UV exposure, heat exposure at 55°C, or incubation at room temperature, as shown in Table 2. ●, RT-PCR; ■, one-step ET RT-PCR; ▲, two-step ET RT-PCR.
Different inactivation rates of F+ RNA phages were achieved by the three treatments when determined by plaque assay. For all four phages, the highest residual titers after partial inactivation, detected by either one-step ET RT-PCR or two-step ET RT-PCR, were observed after innate inactivation at room temperature, followed by heat treatment and then UV irradiation. This was consistent with the results obtained by plaque assay, which indicated that the UV irradiation treatment caused the highest reduction in phage titer, followed by heat treatment and then innate inactivation, for all the phages tested. This confirmed that the ET RT-PCR (either one step or two step) was able to differentiate between infectious and noninfectious phages, albeit to different extents.
Naturally occurring F+ RNA phages.
A total of 16 environmental samples (8 raw sewage and 8 primary treated wastewater samples) were collected and assayed to verify the ability of ET RT-PCR to quantify infectious F+ RNA phages. Phage titers were determined by plaque assay, RT-PCR, and two-step ET RT-PCR, and the results are summarized in Table 3. The four subgroup-specific primer sets were tested individually in each of the samples, and the sum of phage titers obtained by the four primer sets was recorded as the titer of F+ RNA phages in the environmental samples, detected by RT-PCR alone or ET RT-PCR. The average phage titers in raw sewage were 3.32, 4.57, and 3.64 log10 PFU ml−1 when determined by the plaque method, RT-PCR, and two-step ET RT-PCR, respectively, while in primary treated wastewater samples, the average phage titers determined by plaque assay, RT-PCR, and two-step ET RT-PCR were 2.95, 4.59, and 3.57 log10 PFU ml−1, respectively. In both raw sewage and primary treated wastewater, the titers of naturally occurring F+ RNA phages were significantly (P > 0.05) higher when detected by RT-PCR than by the plaque method. When a two-step ET was performed prior to RNA extraction, phage titers decreased significantly (P > 0.05) compared with those obtained by RT-PCR without the enzyme pretreatment, but they were still significantly (P > 0.05) higher than those determined by plaque assay.
TABLE 3.
Naturally occurring F+ RNA phages in environmental samples
| Sample type | Sample no. | Avg (SD) titer of F+ RNA phages (log10 PFU ml−1) |
||
|---|---|---|---|---|
| Plaque assay | RT-PCR | Two-step ET RT-PCR | ||
| Raw sewage | 1 | 3.23 (0.05) | 4.23 (0.15) | 3.68 (0.12) |
| 2 | 3.38 (0.09) | 4.49 (0.18) | 3.94 (0.23) | |
| 3 | 3.42 (0.05) | 4.80 (0.16) | 3.79 (0.19) | |
| 4 | 3.50 (0.06) | 4.61 (0.23) | 3.81 (0.19) | |
| 5 | 3.19 (0.08) | 5.04 (0.17) | 3.61 (0.21) | |
| 6 | 3.16 (0.11) | 5.09 (0.13) | 3.46 (0.13) | |
| 7 | 3.56 (0.05) | 3.91 (0.20) | 3.39 (0.17) | |
| 8 | 3.14 (0.13) | 4.36 (0.16) | 3.41 (0.16) | |
| Primary treated wastewater | 9 | 3.01 (0.13) | 4.26 (0.06) | 3.74 (0.14) |
| 10 | 3.04 (0.14) | 4.79 (0.17) | 3.31 (0.22) | |
| 11 | 2.92 (0.07) | 4.78 (0.23) | 3.89 (0.12) | |
| 12 | 2.93 (0.07) | 4.57 (0.22) | 3.72 (0.14) | |
| 13 | 2.82 (0.05) | 4.99 (0.25) | 3.44 (0.12) | |
| 14 | 2.77 (0.09) | 5.04 (0.22) | 3.56 (0.12) | |
| 15 | 3.20 (0.09) | 3.99 (0.21) | 3.45 (0.12) | |
| 16 | 2.88 (0.05) | 4.33 (0.28) | 3.43 (0.16) | |
DISCUSSION
In this study, we evaluated an ET real-time RT-PCR to quantify infectious F+ RNA phages by the use of proteinase K and RNase A to selectively degrade naked RNA or RNA of inactivated phage particles prior to RNA extraction. F+ RNA phages have been recommended as reliable indicators of fecal contamination, and the phenomenon where subgroups II and III of F+ RNA phages are predominantly associated with fecal contamination of human origin while subgroups I and IV are mainly found in animal feces provides the potential to track the source of fecal contamination by identifying subgroups of F+ RNA phages (22–24). RT-PCR approaches have been used to detect or identify subgroups of F+ RNA phages in order to predict the level or source of fecal contamination by the use of subgroup-specific primers (6, 7, 9). However, RT-PCR signals may result from infectious or inactivated phage particles, or even naked RNA, and thus are not able to assess their infectivity. A previous study indicated that naturally occurring infectious viruses might be overestimated by more than 90-fold when RT-PCR alone was used for their detection (16). Due to the fact that the risks of fecal contamination to human health are caused by the presence of infectious pathogens, differentiation of infectious and inactivated F+ RNA phages provides useful information on the potential presence of infectious enteric pathogens, since F+ RNA phages are transported by the same route as enteric pathogens and have similar persistence (25).
The susceptibilities of infectious F+ RNA phages to RNase A (in the presence or absence of proteinase K) were tested in order to confirm that the RNA of infectious phages would not be degraded by the ET step, which was intended to selectively degrade RNA of inactivated phages (and naked RNA). Confluent bacterial growth on the host cell-seeded plates containing RNase A indicated that F+ RNA phages were not able to infect host cells in the presence of RNase A. However, clear zones formed on the host cell-seeded plates (in the absence of RNase A) where dilutions of RNase A-treated phage suspension (RNase activity was inhibited by RNase inhibitor after treatment) was dispersed indicated that F+ RNA phages were not inactivated by RNase A. A reasonable explanation of this phenomenon is that RNase degrades RNA of F+ RNA phages only during the infection cycle (most probably the attachment stage or RNA-ejecting stage) but has no effect on the RNA of free phages. This explanation agrees with the findings of Toropova et al. (26). These authors showed that the RNA maturation protein complex is poised to leave the protein capsid when F+ RNA phages are attached to the F pili of host cells. During the infection cycle, the genome (probably with the aid of maturation protein) of F+ RNA phages first leaves the protein capsid and enters the host cell through the F pili once contact between phages and F pili is established (26). This process exposes phage RNA to RNase if it is present, while in free phages, the RNA genome is enclosed in and protected by the coat protein capsid and thus is resistant to RNase degradation. The enzymatic activity in the RNase A-treated phage suspension may not be completely inactivated by RNase inhibitor that was added after treatment, and thus, infection of host cells by F+ RNA phages was inhibited on host cell-seeded plates where undiluted RNase A-treated phage suspension was dispersed. Dilution with PBS may decrease the residual RNase to a level that is not sufficient to degrade RNA of phages attaching to host cells. Thus, clear zones were formed on the host cell-seeded plates when dilutions of RNase A-treated phage suspension were dispensed. The inability of RNase A, or a combination of RNase A and proteinase K, to inactivate free infectious F+ RNA phages implies that RNA of infectious phages in partially inactivated phage suspensions or environmental samples is unlikely to be degraded, and thus, this ET real-time RT-PCR would not underestimate the level of infectious F+ RNA phages.
In either partially inactivated phage suspensions or environmental samples, higher phage titers were detected by RT-PCR alone than by ET RT-PCR, which in turn detected higher phage titers than the plaque assay. Since F+ RNA phages are unlikely to aggregate at pH values above their isoelectric points (2.1 to 3.9) (27, 28), results obtained by the plaque assay represent the concentration of infectious single phage particles. Thus, comparison of the results obtained with the plaque assay, RT-PCR, and ET RT-PCR implies that an ET step prior to RNA extraction could reduce signals encountered due to the presence of noninfectious phages (false positive) by the RT-PCR assay alone, although false-positive signals cannot be completely eliminated. However, a previous study by Nuanualsuwan and Cliver (17) reported that vaccine poliovirus 1, feline calicivirus, and hepatitis A virus, which were inactivated by UV irradiation, hypochlorite, or heating at 72°C, could not be detected by RT-PCR when proteinase K and RNase A were added prior to RNA extraction. This may be attributed to the low initial titers (∼103 PFU ml−1) of these viruses, the activity of which might be completely lost after inactivation.
Pecson et al. (18) also reported the ability of one-step ET RT-PCR to reduce, but not eliminate, false-positive signals when partially inactivated phage MS2 was assayed. For the one-step ET, in which proteinase K and RNase A were used simultaneously to degrade RNA of the inactivated phage particles, RNase activity may be lost before all the RNA of the inactivated phage particles is degraded, since degradation of RNase A by proteinase K might occur at the same time (29). Thus, higher phage titers were obtained by one-step ET RT-PCR than by two-step ET RT-PCR. The phenomenon observed in this study, where residual RNase activity inhibited infection of host cells when an undiluted RNase A-treated phage suspension was dispersed on host cell-seeded plates while infection of host cells was not affected when the phage suspension was treated with proteinase K and RNase A, also implied that RNase A was denatured by proteinase K. Inactivation of proteinase K prevented the degradation of RNase A that was subsequently added; thus, the two-step ET was more effective than the one-step ET at reducing false-positive RT-PCR signals caused by the presence of inactivated phage particles. Incomplete digestion of RNA may contribute to the inability of the ET (either one or two step) RT-PCR to completely eliminate false-positive signals, considering the small size of the RT-PCR product. Another possibility is that damage only to the maturation protein also leads to the loss of infectivity, since the maturation protein of F+ RNA phages was responsible for recognizing and attaching to host cells during infection (26, 30, 31), and these phage particles were not readily degraded by ET.
Even though a difference in the reduction of false-positive signals between inactivation treatments was reported in a previous study (18), ET (both one and two step) RT-PCR used in this study did not demonstrate preferential reduction of false-positive signals with one inactivation treatment over another. The extent of false-positive signals reduced by two-step ET RT-PCR in environmental samples was significantly correlated (r2 = 0.72; P < 0.05) with the number of inactivated phages present (Table 3 shows the difference between results of the plaque assay and RT-PCR), and this implies the usefulness of the method to quantify infectious F+ RNA phages. Compared with integrated cell culture RT-PCR, which combines RT-PCR with a cultured cell line and thus is able to determine the infectivity of only culturable viruses (16), ET RT-PCR could be adapted to determine the infectivity of viruses that are difficult to cultivate or those for which a cell line is not currently available. Even though higher phage titers were detected than by the plaque assay, ET RT-PCR, especially two-step ET RT-PCR, significantly reduced false-positive signals caused by the presence of inactivated phages.
ACKNOWLEDGMENTS
Financial support for this research program (USDA Specialty Crops Research Initiative, award number 2009-01208) from the USDA is gratefully acknowledged.
We thank Lawrence D. Goodridge, principal investigator of the research program, for his contributions.
Footnotes
Published ahead of print 21 March 2014
REFERENCES
- 1.van Duin J. 27 January 2006. Bacteriophages with ssRNA. eLS. 10.1038/npg.els.0004286 [DOI] [Google Scholar]
- 2.Bae J, Schwab KJ. 2008. Evaluation of murine norovirus, feline calicivirus, poliovirus, and MS2 as surrogates for human norovirus in a model of viral persistence in surface water and groundwater. Appl. Environ. Microbiol. 74:477–484. 10.1128/AEM.02095-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chung H. 1993. F-specific coliphages and their serogroups, and bacteroides fragilis phages as indicators of estuarine water and shellfish quality. University of North Carolina, Chapel Hill, NC [Google Scholar]
- 4.D'Souza DH, Su X, Roach A, Harte F. 2009. High-pressure homogenization for the inactivation of human enteric virus surrogates. J. Food Prot. 72:2418–2422 [DOI] [PubMed] [Google Scholar]
- 5.Sobsey MD, Love DC, Lovelace GL. 2006. F+ RNA coliphages as source tracking viral indicators of fecal contamination. http://ciceet.unh.edu/news/releases/springReports07/pdf/sobsey.pdf Accessed 11 August 2013 [Google Scholar]
- 6.Haramoto E, Kitajima M, Katayama H, Asami M, Akiba M, Kunikane S. 2009. Application of real-time PCR assays to genotyping of F-specific phages in river water and sediments in Japan. Water Res. 43:3759–3764. 10.1016/j.watres.2009.05.043 [DOI] [PubMed] [Google Scholar]
- 7.Kirs M, Smith DC. 2007. Multiplex quantitative real-time reverse transcriptase PCR for F+-specific RNA coliphages: a method for use in microbial source tracking. Appl. Environ. Microbiol. 73:808–814. 10.1128/AEM.00399-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langlet J, Ogorzaly L, Schrotter JC, Machinal C, Gaboriaud F, Duval JFL, Gantzer C. 2009. Efficiency of MS2 phage and Q[beta] phage removal by membrane filtration in water treatment: applicability of real-time RT-PCR method. J. Memb. Sci. 326:111–116. 10.1016/j.memsci.2008.09.044 [DOI] [Google Scholar]
- 9.Ogorzaly L, Gantzer C. 2006. Development of real-time RT-PCR methods for specific detection of F-specific RNA bacteriophage genogroups: application to urban raw wastewater. J. Virol. Methods 138:131–139. 10.1016/j.jviromet.2006.08.004 [DOI] [PubMed] [Google Scholar]
- 10.Pecson BM, Ackermann M, Kohn T. 2011. Framework for using quantitative PCR as a nonculture based method to estimate virus infectivity. Environ. Sci. Technol. 45:2257–2263. 10.1021/es103488e [DOI] [PubMed] [Google Scholar]
- 11.Hot D, Legeay O, Jacques J, Gantzer C, Caudrelier Y, Guyard K, Lange M, Andreoletti L. 2003. Detection of somatic phages, infectious enteroviruses and enterovirus genomes as indicators of human enteric viral pollution in surface water. Water Res. 37:4703–4710. 10.1016/S0043-1354(03)00439-1 [DOI] [PubMed] [Google Scholar]
- 12.Costán-Longares A, Montemayor M, Payán A, Méndez J, Jofre J, Mujeriego R, Lucena F. 2008. Microbial indicators and pathogens: removal, relationships and predictive capabilities in water reclamation facilities. Water Res. 42:4439–4448. 10.1016/j.watres.2008.07.037 [DOI] [PubMed] [Google Scholar]
- 13.Flannery J, Keaveney S, Dore W. 2009. Use of FRNA bacteriophages to indicate the risk of norovirus contamination in Irish oysters. J. Food Prot. 72:2358–2362 [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez RA, Pepper IL, Gerba CP. 2009. Application of PCR-based methods to assess the infectivity of enteric viruses in environmental samples. Appl. Environ. Microbiol. 75:297–307. 10.1128/AEM.01150-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fittipaldi M, Rodriguez NJP, Codony F, Adrados B, Penuela GA, Morato J. 2010. Discrimination of infectious bacteriophage T4 virus by propidium monoazide real-time PCR. J. Virol. Methods 168:228–232. 10.1016/j.jviromet.2010.06.011 [DOI] [PubMed] [Google Scholar]
- 16.Shieh Y, Wong C, Krantz J, Hsu F. 2008. Detection of naturally occurring enteroviruses in waters using direct RT-PCR and integrated cell culture-RT-PCR. J. Virol. Methods 149:184–189. 10.1016/j.jviromet.2007.12.013 [DOI] [PubMed] [Google Scholar]
- 17.Nuanualsuwan S, Cliver DO. 2002. Pretreatment to avoid positive RT-PCR results with inactivated viruses. J. Virol. Methods 104:217–225. 10.1016/S0166-0934(02)00089-7 [DOI] [PubMed] [Google Scholar]
- 18.Pecson BM, Martin LV, Kohn T. 2009. Quantitative PCR for determining the infectivity of bacteriophage MS2 upon inactivation by heat, UV-B radiation, and singlet oxygen: advantages and limitations of an enzymatic treatment to reduce false-positive results. Appl. Environ. Microbiol. 75:5544–5554. 10.1128/AEM.00425-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Environmental Protection Agency. 2001. Method 1601: male-specific (F+) and somatic coliphage in water by two-step enrichment procedure. EPA 821-R-01-030. Environmental Protection Agency, Washington, DC [Google Scholar]
- 20.Friedman SD, Cooper EM, Casanova L, Sobsey MD, Genthner FJ. 2009. A reverse transcription-PCR assay to distinguish the four genogroups of male-specific (F+) RNA coliphages. J. Virol. Methods 159:47–52. 10.1016/j.jviromet.2009.02.028 [DOI] [PubMed] [Google Scholar]
- 21.Kubista M, Andrade JM, Bengtsson M, Forootan A, Jonák J, Lind K, Sindelka R, Sjöback R, Sjögreen B, Strömbom L. 2006. The real-time polymerase chain reaction. Mol. Aspects Med. 27:95–125. 10.1016/j.mam.2005.12.007 [DOI] [PubMed] [Google Scholar]
- 22.Hsu FC, Shieh Y, Van Duin J, Beekwilder M, Sobsey MD. 1995. Genotyping male-specific RNA coliphages by hybridization with oligonucleotide probes. Appl. Environ. Microbiol. 61:3960–3966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JE, Lee H, Cho YH, Hur HG, Ko GP. 2011. F+ RNA coliphage-based microbial source tracking in water resources of South Korea. Sci. Total Environ. 412:127–131. 10.1016/j.scitotenv.2011.09.061 [DOI] [PubMed] [Google Scholar]
- 24.Schaper M, Jofre J, Uys M, Grabow W. 2002. Distribution of genotypes of F specific RNA bacteriophages in human and non human sources of faecal pollution in South Africa and Spain. J. Appl. Microbiol. 92:657–667. 10.1046/j.1365-2672.2002.01600.x [DOI] [PubMed] [Google Scholar]
- 25.Yousefi Z, Zazouli MA. 2008. Evaluation of bacteriophages methods for detection and isolation of viruses from surface water. World Appl. Sci. J. 3:317–322 [Google Scholar]
- 26.Toropova K, Stockley PG, Ranson NA. 2011. Visualising a viral RNA genome poised for release from its receptor complex. J. Mol. Biol. 408:408–419. 10.1016/j.jmb.2011.02.040 [DOI] [PubMed] [Google Scholar]
- 27.Langlet J, Gaboriaud F, Gantzer C. 2007. Effects of pH on plaque forming unit counts and aggregation of MS2 bacteriophage. J. Appl. Microbiol. 103:1632–1638. 10.1111/j.1365-2672.2007.03396.x [DOI] [PubMed] [Google Scholar]
- 28.Michen B, Graule T. 2010. Isoelectric points of viruses. J. Appl. Microbiol. 109:388–397. 10.1111/j.1365-2672.2010.04663.x [DOI] [PubMed] [Google Scholar]
- 29.Rauber N, Jany KD, Pfleiderer G. 1978. Ribonuclease A digestion by proteinase K. Z. Naturforsch. C 33:660–663 [PubMed] [Google Scholar]
- 30.Paranchych W, Krahn P, Bradley R. 1970. Stages in phage R17 infection. Virology 41:465–473. 10.1016/0042-6822(70)90168-6 [DOI] [PubMed] [Google Scholar]
- 31.Shiba T, Miyake T. 1975. New type of infectious complex of E. coli RNA phage. Nat. Rev. Immunol. 254:157–158 [DOI] [PubMed] [Google Scholar]