

Abstract
Stable isotope probing (SIP) is a cultivation-free methodology that provides information about the identity of microorganisms participating in assimilatory processes in complex communities. In this study, a Herminiimonas-related bacterium was identified as the dominant member of a denitrifying microcosm fed [13C]toluene. The genome of the uncultivated toluene-degrading bacterium was obtained by applying pyrosequencing to the heavy DNA fraction. The draft genome comprised ∼3.8 Mb, in 131 assembled contigs. Metabolic reconstruction of aromatic hydrocarbon (toluene, benzoate, p-cresol, 4-hydroxybenzoate, phenylacetate, and cyclohexane carboxylate) degradation indicated that the bacterium might specialize in anaerobic hydrocarbon degradation. This characteristic is novel for the order Burkholderiales within the class Betaproteobacteria. Under aerobic conditions, the benzoate oxidation gene cluster (BOX) system is likely involved in the degradation of benzoate via benzoyl coenzyme A. Many putative genes for aromatic hydrocarbon degradation were closely related to those in the Rhodocyclaceae (particularly Aromatoleum aromaticum EbN1) with respect to organization and sequence similarity. Putative mobile genetic elements associated with these catabolic genes were highly abundant, suggesting gene acquisition by Herminiimonas via horizontal gene transfer.
INTRODUCTION
Toluene is a useful model compound for investigating the anaerobic metabolism of aromatic compounds (1, 2). Microorganisms that attack toluene use nitrate, Mn(IV), Fe(III), sulfate, and CO2 as terminal electron acceptors (2, 3). Of these different acceptors, the greatest energy yield is obtained with nitrate. The majority of toluene degraders discovered to date that use nitrate as the final electron acceptor belong to the order Rhodocyclales within the Betaproteobacteria: Azoarcus spp. (4–6), Aromatoleum aromaticum EbN1 (formerly Azoarcus sp. EbN1) (7), Georgfuchsia toluolica (8), Thauera aromatica (9), and Dechloromonas aromatica RCB (10). In addition, Magnetospirillum sp. TS-6 within Alphaproteobacteria also degrades toluene using nitrate as the electron acceptor (11). Recently, four toluene-degrading bacterial strains belonging to the Alphaproteobacteria and Gammaproteobacteria were isolated from marine sediment (12), and a non-culture-based, DNA-based stable isotope probing (SIP) study reported the 16S rRNA gene sequences of [13C]toluene degraders in nitrate-reducing microcosms (13). In that study, sequences related to Comamonadaceae bacteria and Thauera bacteria were retrieved from the 13C-labeled heavy DNA fraction (13). The genome sequences of two of the isolated denitrifying toluene degraders, A. aromaticum EbN1 and D. aromatica RCB, are available (14–16), allowing metabolic reconstruction of anaerobic aromatic degradation pathways.
Anaerobically, toluene is activated by fumarate addition, which is mediated by benzylsuccinate synthetase. Benzylsuccinate is converted to benzoyl coenzyme A (CoA), a central intermediate in the anaerobic degradation of many aromatic hydrocarbons (2, 17). The benzene ring of benzoyl-CoA is reduced by benzoyl-CoA reductase (BCR), which is composed of four subunits (a heterotetramer of α, β, γ, and δ subunits). Based on differences in the organization of the gene clusters and gene sequence dissimilarities, BCR clusters into two groups: bcr and bzd types (18). The bcr type is associated with Thauera, Magnetospirillum, and Rhodopseudomonas (18), while the bzd type is associated with Azoarcus (19, 20).
During aerobic metabolism of aromatic compounds, benzoate is degraded by oxygenase systems; however, a novel benzoate degradation pathway that produces benzoyl-CoA as a central intermediate was discovered by Gescher et al. (21). Products of the benzoate oxidation gene cluster (BOX) convert benzoate to benzoyl-CoA. The BOX system is likely widespread: ∼5% of all bacterial species for which genome sequences are available contain putative box genes (17). Both bcr and box cluster genes were found in the genomes of Azoarcus evansii (22, 23), A. aromaticum EbN1 (15), and Magnetospirillum magneticum AMB-1 (NC_007626).
Metagenomic sequencing has successfully revealed novel insights into the physiology and ecology of a variety of intriguing uncultivated microorganisms (e.g., BD1-5, OP11, OD1 bacteria, TM7, and marine group II Euryarchaeota) (24–26). Successful assembly of full and/or partial genomes of uncultivated microorganisms is the result of progress in both high-throughput sequencing and assembly and binning methodologies. As an example, the almost complete genome of Methylotenera mobilis, a novel methylotrophic bacterium, was reconstructed using a combination of SIP and metagenomic analyses (27). Those authors were able to evaluate genome completeness by examining the presence of various metabolic and housekeeping genes (27).
As exemplified by our previous discovery of a novel toluene-degrading, iron-reducing bacterium (28), metagenomic approaches have great potential for the discovery of novel microorganisms with bioremediation potential. Previous studies describing aromatic degradation under nitrate-reducing conditions using metagenomic approaches are scarce, despite many attempts to obtain and characterize axenic cultures. Here, we used metagenomic approaches that preclude some of the biases observed in cultivation-based studies in an attempt to characterize anaerobic aromatic degradation under nitrate-reducing conditions.
MATERIALS AND METHODS
Toluene-degrading microcosm experiments.
Coal tar waste-contaminated sediment was gathered near South Glens Falls, NY, in November 2011, as described by Jeon et al. (29). Replicates of homogenized sediment (∼8.5 g [wet weight]) were anoxically incubated in sterile 125-ml serum bottles containing 50 ml defined bicarbonate-buffered artificial freshwater medium under a headspace of N2 (100%). The components of the artificial freshwater medium were as follows: 0.5 g liter−1 KCl, 0.2 g liter−1 KH2PO4, 1.0 g liter−1 NaCl, 0.5 g liter−1 NH4Cl, 0.1 g liter−1 CaCl2·2H2O, 0.4 g liter−1 MgCl2·6H2O, 1 ml liter−1 nonchelated trace element mixture (30), 1 ml liter−1 vitamin solution (30), 1 ml liter−1 selenite-tungstate solution (31), and 1 mg liter−1 resazurin. For the electron acceptor, 7.5 mM NaNO3 was added. The microcosm-fostering nitrate reduction conditions were designated the CN microcosm. After purging with N2 gas, bicarbonate buffer was added to 20 mM (final concentration), and 1 mM Na2S (final concentration) was used as a reducing agent. The final pH of the medium was adjusted to pH 7.0. To reduce toluene toxic effects during incubation, 0.5 ml of 2,2,4,4,6,8,8-heptamethylnonane (HMN) was added to each bottle as a carrier. Serum bottles were closed with butyl rubber stoppers (Bellco Glass Inc., Vineland, NJ), and 5 μl of either nonlabeled (12C) toluene (Sigma-Aldrich, MO) or ring-13C6-labeled toluene (Cambridge Isotope Laboratories, Inc., Andover, MA) was injected through the stoppers by using a gas-tight syringe. This resulted in a reservoir of ∼0.96 mM toluene in each bottle. The microcosms were incubated statically at 20°C in the dark. Five replicate bottles were prepared for each series to monitor biodegradation activity and successive time-dependent termination. Inoculum-unamended cultures, inoculum-autoclaved cultures, and cultures inoculated without nitrate (nitrate not amended) were used as controls.
Analytical methods.
The time course of toluene concentration changes was measured in headspace gas samples (100 μl) by using a gas chromatograph (GC/FID 2010; Shimadzu, Japan) equipped with an Rtx-1 GC column (film thickness, 0.25 μm; inside diameter, 0.25 mm; length, 30 m; Restek, Bellefonte, PA) and a flame ionization detector. Nitrogen gas was used as carrier, with a column flow rate of 0.9 ml/min. The chromatographic conditions were as follows: injector temperature, 200°C (split ratio of 1:10); oven temperature program consisting of 40°C for 1 min, followed by an increase at a rate of 16°C/min to 145°C and then at a rate of 45°C/min to 300°C, then held for 1 min; detector temperature, 300°C (28). A nitrate test kit was used to measure nitrate concentrations (NECi, Lake Linden, MI). Measurement of the nitrite concentration was performed as described by Shinn (32). Production of 13CO2 gas in the headspace was measured in serum bottles by using a GC fitted with an electron capture detector (GC/ECD-MS 6890A; Agilent, Santa Clara, CA). The GC was equipped with a Varian CP-PoraBOND Q capillary column (5-μm film thickness, 0.32 mm in diameter, 50-m length). The oven was isothermal, maintained at 33°C, and CO2 concentrations were determined three times for each sample. Detailed methods for 13CO2 determinations have been described by Kim et al. (28).
Metagenomic DNA extraction and isopycnic centrifugation.
At a selected time point (8 days; 80% toluene degraded), single [13C]toluene- and [12C]toluene-amended bottles were sacrificed for DNA extraction. The sediment and cells were centrifuged at 8,000 rpm (6,790 × g) at 4°C for 20 min in a Mega17 R centrifuge (Hanil, Republic of Korea). The supernatant was filtered through a 0.2-μm-pore-sized filter (Adventec, Japan) to recover any remaining cells. Harvested cells were combined, immediately frozen, and stored at −80°C until extraction. Total genomic DNA was extracted from frozen cells by using the protocol described by Zhou et al. (33). For isopycnic centrifugation, 5 μg of extracted DNA was loaded into a CsCl solution (1 g ml−1, combining an equal volume of water with an equal weight of CsCl [Sigma]) (34), and 120 μl of a 10-mg ml−1 ethidium bromide (EtBr) solution was added for the detection of heavy and light DNA fractions. The CsCl solution with extracted DNA was transferred to an 8.9-ml polyallomer centrifuge tube (Opti Seal; Beckman Coulter, Krefeld, Germany). The tubes were centrifuged in a type 90 Ti fixed-angle rotor using a Centrikon T-2190 ultracentrifuge (both from Beckman Coulter, Krefeld, Germany) at 55,000 rpm (187,526 × g) at 20°C for 36 h under vacuum (28). Two distinct bands of DNA were observed (one light [12C, density of 1.55 g ml−1] and one heavy [13C, density of 1.59 g ml−1]) in the [13C]toluene-amended sample, while only one band was observed in the samples from the [12C]toluene-amended bottle. The DNA bands were recovered by using a needle and syringe. The DNA was purified by extracting EtBr using 1-butanol-saturated with Tris-EDTA.
T-RFLP fingerprinting, cloning, and sequencing.
DNA isolated by isopycnic centrifugation was used to amplify bacterial 16S rRNA with previously determined primers: 27F (6-carboxyfluorescein [FAM] labeled) and 1492R (35). The PCR conditions were as follows: 5 min at 95°C, followed by 30 cycles of 30 s at 95°C, 30 s at 55°C, and 90 s at 72°C, 10 min at 72°C, and a final hold at 4°C. The PCR products were purified using a PCR purification kit (Solgent, Republic of Korea) in accordance with the manufacturer's recommendations. RsaI (New England BioLabs, Beverly, MA) was used for terminal restriction fragment length polymorphism (T-RFLP) analysis, and the fragmented DNAs were examined using an ABI 3730xL DNA analyzer. The T-RFLP profiles were analyzed using GeneMapper software (version 3.7; Applied Biosystems, Foster, CA).
The amplified 16S rRNA genes from the heavy DNA fraction were cloned using a TA cloning vector kit (RBC Bioscience, Taipei, Taiwan) and sequenced using M13F and M13R primers (36). Terminal restriction fragments (T-RFs; predicted in silico for representative clones) were obtained and their sequences were determined. For phylogenetic analysis, the 16S rRNA gene sequences of related taxa obtained from GenBank were aligned using the SILVA algorithm (http://www.arb-silva.de/aligner) (37). The phylogenetic tree was constructed using the neighbor-joining method with the Kimura two-parameter distance model within the MEGA 5 program (38). The bootstrap values were obtained with 1,000 replications.
Next-generation sequencing of heavy DNA.
Quantified heavy fraction DNA (2 μg) was sequenced using 454 pyrosequencing (Genome Sequencer GLX system; Roche). Preparation and sequencing of samples and analytical processing were performed at the National Instrumentation Center for Environmental Management (NICEM), Republic of Korea, according to the manufacturer's instructions. To reduce the rate of chimerism, minimal filtering was performed for all reads by removing all that were shorter than 100 bp or had an average Phred quality score of <20 (39).
Microbial community composition.
rRNA genes were identified from the metagenomic sequences by comparing in-house data sets with the RDP database (40). All reads that matched an rRNA gene sequence with an alignment length of ≥400 bp with a cut-off E value of 0.001 against the RDP database were collected. When possible, genus-level identifications were assigned based upon shared identity with a known species in the database.
Genome assembly and annotation.
Metagenome sequencing reads were assembled using the Roche De Novo assembler (Newbler assembler v. 2.6). After assembly, 1,241 contigs (ca. 4.2 Mbp) were obtained from the heavy DNA fraction. Putative coding sequences (CDSs) were predicted with the MetaGeneAnnotator (41) and Integrative Services for Genomic Analysis. Predicted CDSs were annotated using the best BLAST hit against the NCBI nonredundant database. Ribosomal RNAs and tRNAs were identified using RNAmmer (42) and tRNAscan-SE (43), respectively. The overall GC content was determined with Artemis software (44). For binning, the oligonucleotide frequencies of the assembled contigs were calculated using the wordfreq program in the EMBOSS package (45). The GC content and frequency of betaproteobacterial genes were used to select betaproteobacterial contigs from the total 1,241 contigs. In addition, we retained only those contigs that were larger than 5 kb in order to increase taxonomic uniformity. This pipeline yielded 131 contigs that could be confidently ascribed to Betaproteobacteria. We also found that the binned contigs had a consistent phylogenetic profile and were hence more likely to originate from a single organism (46). Average nucleotide identity (ANI) was calculated as described previously (47). For functional assignment of the predicted genes, BLAST analysis was performed with the Clusters of Orthologous Groups, GenBank, Pfam, and Kyoto Encyclopedia of Genes and Genomes databases.
Nucleotide sequence accession numbers.
The results of this metagenome shotgun project have been deposited with DDBJ/EMBL/GenBank under the accession number AVCC00000000. The version described in this paper is version AVCC01000000.
RESULTS AND DISCUSSION
Toluene degradation coupled with nitrate reduction.
Toluene degradation was coupled to nitrate reduction in the microcosms (Fig. 1). Following a short lag time the toluene disappeared, and nitrate levels were reduced after 12 days of incubation. The nitrate concentration in the microcosm decreased from 7.4 mM to 0.4 mM during the incubation period, with transient accumulation of nitrite (up to 1 mM). Due to dissolution in HMN, the actual toluene concentrations were lower than the injected toluene concentration (∼0.96 mM). The stoichiometry of toluene degradation with denitrification is as follows: C7H8 + 7.2 NO3− + 0.2 H+ → 7 HCO3− + 3.6 N2 + 0.6 H2O. The actual ratio of toluene degradation relative to denitrification in our experiment was 1:7.3, which matches remarkably well to the expected ratio (1:7.2). When the microcosm was incubated with [13C]toluene as the substrate, 13CO2 production was observed (see Table S1 in the supplemental material). On a mass basis, approximately 85% of the degraded [13C]toluene was converted to 13CO2 after completion of toluene degradation in the microcosms. No toluene loss was detected in the absence of nitrate. Neither decreases in nitrate and toluene concentrations nor increases of 13CO2 levels were observed in the autoclaved microcosms (Fig. 1; see also Table S1).
FIG 1.
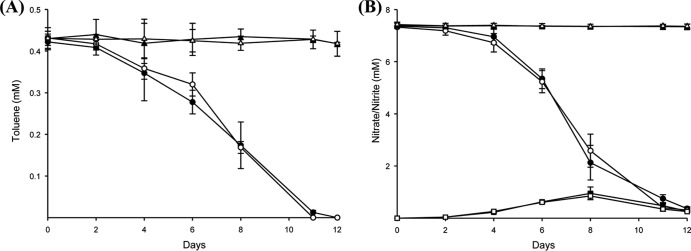
Toluene degradation (A) coupled to nitrate reduction (B) in the CN microcosms. Residual toluene and nitrate concentrations were measured in the [13C]toluene-amended (closed circles), [12C]toluene-amended (open circles), autoclaved [13C]toluene-amended (closed triangles), and autoclaved [12C]toluene-amended (open triangles) microcosms. Nitrite production is indicated by squares: [13C]toluene-amended microcosm, closed squares; [12C]toluene-amended microcosm, open squares. The error bars represent standard deviations from triplicate experiments.
DNA-SIP and T-RFLP analysis.
To identify the microorganisms involved in toluene degradation, total genomic DNA was extracted from [13C]toluene-amended microcosms prior to the complete oxidation of toluene (when approximately 80 to 90% of the toluene had disappeared) to reduce the influence of cross-feeding. Total genomic DNA was separated into light (12C) and heavy (13C) fractions by using isopycnic ultracentrifugation. PCR-based 16S rRNA gene T-RFLP analysis was used to compare the microbial community profiles in the light and heavy DNA fractions treated with and without added nitrate. T-RFLP fingerprinting data indicated that a major peak at 117 bp was present in all toluene-amended samples (see Fig. S1 in the supplemental material). The 117-bp fragment was highly enriched (especially relative to other 16S rRNA signals) in the heavy DNA fraction (see Fig. S1B) and absent in DNA from the no-nitrate-treated samples. Clone libraries of 16S rRNA genes were constructed to identify the terminal restriction fragments (T-RFs). Sequence analysis showed that clones with a T-RF size of 117 bp were affiliated with the 16S rRNA gene sequences of the genus Herminiimonas, which belongs to the family Oxalobacteraceae within the order Burkholderiales (Fig. 2; see also Table S2 in the supplemental material). Although members of the order Burkholderiales are broadly capable of aerobically metabolizing aromatic compounds (48), to our knowledge, anaerobic aromatic metabolism has not previously been reported in this order. Of the 23 clones, 19 belonged to a single phylotype of Herminiimonas, sharing 99.7% 16S rRNA gene identity with each other (these were assumed to be identical, considering PCR-induced sequencing variation and possible multiple copies of rRNA genes [49] [see Table S2]). Only two clones were affiliated with Betaproteobacteria but not in the clades of Oxalobacteraceae. Sequences similar to the Herminiimonas 16S rRNA gene sequence (98.5% and 97.2% identity, respectively) were detected in an anthracene-SIP experiment (50) and in a radionuclide-contaminated aquifer sediment found near well GW-836 at the Oak Ridge National Laboratory in Tennessee (51). The phylogenetic positions of all obtained clone sequences are shown in Fig. S2 in the supplemental material.
FIG 2.

Phylogenetic analysis of the dominant bacterial 16S rRNA gene sequence derived from the heavy DNA fraction. The phylogenetic tree was constructed using the neighbor-joining method. The bootstrap values at each node are the percentage of 1,000 replicates. Bar, 0.2 changes per nucleotide position. Sequences highlighted with a gray box indicate denitrifying toluene degraders.
Metagenome sequencing of 13C-labeled DNA, assembly, and binning.
To characterize the genetic properties of the nitrate-reducing, toluene-degrading microorganisms, the heavy [13C]DNA was sequenced using 454 pyrosequencing (Roche). A half-plate GS-FLX Titanium run yielded 683,701 reads containing 218.5 Mbp of sequence, with an average read length of 320 bp.
The microbial community composition was inferred from single raw reads of the 16S rRNA genes (n = 293) obtained from the metagenome data set. Betaproteobacteria were common (94.9%) (Table 1), with the most abundant family being Oxalobacteraceae (84% of the total 16S rRNA genes identified). Because the genus Herminiimonas belongs to the family Oxalobacteraceae, the tally of metagenomic 16S rRNA genes provided strong independent support of our conclusions derived from the T-RFLP analysis and clone library, namely, that Herminiimonas species were active in the toluene-degrading microcosm.
TABLE 1.
Taxonomic distribution of bacterial 16S rRNA gene sequences in the heavy DNA fraction obtained from the CN microcosm (n = 293)
| Phylogenetic group | Raw read count (%) |
|---|---|
| Alphaproteobacteria | |
| Xanthobacteraceae | 1 (0.3) |
| Betaproteobacteria | |
| Comamonadaceae | 5 (1.7) |
| Oxalobacteraceaea | 246 (84.0) |
| Rhodocyclaceae | 1 (0.3) |
| Unclassified Burkholderiales | 26 (8.9) |
| Deltaproteobacteria | |
| Bacteriovoracaceae | 1 (0.3) |
| Gammaproteobacteria | |
| Chromatiaceae | 1 (0.3) |
| Pseudomonadaceae | 2 (0.7) |
| Flavobacteriia | |
| Flavobacteriaceae | 3 (1.0) |
| Holophagae | |
| Holophagaceae | 5 (1.7) |
| Unclassified Bacteria | 2 (0.7) |
The dominant phylotype and its read count are shown in boldface.
The assembled contigs were sorted into bins based on GC content, tetranucleotide frequency, and open reading frame (ORF) annotation. The results of the principal component analysis of tetranucleotide frequencies are shown in Fig. 3. The ORF sequences of 131 selected contigs yielded consistent BLAST hits to genes affiliated with Betaproteobacteria (average of 88.8% of total ORFs within each contig) and Oxalobacteraceae (average of 59.7% of total ORFs within each contig). The above-described analysis of T-RFLP and the clone library of the 16S rRNA genes, together with the analysis of the metagenomic raw 16S rRNA reads from [13C]DNA, indicated that a single phylotype of Betaproteobacteria (Oxalobacteraceae) was dominant. Thus, the binned contigs likely originated from a single bacterium that was dominant in our microcosm.
FIG 3.

Principal component analysis of tetranucleotide frequencies of the CN genome and related bacterial genomes. The 131 contigs of the CN genome are shown as red circles, and the combined contigs are indicated by a pink circle. Reference genomes are indicated in yellow (Herminiimonas arsenicoxydans ULPAs1; NC_009138), green (Janthinobacterium sp. Marseille; NC_009659), blue (Herbaspirillum seropedicae SmR1; NC_014323), and sky blue (Aromatoleum aromaticum EbN1; NC_006513) circles. The genome size and G+C content are indicated below the appropriate strain name.
Next, we assembled a CN draft Herminiimonas genome (henceforth referred to as the CN genome) for the uncultivated, toluene-degrading, nitrate-reducing bacterium. No 16S rRNA or phylogenetic genes on the contigs were affiliated with other organisms. The CN genome consisted of 131 contigs, amounting to 3.38 Mbp, with a total of 3,196 coding sequences. The mean coverage depth of the CN genome was 52.3×, with an overall GC content of 58.8%. The GC content of Herminiimonas strains ranges from 52.2 to 59.0%. To check the quality completeness and potential contamination of the reads contributing to the CN genome, we examined the draft genome for the presence of 107 key proteins conserved in 95% of all sequenced bacteria (26), and we detected 75 (70.1%). Furthermore, no duplicated single-copy genes were observed. We are confident that the CN genome assembly matched the dominant toluene-degrading bacterium, because the 16S rRNA gene contained in contig 00013 harbored a T-RF fragment (117 bp) matching the major peak shown in Fig. 2. Genomic comparison with Herminiimonas arsenicoxydans and Herbaspirillum seropedicae using reciprocal ANI revealed 73.7 to 74.5% identity. The broad genomic characteristics of the CN genome obtained from our metagenomic data set are compared with the genomic characteristics of three closely related bacteria in Table S3 in the supplemental material. The CN genome size was comparable with that of H. arsenicoxydans but was smaller than that of H. seropedicae. However, the total number of tRNA genes in the CN genome was lower than in both H. arsenicoxydans and H. seropedicae. These results indicate that the CN genome reported here is indeed a draft genome with an incomplete tally of genes, perhaps as a consequence of our strict binning process. Nonetheless, because a substantial proportion of the genome was successfully assembled, we proceeded to analyze aromatic hydrocarbon metabolic pathways to assess the genetic capability of this uncultivated bacterium to metabolize aromatic hydrocarbons.
Aromatic hydrocarbon degradation.
Pathways for aromatic hydrocarbon compound degradation reconstructed from the CN genome are shown in the schematic in Fig. 4. The contigs harboring genes involved in the degradation of aromatic hydrocarbon compounds are summarized in Table S4 in the supplemental material.
FIG 4.

Aromatic hydrocarbon degradation pathways reconstructed from the CN genome. Enzymes: BclA, benzoyl-CoA ligase; BzdNOPQ, benzoyl-CoA reductase class I; Dch, dienoyl-CoA hydratase; Had, hydroxyacyl-CoA dehydrogenase; Oah, oxoacyl-CoA hydrolase; PchCF, p-cresol methylhydroxylase; Pdh, p-hydroxybenzaldehyde dehydrogenase; Hcl, 4-hydroxybenzoate-CoA ligase; HcrABC, 4-hydroxybenzoyl-CoA reductase; BssABC, benzylsuccinate synthase; BbsEF, benzylsuccinate CoA-transferase; BbsG, benzylsuccinyl-CoA dehydrogenase; BbsH, phenylitaconyl-CoA hydratase; BbsCD, alcohol dehydrogenase; BbsAB, benzoylsuccinyl-CoA thiolase; PadJ, phenylacetate-CoA ligase; PadBCD, phenylacetyl-CoA:acceptor oxidoreductase; PadEFGHI, phenylglyoxylate:NAD+ oxidoreductase; BoxAB, benzoyl-CoA oxygenase; BoxC, benzoyl-CoA dihydrodiol lyase; BoxD, NADP+-specific aldehyde dehydrogenase; BoxE, β-ketoadipyl-CoA thiolase; AliA, cyclohexanecarboxylate-CoA ligase; AliB, cyclohexanecarboxylate-CoA dehydrogenase; BadK, cyclohex-1-ene-1-carboxy-CoA hydratase; BadH, 2-hydroxycyclohexane carboxyl-CoA dehydrogenase; BadI, 2-ketocyclohexane carboxyl-CoA hydrolase. The dashed lines indicate reactions involving several steps.
Toluene.
The clusters of putative bss and bbs genes responsible for toluene degradation were located on contig 00056 and contig 00041 of the CN genome, respectively (Fig. 5). The organization of the bss and bbs gene clusters in the CN genome was similar to that in A. aromaticum EbN1 and Azoarcus sp. CIB. However, the sequence of the key putative gene for toluene degradation, bssA, matched most closely to that of Magnetospirillum sp. TS-6 (92% amino acid similarity) within the Alphaproteobacteria. The putative bssH gene was located on another contig (contig 00037; 30.3 kb). BssH has been suggested to export toluene or toluene-derived metabolites (e.g., benzylsuccinate) (52). Since the genomes of Geobacter spp. do not contain the bssH gene, it may be not essential for toluene degradation. In the A. aromaticum EbN1 genome, five characteristic genes are located downstream of bssH. Four of these five genes (homologs of c2B001, c2A200, c2A203, and c2A204) were conserved in contig 00056 and were located downstream of the bss gene cluster. However, the relevance of these genes in anaerobic toluene catabolism and whether they belong to the bss gene cluster remain unknown (52). As in Geobacter metallireducens (an iron-reducing Deltaproteobacteria strain), the bbsJ (encodes a toluene-induced protein of unknown function), and bbsI genes were not found in the bbs gene cluster of the CN genome, despite conservation of these genes in the bss gene clusters of A. aromaticum EbN1, Azoarcus sp. CIB, and T. aromatica K172.
FIG 5.

Comparison of toluene degradation gene clusters between the CN genome and related bacterial genomes.
p-Cresol.
The CN genome harbors single copies of the putative pchFC (coding for p-cresol methylhydroxylase) and pdh (coding for p-hydroxybenzaldehyde dehydrogenase) genes responsible for the degradation of p-cresol. The pchFC gene was identified as involved in p-ethylphenol methylhydroxylase activity in A. aromaticum EbN1 (53). However, homologs of the genes involved in the successive steps for p-ethylphenol degradation in A. aromaticum EbN1 were not found in the CN genome. Although most of the putative genes involved in aromatic compound degradation were highly similar to those of A. aromaticum EbN1 (see Table S4 in the supplemental material), the putative pchFC sequence best matched that of an autotrophic nitrite-oxidizing bacterium (71 to 80% amino acid sequence similarity), Nitrospira defluvii (see Table S4 and Fig. S3 in the supplemental material).
4-Hydroxybenzoate.
HbcL (4-hydroxybenzoate-CoA ligase) is the initial 4-hydroxybenzoate-activating enzyme. Two putative hbcL copies were encoded in a single contig within the CN genome. The second step in 4-hydroxybenzoate degradation is performed by 4-hydroxybenzoyl-CoA reductase (HcrCAB), which is putatively encoded in another contig. Putative ORFs encoding maturation factor of the 4-hydroxybenzoyl-CoA reductase and 4-hydroxybenzoate transporter were located near the hcrCAB genes (see Fig. S4 in the supplemental material). This arrangement is similar to that for the A. aromaticum EbN1 genome, in which the hcrL and hcrABC genes are also not linked in a cluster. The putative korAB genes, which may be involved in regeneration of the ferredoxin of the BCR system, were clustered with putative hcrCAB genes (see Table S4 in the supplemental material; see also “Benzoate and benzoyl-CoA,” below). According to Wöhlbrand et al., the regenerated ferredoxin may be used by both the BCR system (see below) and 4-hydroxybenzoyl-CoA reductase (54).
Phenylacetate.
Phenylacetate degradation is initiated by phenylacetate-CoA ligase. Two copies of putative phenylacetate-CoA ligase genes and two sets of putative phenylacetate ABC transporter genes were identified in the CN genome (see Table S4 and Fig. S5 in the supplemental material). The putative phenylacetate ABC transporter genes were homologous to those in Herbaspirillum spp. and D. aromatica RCB (see Table S4). The CN genome included a pad operon, which included putative padBCD (encoding the phenylacetyl-CoA:acceptor oxidoreductase) and padEFGHI (coding for phenylglyoxylate:acceptor oxidoreductase) genes, while a putative padB gene was observed in another contig. An ORF homologous to thioesterase genes was observed downstream of the putative padC gene; the encoded protein may be involved in the enzymatic release of CoA from phenylglyoxylyl-CoA, which is formed by phenylacetyl-CoA:acceptor oxidoreductase (55). In the A. aromaticum EbN1 genome, the gene encoding thioesterase was located near the padJ gene.
Benzoate and benzoyl-CoA.
The Herminiimonas genus has not been reported to harbor strains that degrade aromatic hydrocarbons under aerobic or anoxic conditions, and no corresponding genes have been noted in their genomes (56). However, we identified both anaerobic and aerobic benzoate degradation pathways in the CN genome (see Table S4).
As the first step of the aerobic benzoate degradation pathway in the CN genome, benzoate is likely catalyzed by benzoate-CoA ligase (encoded by a putative bclA gene) to benzoyl-CoA. A series of aerobic reactions catalyzed by BoxABC degrades benzoyl-CoA to acetyl-CoA and succinyl-CoA. The order of putative box genes in the CN genome cluster resembled that in A. aromaticum EbN1 (Fig. 6). However, the amino acid sequences of putative boxBCZ genes in the CN genome were similar to those of Cupriavidus spp. within the family Burkholderiaceae (75 to 90% similarity [see Table S4]). A. aromaticum EbN1 carries two putative bclA gene copies: one (ebA2757) is within the putative box gene cluster, and the other (ebA5301) is located near the bcr gene cluster. However, T. aromatica and Magnetospirillum strains contain only one bclA gene and may use the same benzoate-CoA ligase (BclA) for both the aerobic and anaerobic pathways (57, 58). While the bclA gene from T. aromatica is located in the box gene cluster for aerobic benzoate degradation (58), the bclA gene in Magnetospirillum strains is not linked to the bcr or box clusters (57) but is instead located within a gene cluster that encodes an ABC transporter. One putative bclA gene was detected in the CN genome, and its amino acid sequence was similar to ebA5301 (80%) of A. aromaticum EbN1. The putative bclA gene of the CN genome was located within a cluster of putative genes encoding a benzoate ABC transporter that matches most closely one carried by Magnetospirillum sp. Therefore, aryl-CoA (benzoyl-CoA) may be a common intermediate of aromatic hydrocarbon degradation under both oxygenated and anoxic conditions. This strategy is considered an adaptation by some facultative denitrifiers to fluctuating oxygenated and anoxic conditions (59). Harboring a ring reducing system that functions under both oxygenated and nitrate-reducing conditions may confer an energetic advantage to strain CN, since the BOX system uses two fewer ATPs than the BCR system (see below).
FIG 6.

Gene clusters involved in the aerobic degradation of benzoyl-CoA identified in the CN, Aromatoleum aromaticum EbN1, and Cupriavidus necator N-1 genomes.
Benzoyl-CoA is a central intermediate in anaerobic aromatic degradation. Benzoyl-CoA is reductively dearomatized by the BCR system under nitrate-reducing conditions. The CN genome contains all the putative genes required for the anaerobic benzoyl-CoA pathway, including those encoding four BCR subunits (bzdNOPQ), ferredoxin (bzdM), the reduced ferredoxin-regenerating system (korAB and bzdV), and a modified β-oxidation pathway (dch, had, and oah). Two types of ATP-dependent BCR enzymes that share an ancestor have been proposed based on amino acid sequence comparison: (i) the bcr-type BCR present in T. aromatica, Rhodopseudomonas palustris, and Magnetospirillum spp.; (ii) the bzd-type BCR present in Azoarcus spp. (18, 60). The sequence of the putative benzoyl-CoA reductase in the CN genome matched closely that of the bzd-type BCR. The BCR system overcomes energetic limitations by using the low-potential electron donor ferredoxin and by coupling ATP-assisted electron transfer to benzoyl-CoA. As described earlier, ORFs encoding 2-oxoglutarate:ferredoxin oxidoreductase (KGOR, regenerating reduced ferredoxins; korAB) are located in the CN genome near the putative hcrCAB instead of near BCR. Although korABC is encoded in a cluster in the A. aromaticum EbN1 genome, only putative korAB genes were found in the CN genome, much like the M. magneticum and T. aromatica genomes (11, 61).
After the formation of cyclohex-1-ene-1-carboxyl-CoA, degradation likely proceeds through subsequent β-oxidation-like steps. Two different β-oxidation types, the Rhodopseudomonas type (BadK, BadH, and BadI) and the Thauera type (Dch, Had, and Oah), have been reported (19). The genomes of A. aromaticum EbN1 and CN contain both types of gene homologs (Fig. 4; see also Table S4 in the supplemental material). The Thauera type and Rhodopseudomonas type of the β-oxidation pathways generate 3-hydroxy-pimelyl-CoA and pimelyl-CoA as the final products, respectively. These final products are further degraded into three molecules of acetyl-CoA and one molecule of CO2, through a dicarboxylic acid β-oxidation pathway with a glutaryl-CoA dehydrogenase and a short-chain fatty acid β-oxidation pathway (2).
The genus Herbaspirillum, a close relative of the genus Herminiimonas, contains a species that harbors genes for dioxygenase systems for benzoate, benzamide, benzonitrile, hydroxyl-benzoate, and vanillate and for further aerobic degradation of the oxygenated products (62). Only two ORFs homologous to those encoding extradiol ring cleavage dioxygenase subunits were detected in the CN genome. No further aerobic degradation pathway genes for oxygenated products were detected. Thus, it appears that the canonical oxygenase-dependent aerobic degradation pathways for aromatic compounds might not be operational in the CN bacterium.
Cyclohexane carboxylate.
Putative genes encoding cyclohexane carboxylate-CoA ligase (AliA) and cyclohexanecarboxyl-CoA dehydrogenase (AliB) were identified in the CN genome. These enzymes activate cyclohexane carboxylate to cyclohexenecarbonyl-CoA. Further degradation of cyclohexenecarbonyl-CoA may be facilitated by the putative badIHR (as in the Rhodopseudomonas-type β-oxidation pathway) and adjacent aliAB genes (see Fig. S6 in the supplemental material). Cyclohexane carboxylate degradation-related genes were also observed in A. aromaticum EbN1, but the amino acid sequences encoded by the corresponding CN genes were more similar to those of Polaromonas naphthalenivorans CJ2 and Leptothrix cholodnii SP 6, which fall taxonomically into the class Burkholderiales (see Table S4).
Horizontal gene transfer.
Most of the core CN housekeeping genes showed greatest similarity to those from genomes in the same family, Oxalobacteraceae. However, the putative aromatic hydrocarbon degradation genes were most similar in sequence and organization to A. aromaticum EbN1, Azoarcus spp., T. aromatica K172, and D. aromatica RCB, which group taxonomically with the family Rhodocyclaceae. The inability of the closest relatives of the CN bacterium, Herminiimonas/Herbaspirillum, to anaerobically degrade aromatic hydrocarbons suggests that the required genes may have been acquired from unrelated taxa via horizontal gene transfer (HGT). In support of this hypothesis, we found that putative mobile elements known to facilitate gene transfer were highly abundant in the 3.38-Mb genome of CN (25 elements/Mb). These included 18 integrases/recombinases, 21 transposases, 3 terminases, 2 resolvases, and 41 phage-related proteins. Particularly germane to this issue, putative transposase and integrase genes were directly linked with the bss and bbs gene clusters (see Table S4 in the supplemental material). A. aromaticum EbN1 similarly exhibits high numbers of transposon-related genes (237; 50/Mb; (14), while the genome of Pseudomonas putida, an aerobic aromatic degrader, contains 96 transposon-related genes in its 6.2-Mb genome (15 elements/Mb) (14). Such trends suggest that HGT is likely a widespread mechanism that confers an aromatic compound degradation ability on many host taxa. Further studies on the nature of gene transfer systems are required to fully understand the transfer and evolution of aromatic hydrocarbon metabolic pathways.
Respiratory system.
Oxygen and nitrate respiratory systems were found in the CN genome (see Table S5 in the supplemental material). Putative genes for complex I (NADH:ubiquinone oxidoreductase), complex II (succinate dehydrogenase), and complex III (cytochrome bc1 complex) in the CN genome are likely involved in oxygen and nitrate respiration. Two terminal cytochrome c oxidase genes were found, allowing oxygen to serve as a terminal electron acceptor. One was an aa3-type cytochrome c oxidase (CoxABC) and the other was a cbb3-type cytochrome c oxidase (CcoNOP). The cytochrome aa3-type cytochrome c oxidase operates at high oxygen concentrations, whereas the cytochrome cbb3-type cytochrome c oxidase works at low oxygen concentrations. The presence of putative terminal oxidases with different affinities for oxygen suggests that the CN genome is capable of adapting to varying external oxygen concentrations. Although the H. arsenicoxydans genome encodes cytochrome bd quinol oxidase (CydAB), which operates in low oxygen, only the putative cydB gene was observed in the CN genome.
To date, the Herminiimonas and Herbaspirillum genera have not been shown to harbor the complete gene set needed for denitrification. For example, the genome of H. arsenicoxydans does not encode nitrous oxide reductase, which is required for the final denitrification, and the only denitrification genes found in H. seropedicae are those encoding nitrate reductases. However, the CN genome contained putative genes encoding the four necessary enzymes that catalyze denitrification: Nar (nitrate reductase), Nir (nitrite reductase), Nor (nitric oxide reductase), and Nos (nitrous oxide reductase) (see Table S5). These genes were most similar to homologs from Thiobacillus denitrificans within the order Hydrogenophilales. Three copies of putative nitrate/nitrite transporters were present in the CN genome, two of which were located upstream of the putative nar gene (dissimilatory nitrate reductase) cluster, while the third was located with the assimilatory nitrite reductase (NirB) genes (see Table S5).
Detoxification systems.
H. arsenicoxydans is resistant to arsenic (via the arsenic efflux pump encoded by ars) and can oxidize arsenite (56). Arsenite oxidation is also viewed as a detoxification mechanism, because arsenate is less toxic than arsenite. No putative aox (coding for energy-generating arsenite oxidase) gene was identified in the CN genome. Putative genes encoding an arsenic efflux pump (arsRDACB) were identified (see Table S6 in the supplemental material). However, a homolog of the arsH gene, encoding flavoprotein (unknown function), was absent. The ars operon of the CN genome is more similar to that of Eshcerichia coli (63) than to those of H. arsenicoxydans (see Fig. S7 in the supplemental material) or A. aromaticum EbN1 (15); notably, other bacteria also carry ars system genes derived from phylogenetically distant taxa (63). In addition to resistance to arsenic, the CN genome carries putative genes encoding proteins responsible for oxidative stress resistance (see Table S6), such as catalase and superoxide dismutase. Other putative reactive oxygen-responsive genes found in the CN genome include two thioredoxin reductases, a hydroperoxide reductase, a hemerythrin, a bacterioferritin, and a bacterioferritin comigratory protein.
Central carbon metabolism.
The CN genome contained the complete set of putative genes for the Embden-Meyerhof-Parnas (EMP) and pentose phosphate pathways. However, the Entner-Doudoroff (ED) pathway may be incomplete, because no putative genes encoding 6-phosphogluconate dehydratase and 4-hydroxy-2-oxoglutarate aldolase were found; this will be resolved by genome completion. The CN genome contained all the putative genes for the tricarboxylic acid cycle and glyoxylate cycle and putative genes for the key gluconeogenesis enzymes phosphoenolpyruvate carboxykinase and pyruvate carboxylase. However, H. arsenicoxydans and H. seropedicae do not harbor a gene coding for pyruvate carboxylase. Most of the central pathway genes in the CN genome were found to be in common with members of the family Oxalobacteraceae.
Conclusions.
The SIP-based metagenomic approach allowed us to reconstruct metabolic pathways for anaerobic degradation in a new, uncultivated, toluene-degrading denitrifying bacterium. More than 93 genes within the CN genome were devoted to the anaerobic catabolism of aromatic compounds, suggesting a possible life strategy for this organism that emphasizes assimilation of a variety of aromatic substrates. Many of the genes in strain CN, including those involved in aromatic hydrocarbon degradation and denitrification, had not previously been reported in the Oxalobacteraceae clade. The absence of these genes among close phylogenetic relatives, together with a high density of mobile genetic elements, suggest that HGT has likely played an important role in the evolution of the CN genome. The ability to link anaerobic degradation of aromatic hydrocarbons to denitrification is a putative key physiological feature that distinguishes the new toluene degrader from other Herminiimonas species.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Mid-Career Researcher Program NRF-2013R1A2A2A05006754 and the Basic Science Research Program (2012R1A1A2A10039384) through a National Research Foundation (NRF) grant, which was funded by the Ministry of Education, Science, and Technology (MEST), National Institute of Biological Resources (NIBR) program (2013-02-055), funded by the Ministry of Environment (MOE) and Chungbuk National University Grant in 2012, and by the BK21 Plus program. E.L.M. was supported by the U.S. National Science Foundation, grant DEB-0841999.
Footnotes
Published ahead of print 14 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03975-13.
REFERENCES
- 1.Weelink SAB, van Eekert MHA, Stams AJM. 2010. Degradation of BTEX by anaerobic bacteria: physiology and application. Rev. Environ. Sci. Biotechnol. (NY) 9:359–385. 10.1007/s11157-010-9219-2 [DOI] [Google Scholar]
- 2.Carmona M, Zamarro MT, Blazquez B, Durante-Rodriguez G, Juarez JF, Valderrama JA, Barragan MJ, Garcia JL, Diaz E. 2009. Anaerobic catabolism of aromatic compounds: a genetic and genomic view. Microbiol. Mol. Biol. Rev. 73:71–133. 10.1128/MMBR.00021-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Washer CE, Edwards EA. 2007. Identification and expression of benzylsuccinate synthase genes in a toluene-degrading methanogenic consortium. Appl. Environ. Microbiol. 73:1367–1369. 10.1128/AEM.01904-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hurek T, Reinhold-Hurek B. 1995. Identification of grass-associated and toluene-degrading diazotrophs, Azoarcus spp., by analyses of partial 16S ribosomal DNA sequences. Appl. Environ. Microbiol. 61:2257–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou J, Fries MR, Chee-Sanford JC, Tiedje JM. 1995. Phylogenetic analyses of a new group of denitrifiers capable of anaerobic growth of toluene and description of Azoarcus tolulyticus sp. nov. Int. J. Syst. Bacteriol. 45:500–506 [DOI] [PubMed] [Google Scholar]
- 6.Fries MR, Zhou J, Chee-Sanford J, Tiedje JM. 1994. Isolation, characterization, and distribution of denitrifying toluene degraders from a variety of habitats. Appl. Environ. Microbiol. 60:2802–2810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rabus R, Widdel F. 1995. Anaerobic degradation of ethylbenzene and other aromatic hydrocarbons by new denitrifying bacteria. Arch. Microbiol. 163:96–103 [DOI] [PubMed] [Google Scholar]
- 8.Weelink SA, van Doesburg W, Saia FT, Rijpstra WI, Roling WF, Smidt H, Stams AJ. 2009. A strictly anaerobic betaproteobacterium Georgfuchsia toluolica gen. nov., sp. nov. degrades aromatic compounds with Fe(III), Mn(IV) or nitrate as an electron acceptor. FEMS Microbiol. Ecol. 70:575–585. 10.1111/j.1574-6941.2009.00778.x [DOI] [PubMed] [Google Scholar]
- 9.Schocher RJ, Seyfried B, Vazquez F, Zeyer J. 1991. Anaerobic degradation of toluene by pure cultures of denitrifying bacteria. Arch. Microbiol. 157:7–12 [DOI] [PubMed] [Google Scholar]
- 10.Coates JD, Chakraborty R, Lack JG, O'Connor SM, Cole KA, Bender KS, Achenbach LA. 2001. Anaerobic benzene oxidation coupled to nitrate reduction in pure culture by two strains of Dechloromonas. Nature 411:1039–1043. 10.1038/35082545 [DOI] [PubMed] [Google Scholar]
- 11.Shinoda Y, Akagi J, Uchihashi Y, Hiraishi A, Yukawa H, Yurimoto H, Sakai Y, Kato N. 2005. Anaerobic degradation of aromatic compounds by Magnetospirillum strains: isolation and degradation genes. Biosci. Biotechnol. Biochem. 69:1483–1491. 10.1271/bbb.69.1483 [DOI] [PubMed] [Google Scholar]
- 12.Alain K, Harder J, Widdel F, Zengler K. 2012. Anaerobic utilization of toluene by marine alpha- and gammaproteobacteria reducing nitrate. Microbiology 158:2946–2957. 10.1099/mic.0.061598-0 [DOI] [PubMed] [Google Scholar]
- 13.Sun W, Cupples AM. 2012. Diversity of five anaerobic toluene-degrading microbial communities investigated using stable isotope probing. Appl. Environ. Microbiol. 78:972–980. 10.1128/AEM.06770-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rabus R. 2005. Functional genomics of an anaerobic aromatic-degrading denitrifying bacterium, strain EbN1. Appl. Microbiol. Biotechnol. 68:580–587. 10.1007/s00253-005-0030-x [DOI] [PubMed] [Google Scholar]
- 15.Rabus R, Kube M, Heider J, Beck A, Heitmann K, Widdel F, Reinhardt R. 2005. The genome sequence of an anaerobic aromatic-degrading denitrifying bacterium, strain EbN1. Arch. Microbiol. 183:27–36. 10.1007/s00203-004-0742-9 [DOI] [PubMed] [Google Scholar]
- 16.Salinero KK, Keller K, Feil WS, Feil H, Trong S, Di Bartolo G, Lapidus A. 2009. Metabolic analysis of the soil microbe Dechloromonas aromatica str. RCB: indications of a surprisingly complex life-style and cryptic anaerobic pathways for aromatic degradation. BMC Genomics 10:351. 10.1186/1471-2164-10-351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuchs G, Boll M, Heider J. 2011. Microbial degradation of aromatic compounds: from one strategy to four. Nat. Rev. Microbiol. 9:803–816. 10.1038/nrmicro2652 [DOI] [PubMed] [Google Scholar]
- 18.Song B, Ward BB. 2005. Genetic diversity of benzoyl coenzyme A reductase genes detected in denitrifying isolates and estuarine sediment communities. Appl. Environ. Microbiol. 71:2036–2045. 10.1128/AEM.71.4.2036-2045.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harwood CS, Burchhardt G, Herrmann H, Fuchs G. 1998. Anaerobic metabolism of aromatic compounds via the benzoyl-CoA pathway. FEMS Microbiol. Rev. 22:439–458 [Google Scholar]
- 20.Lopez Barragán MJ, Carmona M, Zamarro MT, Thiele B, Boll M, Fuchs G, García JL, Díaz E. 2004. The bzd gene cluster, coding for anaerobic benzoate catabolism, in Azoarcus sp. strain CIB. J. Bacteriol. 186:5762–5774. 10.1128/JB.186.17.5762-5774.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gescher J, Zaar A, Mohamed M, Schagger H, Fuchs G. 2002. Genes coding for a new pathway of aerobic benzoate metabolism in Azoarcus evansii. J. Bacteriol. 184:6301–6315. 10.1128/JB.184.22.6301-6315.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gescher J, Eisenreich W, Worth J, Bacher A, Fuchs G. 2005. Aerobic benzoyl-CoA catabolic pathway in Azoarcus evansii: studies on the non-oxygenolytic ring cleavage enzyme. Mol. Microbiol. 56:1586–1600. 10.1111/j.1365-2958.2005.04637.x [DOI] [PubMed] [Google Scholar]
- 23.Ebenau-Jehle C, Boll M, Fuchs G. 2003. 2-Oxoglutarate:NADP(+) oxidoreductase in Azoarcus evansii: properties and function in electron transfer reactions in aromatic ring reduction. J. Bacteriol. 185:6119–6129. 10.1128/JB.185.20.6119-6129.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iverson V, Morris RM, Frazar CD, Berthiaume CT, Morales RL, Armbrust EV. 2012. Untangling genomes from metagenomes: revealing an uncultured class of marine Euryarchaeota. Science 335:587–590. 10.1126/science.1212665 [DOI] [PubMed] [Google Scholar]
- 25.Wrighton KC, Thomas BC, Sharon I, Miller CS, Castelle CJ, VerBerkmoes NC, Wilkins MJ, Hettich RL, Lipton MS, Williams KH, Long PE, Banfield JF. 2012. Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337:1661–1665. 10.1126/science.1224041 [DOI] [PubMed] [Google Scholar]
- 26.Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH. 2013. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 31:533–538. 10.1038/nbt.2579 [DOI] [PubMed] [Google Scholar]
- 27.Kalyuzhnaya MG, Lapidus A, Ivanova N, Copeland AC, McHardy AC, Szeto E, Salamov A, Grigoriev IV, Suciu D, Levine SR, Markowitz VM, Rigoutsos I, Tringe SG, Bruce DC, Richardson PM, Lidstrom ME, Chistoserdova L. 2008. High-resolution metagenomics targets specific functional types in complex microbial communities. Nat. Biotechnol. 26:1029–1034. 10.1038/nbt.1488 [DOI] [PubMed] [Google Scholar]
- 28.Kim SJ, Park SJ, Cha IT, Min D, Kim JS, Chung WH, Chae JC, Jeon CO, Rhee SK. 2014. Metabolic versatility of toluene-degrading, iron-reducing bacteria in tidal flat sediment, characterized by stable isotope probing-based metagenomic analysis. Environ. Microbiol. 16:189–204. 10.1111/1462-2920.12277 [DOI] [PubMed] [Google Scholar]
- 29.Jeon CO, Park W, Padmanabhan P, DeRito C, Snape JR, Madsen EL. 2003. Discovery of a bacterium, with distinctive dioxygenase, that is responsible for in situ biodegradation in contaminated sediment. Proc. Natl. Acad. Sci. U. S. A. 100:13591–13596. 10.1073/pnas.1735529100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Widdel F, Bak F. 1992. Gram-negative mesophilic sulfate-reducing bacteria, p 3352–3378 In Balows A, Tr̈uper HG, Dworkin M, Harder W, Schleifer K-H. (ed), The prokaryotes, 2nd ed, vol 4 Springer, New York, NY [Google Scholar]
- 31.Tschech A, Pfennig N. 1984. Growth yield increase linked to caffeate reduction in Acetobacterium woodii. Arch. Microbiol. 137:163–167 [Google Scholar]
- 32.Shinn MB. 1941. Colorimetric method for determination of nitrite. Ind. Eng. Chem. Anal. Ed. 13:33–35 [Google Scholar]
- 33.Zhou J, Bruns MA, Tiedje JM. 1996. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 62:316–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neufeld JD, Vohra J, Dumont MG, Lueders T, Manefield M, Friedrich MW, Murrell JC. 2007. DNA stable-isotope probing. Nat. Protoc. 2:860–866. 10.1038/nprot.2007.109 [DOI] [PubMed] [Google Scholar]
- 35.Lane D. 1991. 16S/23S rRNA sequencing, p 115–175 In Stackebrandt E, Goodfellow M. (ed), Nucleic acid techniques in bacterial systematics. John Wiley & Sons, Chichester, England [Google Scholar]
- 36.Kim SJ, Koh DC, Park SJ, Cha IT, Park JW, Na JH, Roh Y, Ko KS, Kim K, Rhee SK. 2012. Molecular analysis of spatial variation of iron-reducing bacteria in riverine alluvial aquifers of the Mankyeong River. J. Microbiol. 50:207–217. 10.1007/s12275-012-1342-z [DOI] [PubMed] [Google Scholar]
- 37.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO. 2007. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35:7188–7196. 10.1093/nar/gkm864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kunin V, Copeland A, Lapidus A, Mavromatis K, Hugenholtz P. 2008. A bioinformatician's guide to metagenomics. Microbiol. Mol. Biol. Rev. 72:557–578. 10.1128/MMBR.00009-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. 2009. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37(Database issue):D141–D145. 10.1093/nar/gkn879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Noguchi H, Taniguchi T, Itoh T. 2008. MetaGeneAnnotator: detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res. 15:387–396. 10.1093/dnares/dsn027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. 2007. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35:3100–3108. 10.1093/nar/gkm160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. 2000. Artemis: sequence visualization and annotation. Bioinformatics 16:944–945. 10.1093/bioinformatics/16.10.944 [DOI] [PubMed] [Google Scholar]
- 45.Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16:276–277. 10.1016/S0168-9525(00)02024-2 [DOI] [PubMed] [Google Scholar]
- 46.Ghai R, Pasic L, Fernandez AB, Martin-Cuadrado AB, Mizuno CM, McMahon KD, Papke RT, Stepanauskas R, Rodriguez-Brito B, Rohwer F, Sanchez-Porro C, Ventosa A, Rodriguez-Valera F. 2011. New abundant microbial groups in aquatic hypersaline environments. Sci. Rep. 1:135. 10.1038/srep00135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Konstantinidis KT, Tiedje JM. 2005. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U. S. A. 102:2567–2572. 10.1073/pnas.0409727102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pérez-Pantoja D, Donoso R, Agulló L, Córdova M, Seeger M, Pieper DH, González B. 2012. Genomic analysis of the potential for aromatic compounds biodegradation in Burkholderiales. Environ. Microbiol. 14:1091–1117. 10.1111/j.1462-2920.2011.02613.x [DOI] [PubMed] [Google Scholar]
- 49.von Wintzingerode F, Gobel UB, Stackebrandt E. 1997. Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol. Rev. 21:213–229 [DOI] [PubMed] [Google Scholar]
- 50.Jones MD, Singleton DR, Sun W, Aitken MD. 2011. Multiple DNA extractions coupled with stable-isotope probing of anthracene-degrading bacteria in contaminated soil. Appl. Environ. Microbiol. 77:2984–2991. 10.1128/AEM.01942-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li X, Krumholz LR. 2008. Influence of nitrate on microbial reduction of pertechnetate. Environ. Sci. Technol. 42:1910–1915. 10.1021/es071164j [DOI] [PubMed] [Google Scholar]
- 52.Kube M, Heider J, Amann J, Hufnagel P, Kuhner S, Beck A, Reinhardt R, Rabus R. 2004. Genes involved in the anaerobic degradation of toluene in a denitrifying bacterium, strain EbN1. Arch. Microbiol. 181:182–194. 10.1007/s00203-003-0627-3 [DOI] [PubMed] [Google Scholar]
- 53.Wöhlbrand L, Wilkes H, Halder T, Rabus R. 2008. Anaerobic degradation of p-ethylphenol by “Aromatoleum aromaticum” strain EbN1: pathway, regulation, and involved proteins. J. Bacteriol. 190:5699–5709. 10.1128/JB.00409-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wöhlbrand L, Kallerhoff B, Lange D, Hufnagel P, Thiermann J, Reinhardt R, Rabus R. 2007. Functional proteomic view of metabolic regulation in Aromatoleum aromaticum strain EbN1. Proteomics 7:2222–2239. 10.1002/pmic.200600987 [DOI] [PubMed] [Google Scholar]
- 55.Rhee SK, Fuchs G. 1999. Phenylacetyl-CoA:acceptor oxidoreductase, a membrane-bound molybdenum-iron-sulfur enzyme involved in anaerobic metabolism of phenylalanine in the denitrifying bacterium Thauera aromatica. Eur. J. Biochem. 262:507–515 [DOI] [PubMed] [Google Scholar]
- 56.Muller D, Medigue C, Koechler S, Barbe V, Barakat M, Talla E, Bonnefoy V, Krin E, Arsene-Ploetze F, Carapito C, Chandler M, Cournoyer B, Cruveiller S, Dossat C, Duval S, Heymann M, Leize E, Lieutaud A, Lievremont D, Makita Y, Mangenot S, Nitschke W, Ortet P, Perdrial N, Schoepp B, Siguier P, Simeonova DD, Rouy Z, Segurens B, Turlin E, Vallenet D, Van Dorsselaer A, Weiss S, Weissenbach J, Lett M-C, Danchin A, Bertin PN. 2007. A tale of two oxidation states: bacterial colonization of arsenic-rich environments. PLoS Genet. 3(4):e53. 10.1371/journal.pgen.0030053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kawaguchi K, Shinoda Y, Yurimoto H, Sakai Y, Kato N. 2006. Purification and characterization of benzoate-CoA ligase from Magnetospirillum sp. strain TS-6 capable of aerobic and anaerobic degradation of aromatic compounds. FEMS Microbiol. Lett. 257:208–213. 10.1111/j.1574-6968.2006.00165.x [DOI] [PubMed] [Google Scholar]
- 58.Schuhle K, Gescher J, Feil U, Paul M, Jahn M, Schagger H, Fuchs G. 2003. Benzoate-coenzyme A ligase from Thauera aromatica: an enzyme acting in anaerobic and aerobic pathways. J. Bacteriol. 185:4920–4929. 10.1128/JB.185.16.4920-4929.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fuchs G. 2008. Anaerobic metabolism of aromatic compounds. Ann. N. Y. Acad. Sci. 1125:82–99. 10.1196/annals.1419.010 [DOI] [PubMed] [Google Scholar]
- 60.Boll M. 2005. Dearomatizing benzene ring reductases. J. Mol. Microbiol. Biotechnol. 10:132–142. 10.1159/000091560 [DOI] [PubMed] [Google Scholar]
- 61.Breese K, Boll M, Alt-Mörbe J, Schägger H, Fuchs G. 1998. Genes coding for the benzoyl-CoA pathway of anaerobic aromatic metabolism in the bacterium Thauera aromatica. Eur. J. Biochem. 256:148–154 [DOI] [PubMed] [Google Scholar]
- 62.Pedrosa FO, Monteiro RA, Wassem R, Cruz LM, Ayub RA, Colauto NB, Fernandez MA, Fungaro MH, Grisard EC, Hungria M, Madeira HM, Nodari RO, Osaku CA, Petzl-Erler ML, Terenzi H, Vieira LG, Steffens MB, Weiss VA, Pereira LF, Almeida MI, Alves LR, Marin A, Araujo LM, Balsanelli E, Baura VA, Chubatsu LS, Faoro H, Favetti A, Friedermann G, Glienke C, Karp S, Kava-Cordeiro V, Raittz RT, Ramos HJ, Ribeiro EM, Rigo LU, Rocha SN, Schwab S, Silva AG, Souza EM, Tadra-Sfeir MZ, Torres RA, Dabul AN, Soares MA, Gasques LS, Gimenes CC, Valle JS, Ciferri RR, Correa LC, Murace NK, et al. 2011. Genome of Herbaspirillum seropedicae strain SmR1, a specialized diazotrophic endophyte of tropical grasses. PLoS Genet. 7(5):e1002064. 10.1371/journal.pgen.1002064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mateos LM, Ordóñez E, Letek M, Gil JA. 2006. Corynebacterium glutamicum as a model bacterium for the bioremediation of arsenic. Int. Microbiol. 9:207–215 http://www.im.microbios.org/0903/0903207.pdf [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.