

Abstract
The health benefits of flavonoids for humans are increasingly attracting attention. Because the extraction of high-purity flavonoids from plants presents a major obstacle, interest has emerged in biosynthesizing them using microbial hosts. Eriodictyol is a flavonoid with anti-inflammatory and antioxidant activities. Its efficient synthesis has been hampered by two factors: the poor expression of cytochrome P450 and the low intracellular malonyl coenzyme A (malonyl-CoA) concentration in Escherichia coli. To address these issues, a truncated plant P450 flavonoid, flavonoid 3′-hydroxylase (tF3′H), was functionally expressed as a fusion protein with a truncated P450 reductase (tCPR) in E. coli. This allowed the engineered E. coli to produce eriodictyol from l-tyrosine by simultaneously coexpressing the fusion protein with tyrosine ammonia lyase (TAL), 4-coumarate-CoA ligase (4CL), chalcone synthase (CHS), and chalcone isomerase (CHI). In addition, metabolic engineering was employed to enhance the availability of malonyl-CoA so as to achieve a new metabolic balance and rebalance the relative expression of genes to enhance eriodictyol accumulation. This approach made the production of eriodictyol 203% higher than that in the control strain. By using these strategies, the production of eriodictyol from l-tyrosine reached 107 mg/liter. The present work offers an approach to the efficient synthesis of other hydroxylated flavonoids from l-tyrosine or even glucose in E. coli.
INTRODUCTION
Flavonoids were first found in plants, and their role is to protect plants against certain adverse environmental effects, such as microbial infection and UV radiation (1). More than 9,000 flavonoids have been found in various plants, comprising one of the largest families of natural products (2). In recent years, the health benefits of flavonoids have attracted increasing attention (3–6). The effects of flavonoids on humans can be attributed to antioxidant, antioncogenic, anti-inflammatory, antimicrobial, anticancer, and cardiovascular actions (1, 7). For example, eriodictyol plays critical roles in the pathogenesis of diabetes mellitus, can inhibit immunoglobulin E (IgE)/antigen (Ag)-induced type I hypersensitivity, and has antinociceptive and hyperthermic effects (8–10).
Although flavonoids offer many benefits to health, efforts to obtain high-purity flavonoids by extraction from plants present a major obstacle (7). Chemical synthesis is associated with toxic by-products and requires extreme reaction conditions, as in the application of the Suzuki-Miyaura reaction (11). Therefore, chemical synthesis is not suitable for the production of flavonoids to be incorporated into food ingredients or cosmetics. Among the microorganisms used for the biosynthesis of natural products, Escherichia coli has been the most intensively investigated host because of its genetic tractability and favorable fermentation properties (12).
Eriodictyol is a flavanone, belonging to the family of flavonoids. Previous studies have already demonstrated the feasibility of eriodictyol production in E. coli. By the overexpression of 4CL from Petroselinum crispum, CHS from Petunia X hybrida, and CHI from Medicago sativa in E. coli, 11 mg/liter of eriodictyol was obtained from caffeic acid (13). Furthermore, when Rhizobium trifolii matB and matC were introduced to increase the intracellular malonyl coenzyme A (malonyl-CoA) concentration, and the recombinant strain was cultured in M9 minimal medium (with 1% glucose) with 2 g/liter sodium malonate and 2 mM caffeic acid, eriodictyol production reached 50 mg/liter (14). Increasing malonyl-CoA biosynthesis by tuning the Escherichia coli metabolic network increased the level of eriodictyol production from caffeic acid to 114 mg/liter (15).
l-Tyrosine was used as the precursor of eriodictyol in this study (Fig. 1). Previous studies showed the feasibility of naringenin and pinocembrin production from l-tyrosine in E. coli (16–18). In this study, naringenin was converted to eriodictyol by flavonoid 3′-hydroxylase (F3′H), together with cytochrome P450 reductase (CPR) (19–21). F3′H is a cytochrome P450 protein and has never been expressed in E. coli before. Functional expression of plant cytochrome P450 in E. coli is a challenge due to various factors, such as solubility and cofactor incorporation (22–24). Therefore, one of the major objectives in the present study was the soluble expression of the fusion protein of truncated flavonoid 3′-hydroxylase (tF3′H) and truncated P450 reductase (tCPR) in E. coli.
FIG 1.

Pathway for the synthesis of eriodictyol from l-tyrosine. (A) Six genes are involved in the eriodictyol synthesis pathway: TAL, encoding tyrosine ammonia lyase, from R. glutinis; 4CL, encoding 4-coumarate-CoA ligase, from P. crispum; CHS, encoding chalcone synthase, from P. hybrida; CHI, encoding chalcone isomerase, from M. sativa; F3′H, encoding flavonoid 3′-hydroxylase, from G. hybrida; and CPR, encoding cytochrome P450 reductase, from C. roseus. (B) Four genes were involved in the biosynthesis of malonyl-CoA: the acetyl-CoA carboxylase (ACC) genes accBC and dtsR1, cloned from C. glutamicum; acs, cloned from E. coli BL21(DE3), encoding acetyl-CoA synthase; and ackA, encoding an acetate kinase, which was deleted from E. coli BL21(DE3). The steps that are the focus of this paper are highlighted.
In this study, this fusion protein (tF3′H-tCPR) was functionally expressed together with tyrosine ammonia lyase (TAL), 4-coumarate-CoA ligase (4CL), chalcone synthase (CHS), and chalcone isomerase (CHI) in E. coli. Because a low level of intracellular malonyl-CoA is a major limitation for flavonoid biosynthesis in E. coli, several strategies were applied to enhance the availability of malonyl-CoA (25, 26). By employing these strategies, the final eriodictyol production was obtained directly from l-tyrosine instead of the expensive substrate caffeic acid. The present work offers an approach to the efficient synthesis of hydroxylated flavonoids in E. coli from l-tyrosine or even glucose.
MATERIALS AND METHODS
Genetic material, microbial hosts, cloning vectors, and plasmids.
E. coli JM109 was used for plasmid propagation. E. coli BL21(DE3) was used to construct the flavonoid biosynthesis strains. The pETDuet-1, pACYCDuet-1, pRSFDuet-1, and pCDFDuet-1 expression vectors were obtained from Novagen (Darmstadt, Germany). All of the four Duet expression vectors were designed for the coexpression of two target open reading frames (ORFs). One vector contains two multiple cloning sites (MCS), each of which is preceded by a T7-lac promoter and a ribosome binding site (RBS) (Table 1). All restriction enzymes and DNA ligase were purchased from TaKaRa (DaLian, China). TAL from Rhodotorula glutinis, 4CL from Petroselinum crispum, CHS from Petunia X hybrida, CHI from Medicago sativa, F3′H from Gerbera hybrida, and CPR from Catharanthus roseus were codon optimized for expression in E. coli (http://www.jcat.de/) and were synthesized by GeneScript (Nanjing, China) (16, 27). The acetyl-CoA carboxylase (ACC) genes, accBC and dtsR1, were cloned from Corynebacterium glutamicum ATCC 13032. The acs gene was cloned from E. coli BL21(DE3). Three plasmids involved in the Red recombinase expression system, pKD46, pKD13, and pCP20, were obtained from the CGSC (http://cgsc.biology.yale.edu/) (28).
TABLE 1.
Plasmids
| Plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| pETDuet-1 | Double T7 promoters, two MCS, ColE1 (pBR322) origin; Ampr | Novagen |
| pCDFDuet-1 | Double T7 promoters, two MCS, CloDF13 origin; Smr | Novagen |
| pRSFDuet-1 | Double T7 promoters, two MCS, RSF1030 origin; Knr | Novagen |
| pACYCDuet-1 | Double T7 promoters, two MCS, p15A origin (pACYC184); Cmr | Novagen |
| pCDF-TAL-4CL | pCDFDuet-1 carrying R. glutinis TAL and P. crispum 4CL | 16 |
| pET-CHS-CHI | pETDuet-1 carrying M. sativa CHI and P. hybrida CHS | 16 |
| pRSF-acs | pRSFDuet-1 carrying E. coli acs | This study |
| pRSF-ACC | pRSFDuet-1 carrying C. glutamicum accBC and dtsR1 | This study |
| pRSF-acs-ACC | pRSFDuet-1 carrying E. coli acs and C. glutamicum accBC and dtsR1 | This study |
| pACYC-acs-ACC | pACYCDuet-1 carrying E. coli acs and C. glutamicum accBC and dtsR1 | This study |
| pACYC-tF3′H-tCPR | pACYCDuet-1 carrying tF3′H from G. hybrida and tCPR from C. roseus | This study |
| pACYC-tF3′H | pACYCDuet-1 carrying tF3′H from G. hybrida | This study |
| pACYC- tCPR | pACYCDuet-1 carrying tCPR from C. roseus | This study |
| pRSF-tF3′H-tCPR | pRSFDuet-1 carrying tF3′H from G. hybrida and tCPR from C. roseus | This study |
| pKD46 | Temperature-sensitive plasmid; contains arabinose-inducible phage λ Red recombinase gene for linear DNA exchange; bla (Ampr) | 28 |
| pCP20 | Temperature-sensitive plasmid; contains FLP recombinase gene for removal of antibiotic resistance cassettes; Ampr Cmr | 28 |
| pKD13 | FRT sites flanking the kanamycin resistance gene; Knr | 28 |
Metabolic engineering to enhance the availability of malonyl-CoA.
The acetyl-CoA carboxylase gene in C. glutamicum consists of two subunits, accBC and dtsR1 (29, 30). The accBC gene was amplified with primers accBC-F (containing an EcoRV site) and accBC-R (containing a KpnI site) (Table 2). The dtsR1 gene was amplified by PCR using primers dtsR1-F (KpnI) and dtsR1-R (AvrII) (Table 2). The accBC and dtsR genes were digested and inserted into the EcoRV/KpnI and KpnI/AvrII sites of vector pRSFDuet-1, resulting in pRSF-ACC. The acs gene was PCR amplified with primers acs-F (BamHI) and acs-R (NotI) (Table 2), digested, and inserted into the BamHI/NotI site of pRSF-ACC, resulting in pRSF-acs-ACC. Plasmid pACYC-acs-ACC was assembled in the same manner as plasmid pRSF-acs-ACC.
TABLE 2.
Oligonucleotides used in this study
| Name | Oligonucleotide sequence (5′–3′)a | Target gene | Relevant characteristic |
|---|---|---|---|
| P1 | CTATGGCTCCCTGACGTTTTTTTAGCCACGTATCAATTATAGGTACTTCCATTCCGGGGATCCGTCG | kan | ackA upstream |
| P2 | GATTTGGCGGGTTACAAAACAGCACCGCCAGCTGAGCTGGCGGTGTGAAATGTAGGCTGGAGCTGCTTCG | kan | ackA downstream |
| accBC-F | GGCCGATATCGGTGTCAGTCGAGACTAGGAAGATCAC | accBC | EcoRV |
| accBC-R | CGGGGTACCCTTGATCTCGAGGAGAACAACG | accBC | KpnI |
| acs-F | CGGGATCCGATGAGCCAAATTCACAAACACAC | acs | BamHI |
| acs-R | AAGGAAAAAAGCGGCCGCTTACGATGGCATCGCGATAG | acs | NotI |
| dtsR1-F | CGGGGTACCATGACCATTTCCTCACCTTT | dtsR1 | KpnI |
| dtsR1-R | CGGCCTAGGTTACAGTGGCATGTTGCC | dtsR1 | AvrII |
| F3′H-F | GGGAATTCCATATGCGCAACCCGAACCGTCT | tF3′H | NdeI |
| F3′H-R | CTTGCCACTGCCACTGCTTGTCGACCCCACCTTCGTCGTTTCATACACA | tF3′H | Linker |
| CPR-F | TGTGTATGAAACGACGAAGGTGGGGTCGACAAGCAGTGGCAGTGGCAAG | tCPR | Linker |
| CPR-R | CGGGGTACCTCACCACACATCACGCAGAT | tCPR | KpnI |
Underlined nucleotides indicate homology arms or restriction enzyme sites.
The acetate competition pathway was deleted by knocking out the E. coli chromosomal gene ackA by the Red recombinase method. The ackA gene was PCR amplified with primers P1 and P2 (Table 2) from the E. coli genome. The FLP recombination target (FRT)-flanked antibiotic resistance genes used for selection were deleted by using a temperature-conditional plasmid, pCP20, expressing FLP recombinase from a thermoinducible promoter (28).
Construction and assembly of the l-tyrosine-to-eriodictyol pathway.
pCDF-TAL-4CL and pET-CHS-CHI were constructed in a previous study (16). Naringenin can be transformed to eriodictyol by F3′H and CPR (Fig. 1) (21, 31). Hence, F3′H was functionally expressed in E. coli as a fusion protein with CPR (Fig. 2).
FIG 2.

Functional fusion of F3′H and P450 reductase. Truncated and modified F3′H and CPR were renamed tF3′H and tCPR, respectively. tF3′H and tCPR were fused by PCR. The fusion protein tF3′H-tCPR harbored a Gly-Ser-Thr linker sequence.
Analysis of the secondary structure of F3′H using the MLRC secondary-structure prediction program revealed a helix motif at the first 25 amino acids of the N-terminal region, which was identified as a membrane anchor sequence (32). The truncated F3′H gene (tF3′H), in which the membrane binding domain and stop codon were removed, was PCR amplified from pUC57-F3′H (synthesized by GeneScript, Nanjing, China) with primers F3′H-F and F3′H-R (Table 2). The forward primer F3′H-F starts at the 76th nucleotide of F3′H, and the reverse primer F3′H-R harbors a Gly-Ser-Thr linker sequence (underlined in Table 2) and 18 CPR nucleotides. The Gly-Ser-Thr linker limits secondary-structure formation that can interfere with the interactions between the two proteins. The truncated CPR gene (tCPR), deletions in which removed 71 amino acids from the N terminus (which is the membrane binding domain), was PCR amplified from pUC57-CPR (synthesized by GeneScript, Nanjing, China) using primers CPR-F and CPR-R (Table 2). The forward primer CPR-F contained a Gly-Ser-Thr linker sequence (underlined in Table 2) and 22 nucleotides of F3′H. The two fragments were fused by PCR to form one fragment (tF3′H-tCPR) with primer pair F3′H-F/CPR-R. The tF3′H-tCPR fragment was purified, digested, and inserted into the NdeI/KpnI sites of pACYCDuet-1 and pRSFDuet-1, respectively, resulting in pACYC-tF3′H-tCPR and pRSF-tF3′H-tCPR. All plasmid constructs were verified by Sanger DNA sequencing (Sangon, Shanghai, China).
E. coli BL21(DE3) was transformed with different plasmid combinations to create different engineered strains. The plasmids and strains used in this study are listed in Table 1 and Table 3, respectively.
TABLE 3.
Strains
| Strain | Relevant characteristic(s) | Source |
|---|---|---|
| E. coli BL21(DE3) | F− ompT hsdSB(rB− mB−) gal dcm (DE3) | Novagen |
| E. coli JM109 | endA1 recA1 gyrA96 thi hsdR17(rK− mK+) relA1 supE44 Δ(lac-proAB) [F′ traD36 proAB lacIq lacZΔM15] | Novagen |
| Corynebacterium glutamicum ATCC 13032 | Clone for ACC | ATCC |
| E. coli BA | BL21(DE3) carrying pACYCDuet-1 | This study |
| E. coli BAtF | BL21(DE3) carrying pACYC-tF3′H | This study |
| E. coli BAtC | BL21(DE3) carrying pACYC-tCPR | This study |
| E. coli BAtFC | BL21(DE3) carrying pACYC-tF3′H-tCPR | This study |
| E. coli BM1 | ackA deletion mutant of BL21(DE3) | This study |
| E. coli BM2 | BL21(DE3) carrying pRSF-acs | This study |
| E. coli BM3 | BL21(DE3) carrying pRSF-ACC | This study |
| E. coli BM4 | BL21(DE3) carrying pRSF-acs-ACC | This study |
| E. coli BM | BM1 carrying pRSF-acs-ACC | This study |
| E. coli BF1 | BL21(DE3)carrying pCDF-TAL-4CL, pET-CHS-CHI, and pRSF-tF3′H-tCPR | This study |
| E. coli BF2 | BL21(DE3) carrying pCDF-TAL-4CL, pET-CHS-CHI, and pACYC-tF3′H-tCPR | This study |
| E. coli BMF1 | BM1 carrying pCDF-TAL-4CL, pET-CHS-CHI, pACYC-acs-ACC, and pRSF-tF3′H-tCPR | This study |
| E. coli BMF2 | BM1 carrying pCDF-TAL-4CL, pET-CHS-CHI, pRSF-acs-ACC, and pACYC-tF3′H-tCPR | This study |
Protein identification.
Protein bands in lanes of a 12% polyacrylamide gel were identified by using the Bruker UltraFlex III matrix-assisted laser desorption ionization–tandem time of flight mass spectrometer (MALDI-TOF/TOF MS) (Bruker Daltonics, Karlsruhe, Germany). The peptide fragment ion data acquired from the MALDI-TOF/TOF mass spectrometer were used to search for protein candidates in the NCBI nr database through Mascot (Matrix Science) integrated in BioTools (Bruker Daltonics), which is accessible online (33).
Culture media and microbial growth conditions.
Bacterial cells were inoculated from frozen stocks in the Luria-Bertani plate and were cultured at 37°C for 12 h. Then a single colony was picked up from the plate and was cultured in a 250-ml flask with 25 ml of fresh morpholinepropanesulfonic acid (MOPS) medium at 37°C with orbital shaking at 200 × g (34). After an optical density at 600 nm (OD600) of 1.2 had been reached, an additional 25 ml of fresh medium and a final aliquot of isopropyl-β-d-thiogalactopyranoside (IPTG; final concentration, 0.6 mM) were provided, and cultures were subsequently equilibrated at a lower temperature of 25°C or 30°C for 72 h. The flavonoid, intracellular malonyl-CoA, and intracellular acetyl-CoA concentrations were measured at different postinduction phases during a total fermentation time of 72 h. A final concentration of 1 mM l-tyrosine was added as the substrate. A final concentration of 0.1 mM aminolevulinate was added as a heme precursor to increase cytochrome P450 activity. Various combinations of ampicillin (100 μg/ml), kanamycin (50 μg/ml), chloramphenicol (30 μg/ml), and streptomycin (50 μg/ml) were added to the cultures of plasmid-bearing E. coli strains (16).
Extraction and analysis of intracellular malonyl-CoA and acetyl-CoA.
For the wild-type [WT], BM1, BM2, BM3, BM4, and BM strains, 5 ml of cell culture was removed after induction at 25°C for 36 h in MOPS medium; for the BF1, BF2, BMF1, and BMF2 strains, 5 ml of cell culture was removed every other 12 h in the induction phase. The portions of cell culture removed were chilled immediately on ice and were centrifuged at 5,000 × g and 4°C for 10 min. The cell pellet was resuspended in 1 ml of 6% perchloric acid to facilitate cell lysis. Repeated freezing and dissolution was performed to completely break down the cells. Then 0.3 ml of 3 M potassium carbonate was added while vortexing to neutralize the acid. The solution was centrifuged to pellet the cell debris (15, 35). The supernatant was collected and was stored chilled until analysis of the CoA compounds using quadrupole ion trap-TOF mass spectrometry coupled with liquid chromatography (LCMS-IT-TOF) (Shimadzu, Kyoto, Japan). To determine the dry weight of cells, a 25-ml portion of the same cell culture as that from which portions were removed as described above (WT, BM1, BM2, BM3, BM4, BM, BF1, BF2, BMF1, or BMF2) was spun in a preweighed centrifuge tube at 5,000 × g for 5 min. The cell pellet was washed with deionized water and was dried at 105°C. The differences in weight between empty tubes and tubes with cell residues represented the dry weight of cells.
CoA was analyzed by using a Shimadzu LCMS-IT-TOF consisting of a separation module connected directly to the IT-TOF MS. A Shim-Pack VP-ODS high-performance liquid chromatography (HPLC) column (length, 150 mm; inner diameter, 2.0 mm; Shimadzu, Kyoto, Japan) was used to perform HPLC separation. The samples of malonyl-CoA and acetyl-CoA were eluted at a flow rate of 0.2 ml/min with a gradient of 10 mM ammonium formate (A) and 20% 10 mM ammonium formate in methanol (vol/vol) (B) as follows: at 0 min, 2% B; at 10 min, 60% B; at 20 min, 76% B; at 25 min, 2% B. IT-TOF detection using an electrospray ionization (ESI) source was performed in positive-ion mode using optimized conditions as follows: detector voltage, 1.60 kV; nebulizing gas (N2) flow, 1.5 liters/min; drying gas (N2) flow, 200 kPa; ion accumulation time, 30 ms; collision energy set at 40% for MS2; scan ranges, m/z 750 to 900 for MS1 and m/z 100 to 900 for MS2 (36). The multiple-reaction-monitoring (MRM) mode was used to quantify CoA-thioesters of interest, using mass ions set to detect transitions of the parent ion to the daughter ion specific to the selected analytes. The transitions (m/z for the parent ion → m/z for the daughter ion) for the two CoA-thioesters of interest were as follows: for malonyl-CoA, 854.1 → 428; for acetyl-CoA, 810.1 → 428.
Analysis and quantification of eriodictyol and intermediate metabolites.
To analyze flavonoid production, the entire culture broth was freeze-dried using a VirTis BenchTop 6K manifold freeze dryer (SP Industries, Warminster, PA). Then the freeze-dried culture broth was extracted with dimethyl sulfoxide (DMSO) to obtain eriodictyol and intermediate metabolites. The analysis was performed using a Shimadzu LCMS-IT-TOF consisting of a separation module connected directly to the IT-TOF MS. A Shim-Pack VP-ODS HPLC column (length, 150 mm; inner diameter, 2.0 mm) was used to perform HPLC separation. Eriodictyol and intermediate metabolites were separated by elution with an acetonitrile/water gradient at a flow rate of 0.2 ml/min under the following conditions: 10 to 40% acetonitrile (vol/vol) for 10 min, 40 to 60% acetonitrile (vol/vol) for 5 min, 60 to 10% acetonitrile (vol/vol) for 2 min (14). IT-TOF detection with an ESI source was performed in negative-ion mode using optimized conditions as follows: detector voltage, 1.60 kV; nebulizing gas (N2) flow, 1.5 liters/min; drying gas (N2) flow, 200 kPa; ion accumulation time, 30 ms; collision energy set at 40% for MS2; scan range, m/z 100 to 300 for MS1 and m/z 50 to 300 for MS2. Quantification was performed in the MRM mode in a tandem mass spectrometer (MS-MS) with the mass ions set to detect transitions of the parent ion to the daughter ion specific to the selected analytes. The transitions (m/z for the parent ion → m/z for the daughter ion) were as follows: for eriodictyol, 287 → 151; for naringenin, 271 → 151; for p-coumaric acid, 163 → 119.
Statistical analysis.
Student's t test was employed to investigate statistical differences, and differences with P values of <0.05 were considered significant.
RESULTS
Soluble expression of the translational fusion protein tF3′H-tCPR in E. coli.
In order to investigate soluble expression, several recombinant E. coli strains were developed, including strains BA (blank control), BAtF (expressing tF3′H), BAtC (expressing tCPR), and BAtFC (expressing tF3′H-tCPR) (Table 3). Figure 3 shows the expression profiles of the targeted recombinant proteins. All three proteins were observed in the supernatant (Fig. 3, lanes 2 to 4) but were not found in the cellular debris (lanes 6 to 8).
FIG 3.
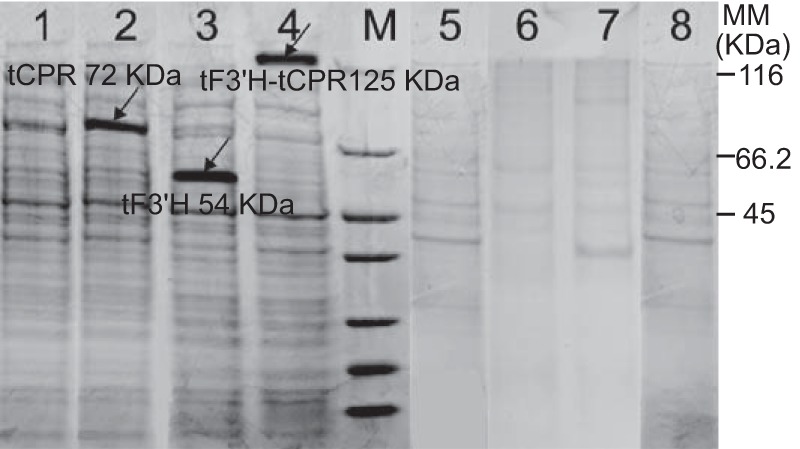
SDS-PAGE analysis of proteins extracted from the supernatant after the disruption of E. coli cells. Proteins in lanes 1 to 4 were extracted from the supernatant after cell disruption; proteins in lanes 5 to 8 were extracted from cellular debris. The extracts obtained from the recombinants were electrophoresed on a 12% polyacrylamide gel under denaturing conditions. The images from SDS-PAGE analyses were captured using the Gel Doc system (Bio-Rad). Lanes 1 and 5, pACYCDuet-1; lanes 2 and 6, pACYC-tCPR; lanes 3 and 7, pACYC-tF3′H; lanes 4 and 8, pACYC-tF3′H-tCPR.
The protein in Fig. 3, lane 2, was identified as CPR (GI 18139; mass, 78,953 Da; score, 1,596). The protein in Fig. 3, lane 3, was identified as F3′H (GI 77176700; mass, 56,315 Da; score, 657). The protein in Fig. 3, lane 4, was identified as F3′H (GI 77176700; mass, 56,315 Da; score, 453) and CPR (GI 18139; mass, 78,953 Da; score, 696). This indicated that soluble expression of the fusion protein (tF3′H-tCPR) was achieved. The matched information for proteins in the database consists of theoretical values, which cannot distinguish truncated or fusion proteins from normal proteins. Therefore, the molecular sizes given for all proteins are larger than those shown in SDS-PAGE, because the data obtained from the database were molecular sizes of whole proteins rather than those of truncated proteins.
Engineering malonyl-CoA availability.
Three strategies were employed in an effort to increase the availability of malonyl-CoA for flavonoid biosynthesis. These strategies include overexpression of acetyl-CoA carboxylase (ACC), overexpression of acetyl-CoA synthase (encoded by acs), an enzyme involved in the acetate assimilation pathway, and deletion of the acetate competition pathway by knockout of the acetate kinase (ackA) gene. Intracellular malonyl-CoA and acetyl-CoA concentrations are shown in Table 4. Overexpression of ACC alone increased the cellular malonyl-CoA concentration by 278% over that in wild-type E. coli. Overexpression of acs to recycle acetate back to acetyl-CoA increased the intracellular malonyl-CoA concentration by 69.5% over that in the WT. By knockout of ackA, the intracellular malonyl-CoA concentration increased by 130% over that in WT E. coli. Coexpression of the acs product with ACC enhanced the intracellular malonyl-CoA concentration by 330% over that in WT E. coli. A combination of the three manipulations gave a total increase in the intracellular malonyl-CoA level of 16.3-fold over that in WT E. coli.
TABLE 4.
Intracellular malonyl-CoA and acetyl-CoA concentrations in E. coli strains
| Strain [characteristic(s)]a | Malonyl-CoA |
Acetyl-CoA |
||
|---|---|---|---|---|
| Concnb | % increase over concn in WT | Concnb | % increase over concn in WT | |
| WT | 0.230 ± 0.01 | 1.86 ± 0.21 | ||
| BM2 (acs overexpression) | 0.390 ± 0.03 | 69.5 | 2.24 ± 0.22 | 20.4 |
| BM3 (ACC overexpression) | 0.870 ± 0.09 | 278 | 1.48 ± 0.17 | −20.5 |
| BM1 (ΔackA) | 0.530 ± 0.08 | 130 | 1.92 ± 0.24 | 3.22 |
| BM4 (acs-ACC overexpression) | 0.990 ± 0.11 | 330 | 1.52 ± 0.14 | −18.3 |
| BM (acs-ACC overexpression, ΔackA) | 3.98 ± 0.16 | 1.63 × 103 | 1.12 ± 0.16 | −39.8 |
The WT strain was included as a reference for comparison.
Expressed as micromoles per gram of cells (dry weight).
Optimization of expression of tF3′H-tCPR.
In order to balance tF3′H-tCPR gene expression, the tF3′H-tCPR fusion gene was cloned into two vectors, pACYCDuet-1, with a low copy number of 10 to 12 (p15A origin of replication), and pRSFDuet-1, with a high copy number of >100 (RSF1030 origin of replication) (35). As shown in Fig. 5, eriodictyol production by strain BF2, carrying a lower copy number of P450 fusion genes, was 21.1% higher than that of strain BF1, which carries a higher copy number of P450 fusion genes. Eriodictyol production by strain BMF2 was 23.5% higher than that by strain BMF1. Transcriptional levels of genes on different vectors measured by quantitative reverse transcription-PCR (qPCR) are shown in the supplemental material.
FIG 5.

Eriodictyol production, intermediate metabolites, and CoA-thioesters accumulated in engineered E. coli strains at different postinduction phases. ■, BF1; ●, BF2; ▲, BMF1; ▼, BMF2. The strains were induced under their optimal culture conditions (30°C for BF1 and BF2; 25°C for BMF1 and BMF2). Data are averages of results for three biological replicates. Error bars represent standard deviations from the means. DCW, dry weight of cells.
Determination of optimal culture conditions.
Four different temperatures were chosen (20°C, 25°C, 28°C, 30°C) for the induction phase. The production of eriodictyol was maximized at 25°C for BMF1 and BMF2 and at 30°C for BF1 and BF2 (Fig. 4A). Strains were induced at OD600 values of 0.6, 0.9, 1.2, 1.5, 1.8, and 2.1. By use of the gradient test described above, an OD600 of 1.2 to 1.8 was taken as representing the optimal condition for all four strains (Fig. 4B). IPTG was added to strains BF1, BF2, BMF1, and BMF2 at concentrations of 0.1, 0.2, 0.4, 0.6, 0.8, 1.0, and 1.2 mM. All four strains exhibited the highest eriodictyol production at an IPTG concentration of 0.6 mM (Fig. 4C). When glucose was used as the substrate, the eriodictyol production of BMF2 was only 5.70 mg/liter. Therefore, l-tyrosine was added for sufficient substrate availability. As shown in Fig. 5B, the eriodictyol production of strains BMF1 and BMF2 reached 86.4 mg/liter and 107 mg/liter from l-tyrosine, respectively, under their optimal culture conditions (25°C in the induction phase, with an OD600 of 1.2 to 1.8 and an IPTG concentration of 0.6 mM). The eriodictyol production of strains BF1 and BF2 reached 35.2 mg/liter and 42.6 mg/liter from l-tyrosine, respectively, under their optimal culture conditions (30°C in the induction phase, with an OD600 of 1.2 to 1.8 and an IPTG concentration of 0.6 mM).
FIG 4.

Determination of optimal culture conditions. (A) Induction temperature at an OD600 of 1.5 and an IPTG concentration of 0.6 mM. (B) Induction OD600 at an IPTG concentration of 0.6 mM and a temperature of 30°C (for BF1 and BF2) or 25°C (for BMF1 and BMF2). (C) Induction IPTG concentration at an OD600 of 1.5 and a temperature of 30°C (for BF1 and BF2) or 25°C (for BMF1 and BMF2). Open bars, strain BF1; light shaded bars, strain BF2; dark shaded bars, strain BMF1; filled bars, strain BMF2. Data are averages of results for three biological replicates. Error bars represent standard deviations from the means.
DISCUSSION
In this work, the soluble expression of the fusion protein (tF3H-tCPR) was found to promote the biosynthesis of eriodictyol from l-tyrosine. Three approaches were involved in this study: (i) soluble fusion expression of tF3′H (cytochrome P450) and tCPR (P450 reductase), (ii) enhancement of the availability of malonyl-CoA, and (iii) balancing of the relative gene expression to enhance eriodictyol accumulation.
In order to solve the problem of the lack of cofactor incorporation into E. coli, the gene for cytochrome P450 reductase (CPR) from C. roseus, which had been successfully expressed in E. coli before, was codon optimized for synthesis of the enzyme. However, previous research showed that eriodictyol cannot be obtained from p-coumaric acid by the functional expression of CPR as a fusion protein with a flavonoid 3′,5′-hydroxylase (F3′5′H) from C. roseus in E. coli (31). Meanwhile, F3′H from G. hybrida, which was functionally expressed in Saccharomyces cerevisiae, was used to hydroxylate naringenin to eriodictyol (21). Therefore, F3′H from G. hybrida and CPR from C. roseus were chosen for the fusion protein (tF3′H-tCPR) in this study. Because a membrane anchorage domain of cytochrome P450 renders the protein insoluble, soluble expression of a translational fusion protein (tF3′H-tCPR) in E. coli was one of the major objectives. By truncation of the first 25 amino acids at the N-terminal end of F3′H, the fusion protein (tF3′H-tCPR) was functionally expressed in E. coli. The fusion protein allowed the efficient synthesis of eriodictyol from l-tyrosine in E. coli.
One molecule of eriodictyol requires three molecules of malonyl-coenzyme A (Fig. 1). Previously, the low intracellular malonyl-CoA concentration restricted the efficient production of flavonoids in E. coli (37, 38). Therefore, it was necessary to engineer the bacteria for enhanced malonyl-CoA availability so as to achieve a metabolic balance between the requirements of malonyl-CoA for cell growth and product synthesis. As shown in Fig. 5, the intracellular malonyl-CoA concentrations of BF1 and BF2 were very low because malonyl-CoA availability was not enhanced in these two strains. In BMF2 and BMF1, malonyl-CoA accumulated and reached its highest concentrations at 48 h, when eriodictyol production achieved its highest level. The increased accumulation of malonyl-CoA indicated that eriodictyol production may achieve its highest level and stop at some point due to the action of other, unknown, limiting factors. Before that point, malonyl-CoA is consumed to synthesize eriodictyol. A biphasic plateau (12 h to 36 h) was observed for malonyl-CoA and p-coumaric acid concentrations, but not for naringenin or eriodictyol, which progress in a linear manner throughout the growth phase. This indicated that the synthesis rate of naringenin is faster than the rate of conversion of naringenin to eriodictyol. Due to the increase in malonyl-CoA availability, although p-coumaric acid production by BMF2 and BMF1 was lower than that by BF2 and BF1 after 48 h of the eriodictyol accumulation phase, the production of eriodictyol by BMF2 was 151% higher than that by BF2. Meanwhile, eriodictyol production by BMF1 was 146% higher than that by BF1. Besides, no direct precursor of malonyl coenzyme A was added here, in contrast to previous work on the pathway of matB and matC (14). Therefore, this approach is more suitable for large-scale industrial production.
Previous studies showed that coumaroyl-CoA inhibits the TAL enzyme and that low-copy-number plasmids could perform better than high-copy-number plasmids for cytochrome P450 (31, 39, 40). Therefore, balancing relative gene expression is a potential route to the rational and efficient regulation of the expression of genes in the overall pathway. Our previous studies showed that coumaroyl-CoA can inhibit TAL enzyme activity, and the combination of pCDF-TAL-4CL and pET-CHS-CHI was shown to be the optimal combination for flavonoid biosynthesis (16, 41). In this study, pACYC-tF3′H-tCPR was shown to be a better choice than pRSF-tF3′H-tCPR for optimization of the expression of tF3′H-tCPR. By this combination of plasmids and genes, gene expression was balanced to enhance eriodictyol accumulation.
The present work demonstrates a way to approach the efficient biosynthesis of eriodictyol via soluble expression of a translational fusion protein, tF3′H-tCPR, in four vectors in E. coli. These four Duet vectors are designed with compatible replicons and drug resistance genes for effective propagation and maintenance of four plasmids in a single cell (42–44). The ability of Duet vectors to be cotransformed, propagated, and induced for robust target protein coexpression makes them ideal for the efficient biosynthesis of eriodictyol (13, 14, 30, 45). Although integration of genes into the chromosome would be more suitable for large-scale industrial production, the current combination of these four vectors can significantly facilitate the process of researching such complicated metabolic pathways. However, the ultimate goal of the current work is to find a strain that can produce these phytochemicals without the addition of antibiotics and inducers, in view of both safety and cost concerns.
Although previous studies required only three genes in the eriodictyol synthesis pathway, they can use only sodium malonate and caffeic acid (which is expensive) as the substrates rather than l-tyrosine or glucose (14, 15). Therefore, the new eriodictyol synthesis pathway, utilizing less expensive precursors, provides greater economic efficiency. However, the dissolubility of l-tyrosine in water is only 2.8 mM (25°C) (46). The poor dissolubility of l-tyrosine is a limitation on higher production of eriodictyol. Since E. coli can synthesize l-tyrosine by its own metabolic pathway, our next plan is to produce eriodictyol directly from glucose. The main goal of the current work is to establish the metabolic pathway from primary metabolites to a heterologous phytochemical. The current method, especially the functional soluble expression of P450 flavonoid hydroxylase and P450 reductase, offers a route for the efficient biosynthesis of other important hydroxylated flavonoid molecules, for use as food ingredients or in cosmetics applications, from l-tyrosine or even glucose in metabolically engineered E. coli.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the Major State Basic Research Development Program of China (973 Program, 2012CB720806), the National Natural Science Foundation of China (31370130), the Natural Science Foundation of Jiangsu Province (BK2011004, BK2010150), the Fundamental Research Funds for the Central Universities (JUSRP51307A), the Foundation for the Author of National Excellent Doctoral Dissertation of People's Republic of China (FANEDD, 2011046), the Program for New Century Excellent Talents in University (NCET-12-0876), and the 111 Project (111-2-06).
Footnotes
Published ahead of print 7 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03986-13.
REFERENCES
- 1.Ververidis F, Trantas E, Douglas C, Vollmer G, Kretzschmar G, Panopoulos N. 2007. Biotechnology of flavonoids and other phenylpropanoid-derived natural products. Part I. Chemical diversity, impacts on plant biology and human health. Biotechnol. J. 2:1214–1234. 10.1002/biot.200700084 [DOI] [PubMed] [Google Scholar]
- 2.Wang Y, Chen S, Yu O. 2011. Metabolic engineering of flavonoids in plants and microorganisms. Appl. Microbiol. Biotechnol. 91:949–956. 10.1007/s00253-011-3449-2 [DOI] [PubMed] [Google Scholar]
- 3.Chemler JA, Lock LT, Koffas MAG, Tzanakakis ES. 2007. Standardized biosynthesis of flavan-3-ols with effects on pancreatic beta-cell insulin secretion. Appl. Microbiol. Biotechnol. 77:797–807. 10.1007/s00253-007-1227-y [DOI] [PubMed] [Google Scholar]
- 4.Broadhurst CL, Polansky MM, Anderson RA. 2000. Insulin-like biological activity of culinary and medicinal plant aqueous extracts in vitro. J. Agric. Food Chem. 48:849–852. 10.1021/jf9904517 [DOI] [PubMed] [Google Scholar]
- 5.Sinclair DA. 2005. Toward a unified theory of caloric restriction and longevity regulation. Mech. Ageing Dev. 126:987–1002. 10.1016/j.mad.2005.03.019 [DOI] [PubMed] [Google Scholar]
- 6.Koopman F, Beekwilder J, Crimi B, van Houwelingen A, Hall RD, Bosch D, van Maris AJ, Pronk JT, Daran JM. 2012. De novo production of the flavonoid naringenin in engineered Saccharomyces cerevisiae. Microb. Cell Fact. 11:155. 10.1186/1475-2859-11-155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mora-Pale M, Sanchez-Rodriguez SP, Linhardt RJ, Dordick JS, Koffas MAG. 2013. Metabolic engineering and in vitro biosynthesis of phytochemicals and non-natural analogues. Plant Sci. 210:10–24. 10.1016/j.plantsci.2013.05.005 [DOI] [PubMed] [Google Scholar]
- 8.Rossato MF, Trevisan G, Walker CIB, Klafke JZ, de Oliveira AP, Villarinho JG, Zanon RB, Royes LFF, Athayde ML, Gomez MV. 2011. Eriodictyol: a flavonoid antagonist of the TRPV1 receptor with antioxidant activity. Biochem. Pharmacol. 81:544–551. 10.1016/j.bcp.2010.11.004 [DOI] [PubMed] [Google Scholar]
- 9.Zhang WY, Lee JJ, Kim Y, Kim IS, Han JH, Lee SG, Ahn MJ, Jung SH, Myung CS. 2012. Effect of eriodictyol on glucose uptake and insulin resistance in vitro. J. Agric. Food Chem. 60:7652–7658. 10.1021/jf300601z [DOI] [PubMed] [Google Scholar]
- 10.Yoo JM, Kim JH, Park SJ, Kang YJ, Kim TJ. 2012. Inhibitory effect of eriodictyol on IgE/Ag-induced type I hypersensitivity. Biosci. Biotechnol. Biochem. 76:1285–1290. 10.1271/bbb.110952 [DOI] [PubMed] [Google Scholar]
- 11.Selepe MA, Van Heerden FR. 2013. Application of the Suzuki-Miyaura reaction in the synthesis of flavonoids. Molecules 18:4739–4765. 10.3390/molecules18044739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pandey RP, Malla S, Simkhada D, Kim B-G, Sohng JK. 2013. Production of 3-O-xylosyl quercetin in Escherichia coli. Appl. Microbiol. Biotechnol. 97:1889–1901. 10.1007/s00253-012-4438-9 [DOI] [PubMed] [Google Scholar]
- 13.Leonard E, Lim KH, Saw PN, Koffas MAG. 2007. Engineering central metabolic pathways for high-level flavonoid production in Escherichia coli. Appl. Environ. Microbiol. 73:3877–3886. 10.1128/AEM.00200-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leonard E, Yan Y, Fowler ZL, Li Z, Lim CG, Lim KH, Koffas MAG. 2008. Strain improvement of recombinant Escherichia coli for efficient production of plant flavonoids. Mol. Pharm. 5:257–265. 10.1021/mp7001472 [DOI] [PubMed] [Google Scholar]
- 15.Fowler ZL, Gikandi WW, Koffas MAG. 2009. Increased malonyl coenzyme A biosynthesis by tuning the Escherichia coli metabolic network and its application to flavanone production. Appl. Environ. Microbiol. 75:5831–5839. 10.1128/AEM.00270-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu J, Du G, Zhou J, Chen J. 2013. Metabolic engineering of Escherichia coli for (2S)-pinocembrin production from glucose by a modular metabolic strategy. Metab. Eng. 16:48–55. 10.1016/j.ymben.2012.11.009 [DOI] [PubMed] [Google Scholar]
- 17.Fowler ZL, Koffas MAG. 2009. Biosynthesis and biotechnological production of flavanones: current state and perspectives. Appl. Microbiol. Biotechnol. 83:799–808. 10.1007/s00253-009-2039-z [DOI] [PubMed] [Google Scholar]
- 18.Kaneko M, Hwang EI, Ohnishi Y, Horinouchi S. 2003. Heterologous production of flavanones in Escherichia coli: potential for combinatorial biosynthesis of flavonoids in bacteria. J. Ind. Microbiol. Biotechnol. 30:456–461. 10.1007/s10295-003-0061-1 [DOI] [PubMed] [Google Scholar]
- 19.Kim DH, Kim BG, Jung N, Ahn JH. 2009. Production of genistein from naringenin using Escherichia coli containing isoflavone synthase-cytochrome P450 reductase fusion protein. J. Microbiol. Biotechnol. 19:1612–1616. 10.4014/jmb.0905.05043 [DOI] [PubMed] [Google Scholar]
- 20.Hotze M, Schröder G, Schröder J. 1995. Cinnamate 4-hydroxylase from Catharanthus roseus and a strategy for the functional expression of plant cytochrome P450 proteins as translational fusions with P450 reductase in Escherichia coli. FEBS Lett. 374:345–350. 10.1016/0014-5793(95)01141-Z [DOI] [PubMed] [Google Scholar]
- 21.Amor I, Hehn A, Guedone E, Ghedira K, Engasser JM, Chekir-Ghedrira L, Ghoul M. 2010. Biotransformation of naringenin to eriodictyol by Saccharomyces cerevisiae functionally expressing flavonoid 3′ hydroxylase. Nat. Prod. Commun. 5:1893–1898 [PubMed] [Google Scholar]
- 22.Harada H, Shindo K, Iki K, Teraoka A, Okamoto S, Yu F, Hattan J, Utsumi R, Misawa N. 2011. Efficient functional analysis system for cyanobacterial or plant cytochromes P450 involved in sesquiterpene biosynthesis. Appl. Microbiol. Biotechnol. 90:467–476. 10.1007/s00253-010-3062-9 [DOI] [PubMed] [Google Scholar]
- 23.Chang MC, Eachus RA, Trieu W, Ro DK, Keasling JD. 2007. Engineering Escherichia coli for production of functionalized terpenoids using plant P450s. Nat. Chem. Biol. 3:274–277. 10.1038/nchembio875 [DOI] [PubMed] [Google Scholar]
- 24.Alonso-Gutierrez J, Chan R, Batth TS, Adams PD, Keasling JD, Petzold CJ, Lee TS. 2013. Metabolic engineering of Escherichia coli for limonene and perillyl alcohol production. Metab. Eng. 19:33–41. 10.1016/j.ymben.2013.05.004 [DOI] [PubMed] [Google Scholar]
- 25.Lim CG, Fowler ZL, Hueller T, Schaffer S, Koffas MAG. 2011. High-yield resveratrol production in engineered Escherichia coli. Appl. Environ. Microbiol. 77:3451–3460. 10.1128/AEM.02186-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu P, Ranganathan S, Fowler ZL, Maranas CD, Koffas MAG. 2011. Genome-scale metabolic network modeling results in minimal interventions that cooperatively force carbon flux towards malonyl-CoA. Metab. Eng. 13:578–587. 10.1016/j.ymben.2011.06.008 [DOI] [PubMed] [Google Scholar]
- 27.Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC, Jahn D. 2005. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 33:W526–W531. 10.1093/nar/gki376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645. 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gande R, Dover LG, Krumbach K, Besra GS, Sahm H, Oikawa T, Eggeling L. 2007. The two carboxylases of Corynebacterium glutamicum essential for fatty acid and mycolic acid synthesis. J. Bacteriol. 189:5257–5264. 10.1128/JB.00254-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyahisa I, Kaneko M, Funa N, Kawasaki H, Kojima H, Ohnishi Y, Horinouchi S. 2005. Efficient production of (2S)-flavanones by Escherichia coli containing an artificial biosynthetic gene cluster. Appl. Microbiol. Biotechnol. 68:498–504. 10.1007/s00253-005-1916-3 [DOI] [PubMed] [Google Scholar]
- 31.Leonard E, Yan Y, Koffas MAG. 2006. Functional expression of a P450 flavonoid hydroxylase for the biosynthesis of plant-specific hydroxylated flavonols in Escherichia coli. Metab. Eng. 8:172–181. 10.1016/j.ymben.2005.11.001 [DOI] [PubMed] [Google Scholar]
- 32.Guermeur Y, Geourjon C, Gallinari P, Deléage G. 1999. Improved performance in protein secondary structure prediction by inhomogeneous score combination. Bioinformatics 15:413–421. 10.1093/bioinformatics/15.5.413 [DOI] [PubMed] [Google Scholar]
- 33.Yamanaka M, Hara K, Kudo J. 2005. Bactericidal actions of a silver ion solution on Escherichia coli, studied by energy-filtering transmission electron microscopy and proteomic analysis. Appl. Environ. Microbiol. 71:7589–7593. 10.1128/AEM.71.11.7589-7593.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neidhardt FC, Bloch PL, Smith DF. 1974. Culture medium for enterobacteria. J. Bacteriol. 119:736–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zha W, Rubin-Pitel SB, Shao Z, Zhao H. 2009. Improving cellular malonyl-CoA level in Escherichia coli via metabolic engineering. Metab. Eng. 11:192–198. 10.1016/j.ymben.2009.01.005 [DOI] [PubMed] [Google Scholar]
- 36.Cheng X, Liu F, Yan T, Zhou X, Wu L, Liao K, Wang G, Hao H. 2012. Metabolic profile, enzyme kinetics, and reaction phenotyping of β-lapachone metabolism in human liver and intestine in vitro. Mol. Pharm. 9:3476–3485. 10.1021/mp300296m [DOI] [PubMed] [Google Scholar]
- 37.Rathnasingh C, Raj SM, Lee Y, Catherine C, Ashok S, Park S. 2012. Production of 3-hydroxypropionic acid via malonyl-CoA pathway using recombinant Escherichia coli strains. J. Biotechnol. 157:633–640. 10.1016/j.jbiotec.2011.06.008 [DOI] [PubMed] [Google Scholar]
- 38.Bhan N, Xu P, Khalidi O, Koffas MAG. 15 November 2013. Redirecting carbon flux into malonyl-CoA to improve resveratrol titers: proof of concept for genetic interventions predicted by OptForce computational framework. Chem. Eng. Sci. 10.1016/j.ces.2012.10.009 [DOI] [Google Scholar]
- 39.Santos CNS, Koffas MAG, Stephanopoulos G. 2011. Optimization of a heterologous pathway for the production of flavonoids from glucose. Metab. Eng. 13:392–400. 10.1016/j.ymben.2011.02.002 [DOI] [PubMed] [Google Scholar]
- 40.Jones KL, Kim S-W, Keasling J. 2000. Low-copy plasmids can perform as well as or better than high-copy plasmids for metabolic engineering of bacteria. Metab. Eng. 2:328–338. 10.1006/mben.2000.0161 [DOI] [PubMed] [Google Scholar]
- 41.Wu J, Liu P, Fan Y, Bao H, Du G, Zhou J, Chen J. 2013. Multivariate modular metabolic engineering of Escherichia coli to produce resveratrol from l-tyrosine. J. Biotechnol. 167:404–411. 10.1016/j.jbiotec.2013.07.030 [DOI] [PubMed] [Google Scholar]
- 42.Novy R, Yaeger K, Held D, Mierendorf R. 2002. Coexpression of multiple target proteins in E. coli. inNovations 15:2–6 [Google Scholar]
- 43.Held D, Yaeger K, Novy R. 2003. New coexpression vectors for expanded compatibilities in E. coli. inNovations 18:4–6 [Google Scholar]
- 44.Selzer G, Som T, Itoh T, Tomizawa J. 1983. The origin of replication of plasmid p15A and comparative studies on the nucleotide sequences around the origin of related plasmids. Cell 32:119–129. 10.1016/0092-8674(83)90502-0 [DOI] [PubMed] [Google Scholar]
- 45.Leonard E, Chemler J, Lim KH, Koffas MA. 2006. Expression of a soluble flavone synthase allows the biosynthesis of phytoestrogen derivatives in Escherichia coli. Appl. Microbiol. Biotechnol. 70:85–91. 10.1007/s00253-005-0059-x [DOI] [PubMed] [Google Scholar]
- 46.Carta R, Tola G. 1996. Solubilities of l-cystine, l-tyrosine, l-leucine, and glycine in aqueous solutions at various pHs and NaCl concentrations. J. Chem. Eng. Data 41:414–417. 10.1021/je9501853 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.