

Abstract
Minimalist secondary structure mimics are typically made to resemble one interface in a protein-protein interaction (PPI), and thus perturb it. We recently proposed suitable chemotypes can be matched with interface regions directly, without regard for secondary structures. This communication describes a modular synthesis of a new chemotype 1, simulation of its solution-state conformational ensemble, and correlation of that with ideal secondary structures and real interface regions in PPIs. Scaffold 1 presents amino acid side-chains that are quite separated from each other, in orientations that closely resemble ideal sheet or helical structures, similar non-ideal structures at PPI interfaces, and regions of other PPI interfaces where the mimic conformation does not resemble any secondary structure. Sixty-eight different PPIs where conformations of 1 matched well were identified. A new method is also presented to determine the relevance of a minimalist mimic crystal structure to its solution conformations. Thus DLD-1faf crystallized in a conformation that is estimated to be 0.91 kcal•mol−1 above the minimum energy solution state.
Keywords: amino acids, helical structures, protein-protein interactions, peptidomimetics, computational chemistry, secondary structures
Minimalist mimics of secondary structures display amino acid side-chains on backbones more rigid than, and structurally different to, polypeptide chains.[1–3] Mimics that can be made conveniently with a variety of amino acid side-chains tend to be the most valuable, hence modular syntheses involving amino acid-derived starting materials are ideal.[4–6] Besides these synthetic considerations, it is also important to gain an understanding of how well ideal secondary structures can be represented by the best-fitting accessible conformer from the mimic’s conformational ensemble. Such analyses indicate how a minimalist mimic can optimally fit on one secondary structure relative to another, and which side-chain spacings are best represented. This is valuable information because otherwise, for example, it is unclear which of the so-called ideal α-helical mimics actually fit better on other secondary structures, and which ones best present a given side-chain combination (eg i, i+3, i+4 or i, i+4, i+5).[7]
Reported here are modular syntheses of novel minimalist mimics 1 from amino acid-derived starting materials. We hypothesized scaffold 1 could present three amino acid side-chains in orientations resembling extended secondary structures with wide side-chain separations. To test our hypothesis, a large number of accessible (<3 kcal•mol−1) conformations for each stereomer of 1 were simulated, then systematically matched with the common secondary structures via the data mining technique we call Exploring Key Orientations on Secondary structures (EKOS).[7] A solid state structure of a compound 1 was also obtained, and EKOS was used to relate that conformation to ideal secondary structures, and to the predicted conformational ensemble for scaffold 1 in solution. Finally, some preferred conformations of 1 were statistically related to more than 120,000 structurally characterized protein-protein interfaces, the 68 that best matched side-chain orientations of any accessible conformer of 1 were selected, and the secondary structures at these interfaces were analyzed[8] to explore how this structure is predicted to best match side-chain orientations at PPI interfaces. Throughout, these data are compared with the previously reported[8–10] mimics A.
A vulnerability in syntheses of substrates A is the enolizable, and therefore stereochemically delicate, CH.[9] Conversely, an attribute of scaffolds 1 is that they are based on β-substituted β-amino acids where a methylene group insulates the chiral center from stereomutation. Another problem in syntheses of scaffolds A is that fragments bearing bulkier amino acid side-chains tend to retard coupling reactions. We postulated this steric effect would be less pronounced in construction of 1 where the side-chains are one methylene removed from the electrophilic coupling site.
Syntheses of molecules 1 began with preparation of β-substituted β-amino acid derivatives 2 using a novel combination of literature procedures[11, 12] that allowed us to obtain multi-gram amounts, without chromatography in most cases (see supporting). These amino acids 2 were reacted with Meldrum’s acid, a known reaction for α-amino acids,[13] to form six-membered homologs of tetramic acids (Scheme 1). Removal of the Boc-protecting group gave the piperidinediones 3 that were converted to the vinylic chlorides 4. Those analogs with threonine side-chains have an inherent marker for loss of stereochemical fidelity; as anticipated, no epimerization was observed, at least in the syntheses of the compounds containing Thr.
Scheme 1.

Syntheses of the electrophilic N-caps 4.
Nitriles 5, intermediates in the syntheses of the β-amino acids 2, were simultaneously N-deprotected and hydrolyzed, then reductively coupled with the known, and commercially available, synthons B[14] or C[15] to give the amines 6 (Scheme 2). Reaction of these β-amino esters with Bestmann’s ylide[16] gave the protected intermediates 7. Compounds 5 to 7 were isolated, without chromatography, on multigram scales. N-Deprotection of the intermediates 7 gave the nucleophiles 8. Amines 8 were then condensed with the electrophiles 4 to give the scaffolds 9 bearing two side-chains. As anticipated, this process seems largely unaffected by steric demands of the side-chains since the coupling yields were uniformly high.
Scheme 2.

Syntheses of the target materials 1.
In a divergent step, intermediates 7 were C-deprotected, then converted to the vinylogous chlorides 10. Convergence was then possible via coupling of the nucleophiles 8 with the electrophiles 10 to give the extended systems 11 that were then N-deprotected and capped with the electrophiles 4.
The syntheses of mimics 1 may seem complicated, but they are driven by the simple modularity concept expressed in Figure 2. Overall, the divergent-convergent syntheses of compounds 1 pivot around synthons 7; these were converted to nucleo- and electrophiles that were joined to elongate the scaffold. Fragments 8 and 10 in Scheme 2 are similar to C- and N-protected amino acids in peptide syntheses.
Figure 2.
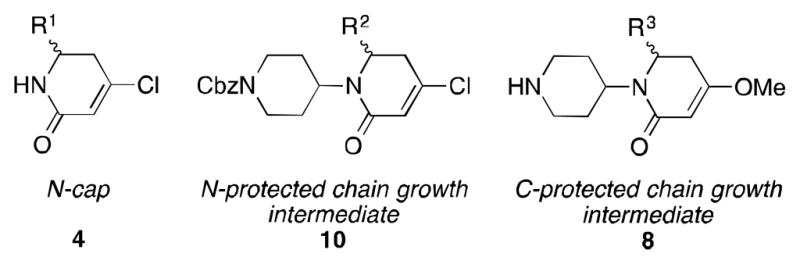
N-Capping, N-protected, and C-protected synthons like 4, 10, and 8, are pivotal in divergent-convergent routes to scaffolds 1.
No spectroscopic technique can characterize conformational ensembles of small molecules like 1 or A because they equilibrate between hundreds solution states that have significantly different side-chain orientations. Techniques like CD and NOESY/ROESY NMR give a conformational average, that does not correspond to any real state.[6] An X-ray structure shows whatever mimic conformation happened to crystallize, where part of the shape is governed by crystal packing forces. Those packing forces are significant enough that some solid-state conformations may not be preferred in solution. Indeed, a solid state mimic conformation that does not resemble the target secondary structure has been reported by Hamilton et al,[17] and this type of occurrence should not be surprising because crystal packing forces are relatively large.
A problem that has not been addressed in studies of minimalist mimics is how to evaluate the relevance of a solid-state structure to a conformational ensemble in solution. Here we suggest EKOS can be used to determine if a solid-state conformation from an X-ray structure is present in the simulated conformational ensemble and, if it is, provide an estimate of the energy difference between the simulated solution-state conformer closest to the solid-state structure and the lowest energy one overall. EKOS also makes it possible to overlay the single conformer represented in a solid-state structure on all ideal secondary structures to find the one it best matches and the preferred side-chain combination. This information is generated free from the bias of the researcher(s) who may have designed the scaffold to mimic a particular secondary structure and side-chain conformation.
An example of the use of EKOS as described above is as follows. Figure 3a shows data for a solid-state structure of DLD-1faf. That structure overlaid on an ideal α-helix with an RMSD (root-mean-square deviation) of 1.5 Å (based on the six Cα + Cβ coordinates of the side-chains; Figure 3b). To estimate the energy of the conformer represented by the X-ray structure, but in solution, the EKOS was used to systematically overlay the simulated conformational ensemble on the X-ray structure to find the conformer with the best fit. In fact, that conformer (matched to 0.46 Å RMSD; Figure 3c) had a simulated energy of 0.91 kcal•mol−1 relative to the lowest one generated, ie it is predicted to be significantly populated in solution. The simulated conformer that overlaid most closely on the ideal α-helix did so with a similar RMSD and energy (0.50 Å, 0.84 kcal•mol−1, Figure 3d). Overall, these data indicate there is a conformer in the simulated ensemble that represents an ideal α-helix better than the one that crystallized. Moreover, the simulated solution-state conformers closest to the solid state structure and to an ideal α-helix seem to have about the same relative energies. The conformer from the X-ray and the most α-helical one from the simulation both had the same side-chain correspondence: i, i+5, i+10.
Figure 3.

a X-Ray crystal structure of DLD-1faf. That structure is shown overlaid upon: b an ideal α-helix; and, c the simulated conformer best matching the X-ray structure. d The simulated conformer best matching an ideal α-helix is overlaid on an ideal α-helix. e Two copies of the X-ray structure overlapping on the C- and N-termini illustrates the helical structure is anticipated to repeat every 3 nm and 5 residues.
Figure 3e shows copies of the X-ray structure arranged such that the C-terminal piperidinone of one is superimposed on the N-terminal piperidinone of the other. This illustrates how the solid-state conformer is anticipated to coil in a helical way, repeating every 5 residues and 3 nm. Parenthetically, Whitesides and coworkers communicated repeated reductive couplings of unfunctionalized 4-piperidinones to give “linker rods”.[18] A “rod” with four piperidine repeats, was 1.6 nm long in the solid state (X-ray), comparable to 1.7 nm for 1faf which has five rings, Figure 3a. Whiteside’s structure is like a straight rod whereas 1 twists to accommodate the chiral piperidinones making it shorter between the N- and C-termini.
EKOS was also used to systematically overlay simulated conformers in the ensemble of 1aaa (all stereomers) on the most common ideal secondary structure elements, as previously described.[7] Figure 4 features data for two stereomers of 1; it shows the RMSDs for the best fitting conformers corresponding to each element of secondary structure (complete data is given in the supporting Figure S1). These data are calibrated relative to the average RMSDs for all the best fitting conformers for a given stereomer. Thus for LLL-1aaa 0.65 Å was the average for the seven best fitting conformers on the seven secondary structures indicated. This graphic indicates the LLL-isomer is predicted to mimic a sheet-turn-sheet closely, ideal helical structures less well, and extended parallel β-sheet- and -strand structures very poorly (Figure 4a). Comparing this with the plot for the DLD-isomer (selected because the X-ray data in Figure 3 was accumulated for that isomer) shows a similar tendency towards helical conformations. The fact that the overlays on sheet-turn-sheet and antiparallel β-sheet structures have identical RMSD values is indicative of the fact that the antiparallel β-sheet region is part of the sheet-turn-sheet motif, and this is where the mimic overlaid.
Figure 4.

Predicted mimicry of secondary structures for: a, b two selected stereomers of 1aaa (full data is given in supporting) and c LLL-Aaaa.
X-ray structures of two other analogs in this series were also obtained and showed variance at the piperidine-piperidone dihedral angles (see supporting Figure S3 and S4). This is consistent with the mimics 1 populating a diversity of conformational states.
Figure 4c shows the corresponding data for LLL-Aaaa. Scaffold A has a strong tendency to mimic β-sheet structures, but not helical ones, and that trend is in fact consistent for all the stereomers of A.
Minimalist mimics are usually designed to perturb real protein-protein interactions (PPIs) by displacing a secondary structure at the interface. However, interfaces rarely have ideal secondary structures, and many do not feature secondary structures at all. Exploring Key Orientations (EKO) is data mining to compare simulated conformational ensembles of minimalist mimics with crystallographically characterized PPI regions, irrespective of the secondary structures involved.[8] Here EKO analyses for LLL-1aaa and LLL-Aaaa were performed to determine if the biases for ideal secondary structures predicted above (using EKOS) are consistent with the statistical match of accessible conformations on real interface secondary structures. To do this we developed a script to assess the secondary structure content of the interface regions that best match simulated conformers. After analyses of tens of thousands of PPI structures, EKO found 68 interface regions that matched a simulated conformer of 1 with RMSDs of <0.3 Å, and 32 that similarly matched conformers of A. Figure 5 shows only LLL-1aaa matched on helical regions at PPI interfaces though it found relatively more sheet interface regions. LLL-1aaa and LLL-Aaaa had a similar proclivity to match with contiguous or non-contiguous motifs of no particular secondary structure (called “single segment” and “multiple segments” in Figure 5). Overall, these observations are consistent with those from comparisons of the simulated conformational ensembles with ideal secondary structures in Figure 4. Four representative overlays for LLL-1aaa at interfaces are illustrated in the supporting material (Figure S5).
Figure 5.
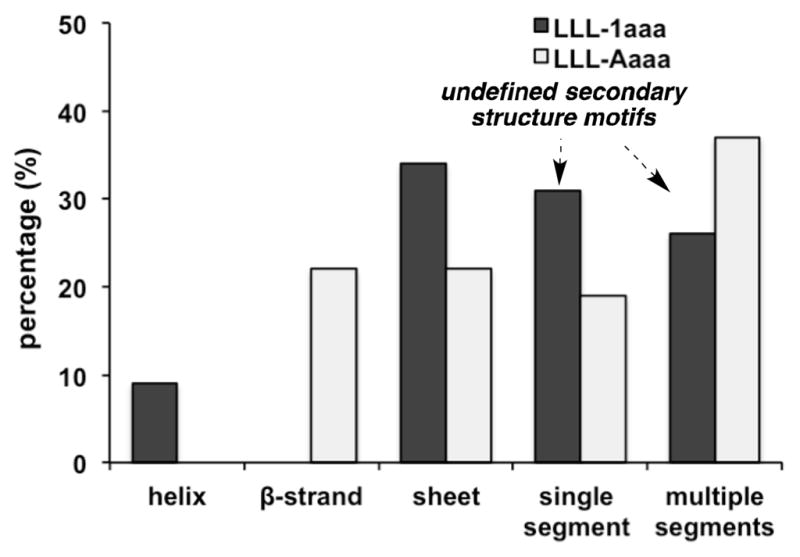
The most favorable PPI matching regions from EKO analyses of LLL-1aaa and LLL-Aaaa compared in terms of the secondary structure motifs that they overlay upon. “Single segment” means the side-chains are three residues on a single chain segment, whereas “multiple segments” refers to situations for which the three important side-chains are on non-contiguous regions of the protein.
In summary, a route to mimics 1 that is not vulnerable to epimerization, and involves facile couplings, was devised. Only six methylene groups differentiate the structures of 1aaa and Aaaa but that drastically changes side-chain orientations in their solution conformers. Overall, we assert that even though none of the conclusions about the secondary structure preferences for 1 in solution can be confirmed spectroscopically, and limited inferences can be drawn from X-ray data, it is much better to have insight from EKOS and EKO than to have none at all. EKOS indicated there are significant differences between the ways simulated preferred conformers of the featured scaffolds overlay on secondary structures; 1aaa is predicted to be more linear, and has a bias towards helical motifs whereas Aaaa best mimics β-strands. Conformers of scaffold 1 have been identified that resemble several different secondary structures, and that gave one referee the impression that this chemotype was more plastic than many other minimalist mimics. We do not think that is true because, as we recently pointed out,[7] all secondary structure mimics tend to adopt many conformers, and some of these can overlay closely on other secondary structures in unexpected ways. Overall, simulated preferred conformers of molecules 1 and A optimally superimpose on different interface regions and proteins when they are systematically overlaid on a huge database of PPI structures using EKO; in other words, these two scaffold designs complement each other. Applications of mimics 1 in perturbation of a particular PPI based on EKO analyses will be reported in due course.
Supplementary Material
Figure 1.

Pyrrolidine-pyrrolidinone mimic A can snake between various S- and crescent-shaped conformers, whereas all the inter-ring σ-rotations in 1 together represent only one significant degree of freedom making the scaffold overall more linear.
Acknowledgments
Financial support for this work was provided by The National Institutes of Health (GM087981) and The Robert A. Welch Foundation (A-1121). We gratefully acknowledge Dr Joe H. Reibenspies contribution of the X-ray crystallographic data.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Additional Information
Experimental procedures and spectroscopic data for compounds 1-11, procedures and results of the overlays on secondary structures, data of protein database mining and X-ray crystallographic data for compound 1faf, 9gg and 11af can be found in the supporting information.
Contributor Information
Dongyue Xin, Department of Chemistry, Texas A & M University, Box 30012, College Station, TX 77841-3012 (USA), Fax: (+1) 979-845-8839.
Dr. Lisa M. Perez, Laboratory for Molecular Simulation, Texas A & M University, Box 30012, College Station, TX 77842
Prof. Thomas R. Ioerger, Department of Computer Science, Texas A & M University, College Station, TX 77843-3112
Prof. Kevin Burgess, Email: Burgess@tamu.edu, Department of Chemistry, Texas A & M University, Box 30012, College Station, TX 77841-3012 (USA), Fax: (+1) 979-845-8839
References
- 1.Yin H, Hamilton AD. Angew Chem, Int Ed. 2005;44:4130–4163. doi: 10.1002/anie.200461786. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2005;117:4200–4235. [Google Scholar]
- 2.Eguchi M, Kahn M. Mini-Rev Med Chem. 2002;2:447–462. doi: 10.2174/1389557023405783. [DOI] [PubMed] [Google Scholar]
- 3.Ko E, Liu J, Burgess K. Chem Soc Rev. 2011;40:4411–4421. doi: 10.1039/c0cs00218f. [DOI] [PubMed] [Google Scholar]
- 4.Angelo NG, Arora PS. J Am Chem Soc. 2005;127:17134–17135. doi: 10.1021/ja056406z. [DOI] [PubMed] [Google Scholar]
- 5.Angelo NG, Arora PS. J Org Chem. 2007;72:7963–7967. doi: 10.1021/jo701292h. [DOI] [PubMed] [Google Scholar]
- 6.Tosovska P, Arora PS. Org Lett. 2010;12:1588–1591. doi: 10.1021/ol1003143. [DOI] [PubMed] [Google Scholar]
- 7.Xin D, Ko E, Perez LM, Ioerger TR, Burgess K. Org Biomol Chem. 2013;11:7789–7801. doi: 10.1039/c3ob41848k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ko E, Raghuraman A, Perez LM, Ioerger TR, Burgess K. J Am Chem Soc. 2013;135:167–173. doi: 10.1021/ja3067258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raghuraman A, Xin D, Perez LM, Burgess K. J Org Chem. 2013;78:4823–4833. doi: 10.1021/jo400323k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raghuraman A, Ko E, Perez LM, Ioerger TR, Burgess K. J Am Chem Soc. 2011;133:12350–12353. doi: 10.1021/ja2033734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodriguguez ML, Doulut M, Heitz SA, Martinez J. Tetrahedron Lett. 1991;32:923–926. [Google Scholar]
- 12.Mosa F, Thirsk C, Vaultier M, Maw G, Whiting A. Org Synth. 2008;85:219–230. [Google Scholar]
- 13.Guichard G, Chaloin O, Cabart F, Marin J, Zhang H, Mihara H. Org Synth. 2008;85:147–157. [Google Scholar]
- 14.Cooper CS, Klock PL, Chu DTW, Hardy DJ, Swanson RN, Plattner JJ. J Med Chem. 1992;35:1392–1398. doi: 10.1021/jm00086a007. [DOI] [PubMed] [Google Scholar]
- 15.Wang Z, Miller EJ, Scalia SJ. Org Lett. 2011;13:6540–6543. doi: 10.1021/ol202840s. [DOI] [PubMed] [Google Scholar]
- 16.Schobert R. Org Synth. 2005;82 No pp given. [Google Scholar]
- 17.Sutherell CL, Thompson S, Scott RTW, Hamilton AD. Chem Commun. 2012;48:9834–9836. doi: 10.1039/c2cc34791a. [DOI] [PubMed] [Google Scholar]
- 18.Semetey V, Moustakas D, Whitesides George M. Angew Chem Int Ed. 2006;45:588–591. doi: 10.1002/anie.200502991. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2006;118:602–605. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.