

Abstract
Spinal cord injury (SCI) is a major unsolved challenge in medicine. Impact trauma to the spinal cord shears blood vessels, causing an immediate ‘primary hemorrhage’. During the hours following trauma, the region of hemorrhage enlarges progressively, with delayed or ‘secondary hemorrhage’ adding to the primary hemorrhage, and effectively doubling its volume. The process responsible for the secondary hemorrhage that results in early expansion of the hemorrhagic lesion is termed ‘progressive hemorrhagic necrosis’ (PHN). PHN is a dynamic process of auto destruction whose molecular underpinnings are only now beginning to be elucidated. PHN results from the delayed, progressive, catastrophic failure of the structural integrity of capillaries. The resulting ‘capillary fragmentation’ is a unique, pathognomonic feature of PHN. Recent work has implicated the Sur1-Trpm4 channel that is newly upregulated in penumbral microvessels as being required for the development of PHN. Targeting the Sur1-Trpm4 channel by gene deletion, gene suppression, or pharmacological inhibition of either of the two channel subunits, Sur1 or Trpm4, yields exactly the same effects histologically and functionally, and exactly the same unique, pathognomonic phenotype – the prevention of capillary fragmentation. The potential advantage of inhibiting Sur1-Trpm4 channels using glibenclamide is a highly promising strategy for ameliorating the devastating sequelae of spinal cord trauma in humans.
Keywords: Spinal cord injury, Sur1-Trpm4 Channel, Glibenclamide, Riluzole
Introduction
Spinal cord injury (SCI) is a major unsolved challenge in medicine. Worldwide, the incidence of SCI ranges from 10 to 83 per million people per year, with half of these patients suffering a complete lesion and one-third becoming tetraplegic [1]. In the United States, 250,000 people live with SCI and 11,000 new cases are added yearly. At present, little can be done to undo or repair the initial damage to spinal cord tissues, but great hope lies in reducing secondary injury processes triggered by the trauma that increase the damage and worsen clinical outcome.
Impact trauma to the spinal cord shears blood vessels, causing an immediate ‘primary hemorrhage’. The volume of the primary hemorrhage is directly related to the severity of the impact [2]. Numerous secondary injury mechanisms are then initiated, on a time scale from seconds to days, with microvascular dysfunction and endothelial cell loss being among the earliest pathophysiological responses observed [3]. During the hours following trauma, the region of hemorrhage enlarges progressively, with delayed or ‘secondary hemorrhage’ adding to the primary hemorrhage, and effectively doubling its volume. The process responsible for the secondary hemorrhage that results in early expansion of the hemorrhagic lesion is termed ‘progressive hemorrhagic necrosis’ (PHN). PHN is a dynamic process of autodestruction whose molecular underpinnings are only now beginning to be elucidated.
PHN results from the delayed, progressive, catastrophic failure of the structural integrity of capillaries [4]. As capillaries in the vicinity of the lesion fail, numerous microhemorrhages (petechial hemorrhages) form and coalesce, resulting in hemorrhagic lesion expansion. This dynamic process wherein the hemorrhagic contusion enlarges progressively results in the autodestruction of spinal cord tissues [4-8]. PHN is particularly damaging because it expands the volume of neural tissue destroyed by the primary injury. The capillary dysfunction implicit with PHN causes tissue ischemia and hypoxia [9], and the extravasated blood resulting from PHN is toxic to CNS cells, especially to the myelin-forming oligodendrocytes of white matter [10], resulting in further injury to neural tissues due to oxidative stress, lipid peroxidation and inflammation. Together, these processes render PHN one of the most destructive mechanisms of secondary injury identified following trauma to the spinal cord.
Early lesion expansion due to PHN was described more than 4 decades ago in an animal model of SCI [11], but has not been well characterized in humans, due largely to the enormous medical and technical challenges that are incurred when performing magnetic resonance imaging (MRI) on severely injured, often medically unstable patients early after trauma. Notwithstanding such difficulties, emerging evidence supports the concept of lesion expansion in humans with SCI [12,13], making this mechanism of secondary injury highly relevant clinically. The importance of these observations lies in the hope that, if early expansion of the hemorrhagic lesion can be halted, patients with acute SCI may suffer the least overall injury.
Progressive Hemorrhagic Necrosis – A Unique Phenotype of Capillary Fragmentation
Early lesion expansion
Histological and MRI studies on animal models of SCI have shown that early expansion of a hemorrhagic contusion is a common feature following trauma to the spinal cord. To our knowledge, the earliest study (weight drop; midline, lower thoracic / upper lumbar) quantifying early lesion expansion reported that, on H&E-stained sections, intramedullary hemorrhages involved an aggregate of 11% of the spinal cord area at the level of maximal bleeding immediately after trauma, and that this increased 2.5-fold to 28% after 8 hr [11]. In our previous study (weight drop; lateral C7), we reported a 2-fold increase in the amount of extravasated blood in tissues from the epicenter during the first 12 hr after trauma [4]. In an MRI study (0.5 mm compression for 30 msec; T7), the T2 lesion volume was found to expand∼1.5-fold over 5.5 hr [14]. In our recent study (weight drop; lateral C7) based on MRI T2 lesion volumes and measurements of hemorrhagic lesion areas, we found a 2–2.5-fold increase in the hemorrhagic contusion that takes place during the first 24 hr after blunt impact trauma to the spinal cord [15].
Together, the animal studies from different laboratories and from different epochs, using different methods to induce trauma, and different approaches to document lesion expansion, establish the existence of significant lesion expansion during the early hours after blunt impact trauma to the spinal cord.
Capillary fragmentation
The secondary hemorrhage that develops following the trauma arises from individual, discrete microscopic (petechial) hemorrhages that appear first near the site of injury then in more distant areas rostrally and caudally, mostly in the gray matter [4,16]. As microscopic hemorrhages form and coalesce, the lesion gradually expands, with a characteristic region of hemorrhage that ‘caps’ the advancing front of the lesion [6,8]. A small hemorrhagic lesion that initially involves primarily the capillary-rich gray matter enlarges several-fold during 3–24 hr after injury (Figure 1A) [11,17].
Figure 1. Hemorrhagic necrosis in rat and human spinal cord after trauma.
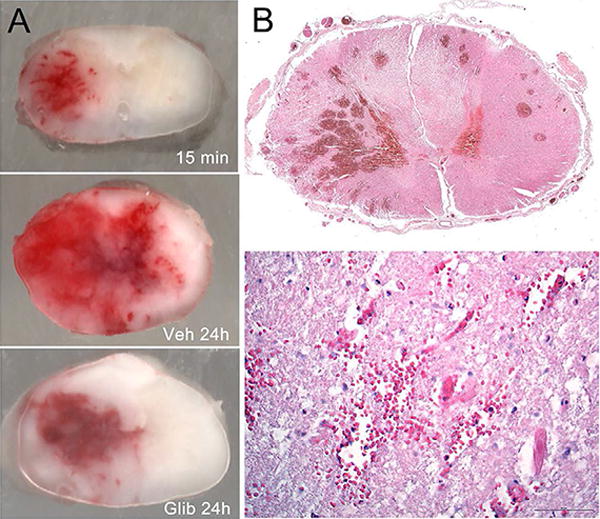
A: Cross sections of rat spinal cords harvested 15 min and 24 hr after a cervical hemi-cord blunt impact trauma, with the rats at 24 hr administered either vehicle (Veh) or glibenclamide (Glib); the rats were perfused to remove intravascular blood. These images show: (i) grossly intact spinal cords with intact pia and no mechanical disruption of tissues; (ii) primary hemorrhage at 15 min that is confined mostly to ipsilateral grey matter; (iii) primary plus secondary hemorrhage at 24 hr, after the hemorrhage has expanded into contralateral grey and white matter in the vehicle-treated rat; (iv) minimal expansion of the hemorrhage at 24 hr in the glibenclamide-treated rat; from Simard et al. [26]. B: Cross sections of human spinal cord 3 days after trauma showing numerous microscopic (petechial) hemorrhages (upper) and extravasated blood near damaged microvessels (lower); from Simard et al. [19].
The formation of discrete microscopic hemorrhages is linked to the delayed progressive catastrophic failure of the structural integrity of capillaries. In static histological tissue sections, vimentin- or CD-31-positive capillaries appear foreshortened, as small segments of nearly the same length and width, a phenomenon termed ‘capillary fragmentation’ (Figure 2A) [4,18-20]. In some cases, including in humans (Figure 1B), extravasated erythrocytes are observed near microvessels that appear broken [19]. The presence of fragmented capillaries in the penumbra of injury is a pathognomonic feature of PHN. As discussed below, when the molecular antecedent of PHN is blocked, capillary fragmentation is absent (Figure 2B,2C,2D), early expansion of the hemorrhagic lesion is halted, and secondary hemorrhage is prevented, i.e., the volume of extravasated blood measured at 24 hr is nearly the same as in the primary hemorrhage, measured 15 min after trauma (Figure 1A) [4,15].
Figure 2. Capillary fragmentation is pathognomonic for progressive hemorrhagic necrosis.
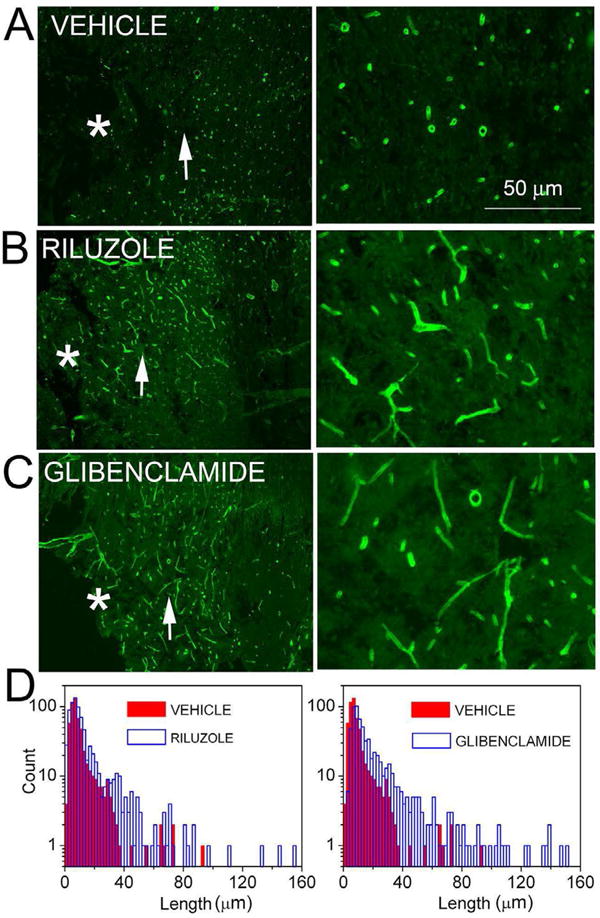
A–C: Images at low (left panels) and at high (right panels) magnification of spinal cord sections in the region of the penumbra immunolabeled for laminin, from rats administered vehicle (A), riluzole (B) or glibenclamide (C); the epicenter of injury is denoted by an asterisk; the region shown at high magnification in the panel on the right is indicated by an arrow in the panel on the left. D: Distribution histograms of the lengths of penumbral microvessels in rats administered vehicle, riluzole or glibenclamide. From Simard et al. [20].
Expression of the Sur1-Trpm4 channel in SCI
Accumulating evidence indicates that the molecular antecedent of PHN is the Sur1-Trpm4 channel. This ion channel is not expressed constitutively, but is transcriptionally upregulated in endothelial and other cells after spinal cord trauma. In mice, rats and humans, upregulation of the regulatory subunit of the channel, Sur1 protein and its mRNA (Abcc8), has been demonstrated in microvessels, neurons and white matter using immunohistochemistry, immunoblot analysis, and in situ hybridization [4,19,21]. In mice and rats, upregulation of the pore forming subunit of the channel, Trpm4 protein and its mRNA (Trpm4), has been shown in microvessels and neurons using immunohistochemistry, quantitative RT-PCR and in situ hybridization [18,21].
Co-association of the regulatory and pore-forming subunits to form functional Sur1-Trpm4 channels recently was shown using Förster resonance energy transfer (FRET) imaging microscopy and co-immunoprecipitation [21]. Antibody-based FRET was used to evaluate rat spinal cord tissues before and after injury. In uninjured spinal cord, Sur1 and Trpm4 immunolabeling was minimal [4,18,19], and FRET signals were absent. However, 24 hours after spinal cord trauma, Sur1 and Trpm4 immunolabeling was prominent, immunolabeling for Sur1 and Trpm4 co-localized, and FRET signals were detected in various cellular structures, including microvessels (Figure 3A).
Figure 3. The Sur1-Trpm4 channel in spinal cord injury.
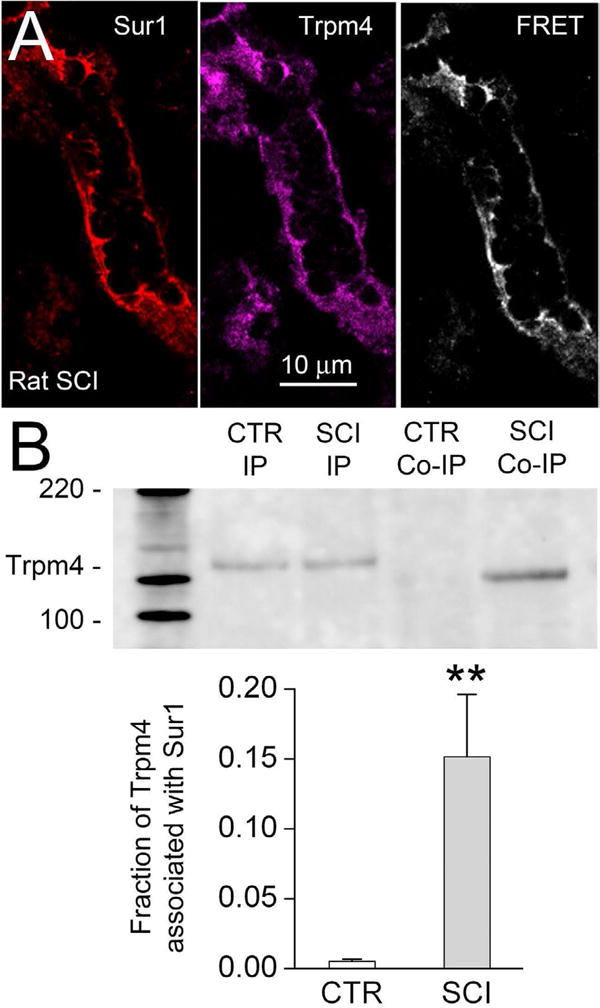
A: Immunolabeling for Sur1 and Trpm4, as indicated, and FRET image of a microvessel 24 hr after spinal cord injury showing the co-association of Sur1 and Trpm4 to form functional Sur1-Trpm4 channels. B: Immunoblot using anti-Trpm4 antibody of protein immuno-isolated from uninjured spinal cord (CTR) or spinal cord 6 hours after injury (SCI) using anti-Trpm4 antibody (IP) or anti-Sur1 antibody (co-IP); note that co-associated Sur1-Trpm4 is found only after spinal cord injury. The bar graph shows the result of densitometric analysis (mean ± S.E.) of the abundance of Trpm4 associated with Sur1 in the uninjured spinal cord versus 6 hr after spinal cord injury (SCI) (n=3; **, P<0.01). From Woo et al. [21].
In the same report [21], co-immunoprecipitation experiments showed abundant Sur1 and Trpm4 after spinal cord trauma. Co-immunoprecipitation using anti-Trpm4 antibody yielded Sur1, and co-immunoprecipitation using anti-Sur1 antibody yielded Trpm4. Importantly, coimmunoprecipitation using anti-Sur1 antibody yielded Trpm4 only after spinal cord injury, not in uninjured spinal cord (Figure 3B). Together, these findings with co-immunoprecipitation and FRET demonstrate that Sur1 and Trpm4 co-assemble in vivo to form functional Sur1-Trpm4 channels following spinal cord injury.
Progressive hemorrhagic necrosis – role of the Sur1-Trpm4 channel
Various cell types exhibit de novo upregulation of Sur1-Trpm4 channels after spinal cord trauma, but secondary hemorrhage due to PHN is linked specifically to channel upregulation in microvessels [4,18,19]. Sur1-Trpm4 channels have been shown to be responsible for the necrotic death of endothelial cells that results in delayed fragmentation of capillaries and formation of microhemorrhages.
Evidence for involvement of the Sur1-Trpm4 channel in PHN comes from an analysis of the effects of gene deletion, gene suppression, or pharmacological inhibition of the two channel subunits, Sur1 and Trpm4. Remarkably, interfering with the function of either of the two channel subunits yields exactly the same effect histologically and functionally.
The phenotype observed after spinal cord injury in Abcc8–/– mice is exactly the same as in Trpm4–/– mice [18,19]. Both genotypes show the same post-SCI phenotype, and both are equally protected from PHN. Both genotypes show minimal secondary hemorrhage and lesion expansion, and the absence of capillary fragmentation, the hallmark of PHN. In a model of unilateral trauma (T9), functional outcomes, measured using the Basso mouse scale, and lesion volumes at 1 week are significantly better in both knockout mice compared to wild type mice.
Similarly, the phenotype observed after spinal cord injury in rats administered antisense oligodeoxynucleotide (AS-ODN) targeting Abcc8 is exactly the same as in rats administered AS-ODN targeting Trpm4. Both show the same phenotype, and both are equally protected from PHN. Both show minimal secondary hemorrhage and lesion expansion, and the absence of capillary fragmentation, the hallmark of PHN. In a model of unilateral trauma (C7), functional outcomes, measured using the Basso, Beattie, Bresnahan scale, and lesion volumes at 6 weeks are significantly better in rats administered either AS-ODN compared to rats administered scrambled ODN.
Pharmacological blockade of Sur1
Pharmacological blockade of Sur1 has been studied using two highly selective agents, glibenclamide and repaglinide [4]. Glibenclamide is a second generation sulfonylurea drug that binds to Sur1 with subnanomolar or nanomolar affinity (0.4–4.0 nM) [22] and potently inhibits the Sur1-Trpm4 channel (EC50 = 48 nM) [21,23]. Repaglinide is a member of a distinct class of insulin secretagogues that are structurally unrelated to sulphonylureas and whose binding site on Sur1 may differ from that of sulfonylureas [24]. Like glibenclamide, repaglinide produces high-affinity block of both native and recombinant β-cell KATP channels (IC50 = 0.9–7 nM), and shows higher potency in inhibiting pancreatic Sur1-regulated KATP channels than cardiovascular Sur2-regulated channels [25].
In rat models of SCI, glibenclamide and repaglinide exert beneficial effects that exhibit the signature features of inhibition of PHN. Rats treated with either glibenclamide or repaglinide show minimal secondary hemorrhage and lesion expansion, and the absence of capillary fragmentation, the hallmark of PHN (Figure 2C,2D). In models of unilateral or bilateral trauma (C7), functional outcomes, measured using the Basso, Beattie, Bresnahan scale, and lesion volumes at 6 weeks are significantly better in rats administered either compound, compared to controls [4,26]. Notably, direct comparison between glibenclamide and AS-ODN directed against Abcc8 shows equivalent effects in rats, in terms of functional outcomes and lesion volumes at 6 weeks, with neither compound exhibiting any observable toxicity [19].
In 4 separate series on rats from our laboratory [4,15,19,26], and in one series from an independent laboratory [27], glibenclamide treatment beginning shortly after trauma was found to be highly effective in reducing lesion size and improving neurological function. In a 6th series of rats, treatment at the clinically more relevant time of 3 hr after trauma also was found to be highly beneficial [20]. As might be expected, the magnitude of the benefit observed with glibenclamide depends on the magnitude of the primary injury [26], but all studies to date examining functional outcome and lesion size at 6 weeks have demonstrated a significant treatment effect, regardless of the initial severity [19,20,26].
Pharmacological blockade of Trpm4
At present, specific pharmacological blockade of Trpm4 is not feasible, possibly because of structural similarities of Trpm4 to other ion channels. Drugs that block Trpm4 are pleotropic, affecting other molecular targets. To date, pharmacological blockade of Trpm4 in SCI models has been pursued using flufenamic acid or riluzole. Flufenamic acid is an open-channel blocker of Trpm4 and of numerous other nonselective cationic channels, a widely expressed, heterogeneous family of channels of diverse molecular origins [28]. The benzothiazole, riluzole, recently was shown to block Trpm4 currents (IC50 = 31 μM) [20], but it also blocks other molecules. Riluzole was first proposed to inhibit glutamate release [29-32], thus protecting neurons from excitotoxic damage [33,34]. At micromolar concentrations (IC50, 3–10 μM), riluzole is considered to be a relatively selective blocker of ‘persistent sodium currents’ in cardiac myocytes and CNS neurons, including spinal cord neurons, where the molecular identity of the channel(s) responsible for these currents is not known [35-40].
In rat models of SCI, flufenamic acid and riluzole exert beneficial effects that exhibit the signature features of inhibition of PHN. The effects of flufenamic acid and riluzole are qualitatively similar to those of glibenclamide, including reduced necrotic lesion volumes and better functional outcomes [4,20,41,42]. Although a number of molecular targets could be involved, it is striking that both compounds show the same phenotype as gene suppression of Sur1 [19] or gene suppression of Trpm4 [18]. Rats treated with either flufenamic acid or riluzole show minimal secondary hemorrhage and lesion expansion, and the absence of capillary fragmentation, the hallmark of PHN (Figure 2B,2D). In a model of unilateral trauma (C7), functional outcomes, measured using the Basso, Beattie, Bresnahan scale, and lesion volumes at 6 weeks are significantly better in rats administered either compound, compared to controls. To our knowledge, the inhibition of other molecular targets potentially affected by flufenamic acid and riluzole would not be expected to manifest the unique phenotype of inhibiting capillary fragmentation, since this effect is specific for the Sur1-Trpm4 channel.
Glibenclamide vs. riluzole
Riluzole has been found to be efficacious in preclinical models of SCI [20,43,44], may have a beneficial effect on motor outcome in cervical SCI, as recently reported in a small open-label Phase I clinical trial [45], and currently is the only drug that is anticipated for study in a Phase II clinical trial of acute SCI (Clinical Trials.gov identifier, NCT01597518). In the anticipated clinical trial, riluzole will be administered enterally at a dose of 2 × 100 mg the first 24 hours followed by 2 × 50 mg for the following 13 days after injury.
A recent preclinical study using a rat model of SCI, which was so severe as to have attendant mortality, compared treatment with riluzole (2.5 mg/kg IP every 12 hr × 1 week) vs. glibenclamide (10 μg/kg IP loading dose plus 200 ng/hr continuous subcutaneous infusion × 1 week), starting 3 hr after trauma [20]. This study found that glibenclamide is superior to riluzole in terms of both toxicity and efficacy. During the acute phase after trauma, both drugs reduced capillary fragmentation and PHN (Figure 2), and both prevented death. At 6 weeks, modified (unilateral) Basso, Beattie, Bresnahan locomotor scores were similar, but measures of complex function (grip strength, rearing, accelerating rotarod) and tissue sparing were significantly better with glibenclamide than with riluzole. Note that in this preclinical study, riluzole was administered parenterally, which yields better bioavailability than enteral administration, and it was administered at a higher dose (on a ‘per kilogram’ basis), compared to the dose proposed for the anticipated clinical trial.
Apart from inhibiting PHN via blockade of Sur1-Trpm4, riluzole exerts other biological effects. At micromolar concentrations (IC50, 3–10 μM), riluzole blocks ‘persistent sodium currents’ in cardiac myocytes and CNS neurons, including spinal cord neurons, where the molecular identity of the channel(s) responsible for these currents is not known [35-40]. Riluzole blocks several molecularly identified potassium channels [46-50], and calcium channels [51-53]. Riluzole also inhibits glutamate release [29,30-32], and it interacts with γ-aminobutyric acid A and glycine receptor-activated channels [30,54-56]. Riluzole also directly binds to and inhibits protein kinase C [57]. The role of any of these molecular targets in PHN is not known. It is possible that riluzole exerts part of its salutary effects via one these mechanisms, in addition to Sur1-Trpm4 inhibition, but specific involvement of any of these mechanisms has not been shown. Notably, involvement of glutamate antagonism is now thought to be unlikely [58,59].
A high degree of non-specificity in a drug generally is undesirable, since this can lead to untoward side-effects and undue toxicity. Non-specificity may account for the acute toxicity observed with high doses of riluzole, which includes somnolence, coma or a moribund state [60,61]. In addition, riluzole exhibits an unusual, dose-limiting CNS toxicity that is present only in CNS trauma, not in uninjured controls: mortality rates of 0%, 8% and 70% are observed with 4, 6 and 8 mg/kg IP every 12 hr, respectively, in rats after SCI, whereas these doses are well tolerated in normal uninjured rats [44].
Drug specificity is less problematic with glibenclamide. Apart from high-potency block of Sur1-Trpm4 channels (EC50 = 48 nM) [21,23], glibenclamide also blocks Sur1-regulated KATP channels in pancreatic β cells [62]. However, at the doses used in CNS ischemia and trauma, the potential consequence of block of pancreatic KATP channels – hypoglycemia – is not observed; infusion of 200 ng/hr of glibenclamide in rats has a minimal effect on serum glucose [4,63,64]. Sur2-regulated KATP channels in cardiac and smooth muscle cells are less sensitive to block by glibenclamide, by a factor of 10 times or more [62,65]. Other ATP-binding cassette proteins of the ABC gene family may be blocked by glibenclamide, but only at micromolar concentrations [66], far greater than the concentrations acheived with infusion of 200 ng/hr in rats.
In the 6 series of rats reported to date on glibenclamide in SCI (see above), the drug was delivered by constant subcutaneous infusion. From a pharmacokinetic perspective, cutaneous delivery of glibenclamide is highly effective for maintaining steady plasma levels, is superior to enteral administration, and appears to be equivalent to intravenous (IV) administration [67]. Constant subcutaneous infusion of glibenclamide was used in the preclinical studies as a convenient alternative to constant IV infusion, as is used with injectable glibenclamide (RP-1127) in clinical trials for other CNS indications (ClinicalTrials.gov identifiers: NCT01454154; NCT01268683; NCT01794182). In the animal studies, no clinically relevant hypoglycemia or other toxicity has been detected with infusions of 200 ng/hr [4,63,64] or 400 ng/hr [68]. In a Phase I trial of RP-1127 in 16 normal subjects (ClinicalTrials.gov identifier: NCT01132703), a 3-day IV infusion (125 μg/hr) produced no clinically significant hypoglycemia or other serious adverse event (S. Jacobson, personal communication).
From the perspective of efficacy in targeting the Sur1-Trpm4 channel for reducing PHN, as well as from the perspective of safety and tolerability, glibenclamide may be a better choice than riluzole for the treatment of acute spinal cord injury.
Discussion
Each year, traumatic injury to the spinal cord devastates the lives of thousands of people worldwide. Short of preventing primary injury, the best hope for reducing the life-long impact of SCI rests with decreasing the secondary injury that results from PHN occurring during the acute phase after trauma. Although progress has been made in axonal and dendritic remodeling, cell replacement therapies, and rehabilitation, it is generally acknowledged that these treatments work best when administered to patients with the smallest possible lesion. To date, clinical trials with agents such as methylprednisolone (NASCIS II and III) and GM-1 ganglioside have shown risk-benefit profiles that are not sufficiently favorable to warrant routine clinical use, and other therapies intended for treatment in the acute phase have yet to be proven [69].
As reviewed here, emerging data indicate that in the earliest phase after trauma, the Sur1-Trpm4 channel is newly upregulated in the penumbra of injury, and that the expression of this channel in penumbral microvessels is integral to the subsequent development of PHN, which is responsible to early expansion of the hemorrhagic lesion. Also, as reviewed here, considerable data have now been published showing that targeting this channel by gene deletion, by gene suppression, or by pharmacological inhibition of either of the two channel subunits yields exactly the same effect histologically and functionally, and the exactly same unique, pathognomonic phenotype – the prevention of capillary fragmentation in the penumbra. The possibility of inhibiting the Sur1-Trpm4 channel using glibenclamide is a promising strategy for ameliorating the devastating sequelae of spinal cord trauma in humans.
Acknowledgments
This work was supported by grants to JMS from the Veterans Administration (Baltimore), the National Institute of Neurological Disorders and Stroke (NINDS) (NS060801), and the Department of the Army (W81XWH 1010898); to VG from NINDS (NS061934).
Footnotes
Conflict of interest statement: JMS holds a US patent (#7,872,048), “Methods for treating spinal cord injury with a compound that inhibits a NC (Ca-ATP) channel “. JMS is a member of the scientific advisory board and holds shares in Remedy Pharmaceuticals. No support, direct or indirect, was provided to JMS, or for this project, by Remedy Pharmaceuticals. All other authors declare no conflict of interest.
References
- 1.Wyndaele M, Wyndaele JJ. Incidence, prevalence and epidemiology of spinal cord injury: what learns a worldwide literature survey? Spinal Cord. 2006;44:523–529. doi: 10.1038/sj.sc.3101893. [DOI] [PubMed] [Google Scholar]
- 2.Mautes AE, Weinzierl MR, Donovan F, Noble LJ. Vascular events after spinal cord injury: contribution to secondary pathogenesis. Phys Ther. 2000;80:673–687. [PubMed] [Google Scholar]
- 3.Fassbender JM, Whittemore SR, Hagg T. Targeting microvasculature for neuroprotection after SCI. Neurotherapeutics. 2011;8:240–251. doi: 10.1007/s13311-011-0029-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simard JM, Tsymbalyuk O, Ivanov A, Ivanova S, Bhatta S, et al. Endothelial sulfonylurea receptor 1-regulated NC Ca-ATP channels mediate progressive hemorrhagic necrosis following spinal cord injury. J Clin Invest. 2007;117:2105–2113. doi: 10.1172/JCI32041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noble LJ, Wrathall JR. Correlative analyses of lesion development and functional status after graded spinal cord contusive injuries in the rat. Exp Neurol. 1989;103:34–40. doi: 10.1016/0014-4886(89)90182-9. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Z, Krebs CJ, Guth L. Experimental analysis of progressive necrosis after spinal cord trauma in the rat: etiological role of the inflammatory response. Exp Neurol. 1997;143:141–152. doi: 10.1006/exnr.1996.6355. [DOI] [PubMed] [Google Scholar]
- 7.Steward O, Schauwecker PE, Guth L, Zhang Z, Fujiki M, et al. Genetic approaches to neurotrauma research: opportunities and potential pitfalls of murine models. Exp Neurol. 1999;157:19–42. doi: 10.1006/exnr.1999.7040. [DOI] [PubMed] [Google Scholar]
- 8.Guth L, Zhang Z, Steward O. The unique histopathological responses of the injured spinal cord. Implications for neuroprotective therapy. Ann N Y Acad Sci. 1999;890:366–384. doi: 10.1111/j.1749-6632.1999.tb08017.x. [DOI] [PubMed] [Google Scholar]
- 9.Tator CH, Koyanagi I. Vascular mechanisms in the pathophysiology of human spinal cord injury. J Neurosurg. 1997;86:483–492. doi: 10.3171/jns.1997.86.3.0483. [DOI] [PubMed] [Google Scholar]
- 10.Regan RF, Guo Y. Toxic effect of hemoglobin on spinal cord neurons in culture. J Neurotrauma. 1998;15:645–653. doi: 10.1089/neu.1998.15.645. [DOI] [PubMed] [Google Scholar]
- 11.Balentine JD. Pathology of experimental spinal cord trauma. I. The necrotic lesion as a function of vascular injury. Lab Invest. 1978;39:236–253. [PubMed] [Google Scholar]
- 12.Fleming JC, Norenberg MD, Ramsay DA, Dekaban GA, Marcillo AE, et al. The cellular inflammatory response in human spinal cords after injury. Brain. 2006;129:3249–3269. doi: 10.1093/brain/awl296. [DOI] [PubMed] [Google Scholar]
- 13.Aarabi B, Simard JM, Kufera JA, Alexander M, Zacherl KM, et al. Intramedullary lesion expansion on magnetic resonance imaging in patients with motor complete cervical spinal cord injury. J Neurosurg Spine. 2012;17:243–250. doi: 10.3171/2012.6.SPINE12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bilgen M, Abbe R, Liu SJ, Narayana PA. Spatial and temporal evolution of hemorrhage in the hyperacute phase of experimental spinal cord injury: in vivo magnetic resonance imaging. Magn Reson Med. 2000;43:594–600. doi: 10.1002/(sici)1522-2594(200004)43:4<594::aid-mrm15>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 15.Simard JM, Popovich PG, Tsymbalyuk O, Caridi J, Gullapalli RP, et al. MRI evidence that glibenclamide reduces lesion expansion in a rat model of spinal cord injury. Spinal Cord. 2013 doi: 10.1038/sc.2013.99. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawata K, Morimoto T, Ohashi T, Tsujimoto S, Hoshida T, et al. Experimental study of acute spinal cord injury: a study of spinal blood flow. No Shinkei Geka. 1993;21:239–245. [PubMed] [Google Scholar]
- 17.Iizuka H, Yamamoto H, Iwasaki Y, Yamamoto T, Konno H. Evolution of tissue damage in compressive spinal cord injury in rats. J Neurosurg. 1987;66:595–603. doi: 10.3171/jns.1987.66.4.0595. [DOI] [PubMed] [Google Scholar]
- 18.Gerzanich V, Woo SK, Vennekens R, Tsymbalyuk O, Ivanova S, et al. De novo expression of Trpm4 initiates secondary hemorrhage in spinal cord injury. Nat Med. 2009;15:185–191. doi: 10.1038/nm.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simard JM, Woo SK, Norenberg MD, Tosun C, Chen Z, et al. Brief suppression of Abcc8 prevents autodestruction of spinal cord after trauma. Sci Transl Med. 2010;2:28ra29. doi: 10.1126/scitranslmed.3000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simard JM, Tsymbalyuk O, Keledjian K, Ivanov A, Ivanova S, et al. Comparative effects of glibenclamide and riluzole in a rat model of severe cervical spinal cord injury. Exp Neurol. 2012;233:566–574. doi: 10.1016/j.expneurol.2011.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woo SK, Kwon MS, Ivanov A, Gerzanich V, Simard JM. The sulfonylurea receptor 1 (Sur1)-transient receptor potential melastatin 4 (Trpm4) channel. J Biol Chem. 2013;288:3655–3667. doi: 10.1074/jbc.M112.428219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aguilar-Bryan L, Nelson DA, Vu QA, Humphrey MB, Boyd AE., 3rd Photoaffinity labeling and partial purification of the beta cell sulfonylurea receptor using a novel, biologically active glyburide analog. J Biol Chem. 1990;265:8218–8224. [PubMed] [Google Scholar]
- 23.Chen M, Dong Y, Simard JM. Functional coupling between sulfonylurea receptor type 1 and a nonselective cation channel in reactive astrocytes from adult rat brain. J Neurosci. 2003;23:8568–8577. doi: 10.1523/JNEUROSCI.23-24-08568.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hansen AM, Christensen IT, Hansen JB, Carr RD, Ashcroft FM, et al. Differential interactions of nateglinide and repaglinide on the human beta-cell sulphonylurea receptor 1. Diabetes. 2002;51:2789–2795. doi: 10.2337/diabetes.51.9.2789. [DOI] [PubMed] [Google Scholar]
- 25.Stephan D, Winkler M, Kühner P, Russ U, Quast U. Selectivity of repaglinide and glibenclamide for the pancreatic over the cardiovascular K(ATP) channels. Diabetologia. 2006;49:2039–2048. doi: 10.1007/s00125-006-0307-3. [DOI] [PubMed] [Google Scholar]
- 26.Simard JM, Popovich PG, Tsymbalyuk O, Gerzanich V. Spinal cord injury with unilateral versus bilateral primary hemorrhage--effects of glibenclamide. Exp Neurol. 2012;233:829–835. doi: 10.1016/j.expneurol.2011.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Popovich PG, Lemeshow S, Gensel JC, Tovar CA. Independent evaluation of the effects of glibenclamide on reducing progressive hemorrhagic necrosis after cervical spinal cord injury. Exp Neurol. 2012;233:615–622. doi: 10.1016/j.expneurol.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peña F, Ordaz B. Non-selective cation channel blockers: potential use in nervous system basic research and therapeutics. Mini Rev Med Chem. 2008;8:812–819. doi: 10.2174/138955708784912166. [DOI] [PubMed] [Google Scholar]
- 29.Benavides J, Camelin JC, Mitrani N, Flamand F, Uzan A, et al. 2-Amino-6-trifuoromethoxy benzothiazole, a possible antagonist of excitatory amino acid neurotransmission--II. Biochemical properties. Neuropharmacology. 1985;24:1085–1092. doi: 10.1016/0028-3908(85)90196-0. [DOI] [PubMed] [Google Scholar]
- 30.Martin D, Thompson MA, Nadler JV. The neuroprotective agent riluzole inhibits release of glutamate and aspartate from slices of hippocampal area CA1. Eur J Pharmacol. 1993;250:473–476. doi: 10.1016/0014-2999(93)90037-i. [DOI] [PubMed] [Google Scholar]
- 31.Zona C, Cavalcanti S, De Sarro G, Siniscalchi A, Marchetti C, et al. Kainate-induced currents in rat cortical neurons in culture are modulated by riluzole. Synapse. 2002;43:244–251. doi: 10.1002/syn.10040. [DOI] [PubMed] [Google Scholar]
- 32.Coderre TJ, Kumar N, Lefebvre CD, Yu JS. A comparison of the glutamate release inhibition and anti-allodynic effects of gabapentin, lamotrigine, and riluzole in a model of neuropathic pain. J Neurochem. 2007;100:1289–1299. doi: 10.1111/j.1471-4159.2006.04304.x. [DOI] [PubMed] [Google Scholar]
- 33.Azbill RD, Mu X, Springer JE. Riluzole increases high-affinity glutamate uptake in rat spinal cord synaptosomes. Brain Res. 2000;871:175–180. doi: 10.1016/s0006-8993(00)02430-6. [DOI] [PubMed] [Google Scholar]
- 34.Dunlop J, Beal McIlvain H, She Y, Howland DS. Impaired spinal cord glutamate transport capacity and reduced sensitivity to riluzole in a transgenic superoxide dismutase mutant rat model of amyotrophic lateral sclerosis. J Neurosci. 2003;23:1688–1696. doi: 10.1523/JNEUROSCI.23-05-01688.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Urbani A, Belluzzi O. Riluzole inhibits the persistent sodium current in mammalian CNS neurons. Eur J Neurosci. 2000;12:3567–3574. doi: 10.1046/j.1460-9568.2000.00242.x. [DOI] [PubMed] [Google Scholar]
- 36.Tazerart S, Viemari JC, Darbon P, Vinay L, Brocard F. Contribution of persistent sodium current to locomotor pattern generation in neonatal rats. J Neurophysiol. 2007;98:613–628. doi: 10.1152/jn.00316.2007. [DOI] [PubMed] [Google Scholar]
- 37.Lamanauskas N, Nistri A. Riluzole blocks persistent Na+ and Ca2+ currents and modulates release of glutamate via presynaptic NMDA receptors on neonatal rat hypoglossal motoneurons in vitro. Eur J Neurosci. 2008;27:2501–2514. doi: 10.1111/j.1460-9568.2008.06211.x. [DOI] [PubMed] [Google Scholar]
- 38.Lamas JA, Romero M, Reboreda A, Sánchez E, Ribeiro SJ. A riluzole-and valproate-sensitive persistent sodium current contributes to the resting membrane potential and increases the excitability of sympathetic neurones. Pflugers Arch. 2009;458:589–599. doi: 10.1007/s00424-009-0648-0. [DOI] [PubMed] [Google Scholar]
- 39.Weiss SM, Saint DA. The persistent sodium current blocker riluzole is antiarrhythmic and anti-ischaemic in a pig model of acute myocardial infarction. PLoS One. 2010;5:e14103. doi: 10.1371/journal.pone.0014103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xie RG, Zheng DW, Xing JL, Zhang XJ, Song Y, et al. Blockade of persistent sodium currents contributes to the riluzole-induced inhibition of spontaneous activity and oscillations in injured DRG neurons. PLoS One. 2011;6:e18681. doi: 10.1371/journal.pone.0018681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ates O, Cayli SR, Gurses I, Turkoz Y, Tarim O, et al. Comparative neuroprotective effect of sodium channel blockers after experimental spinal cord injury. J Clin Neurosci. 2007;14:658–665. doi: 10.1016/j.jocn.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 42.Schwartz G, Fehlings MG. Evaluation of the neuroprotective effects of sodium channel blockers after spinal cord injury: improved behavioral and neuroanatomical recovery with riluzole. J Neurosurg. 2001;94:245–256. doi: 10.3171/spi.2001.94.2.0245. [DOI] [PubMed] [Google Scholar]
- 43.Wahl F, Stutzmann JM. Neuroprotective effects of riluzole in neurotrauma models: a review. Acta Neurochir Suppl. 1999;73:103–110. doi: 10.1007/978-3-7091-6391-7_18. [DOI] [PubMed] [Google Scholar]
- 44.Wu Y, Satkunendrarajah K, Teng Y, Chow DS, Buttigieg J, et al. Delayed post-injury administration of riluzole is neuroprotective in a preclinical rodent model of cervical spinal cord injury. J Neurotrauma. 2013;30:441–452. doi: 10.1089/neu.2012.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grossman RG, Fehlings M, Frankowski R, Burau KD, Chow D, et al. A Prospective Multicenter Phase 1 Matched Comparison Group Trial of Safety, Pharmacokinetics, and Preliminary Efficacy of Riluzole in Patients with Traumatic Spinal Cord Injury. J Neurotrauma. 2013 doi: 10.1089/neu.2013.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zona C, Siniscalchi A, Mercuri NB, Bernardi G. Riluzole interacts with voltage-activated sodium and potassium currents in cultured rat cortical neurons. Neuroscience. 1998;85:931–938. doi: 10.1016/s0306-4522(97)00604-0. [DOI] [PubMed] [Google Scholar]
- 47.Duprat F, Lesage F, Patel AJ, Fink M, Romey G, et al. The neuroprotective agent riluzole activates the two P domain K(+) channels TREK-1 and TRAAK. Mol Pharmacol. 2000;57:906–912. [PubMed] [Google Scholar]
- 48.Cao YJ, Dreixler JC, Couey JJ, Houamed KM. Modulation of recombinant and native neuronal SK channels by the neuroprotective drug riluzole. Eur J Pharmacol. 2002;449:47–54. doi: 10.1016/s0014-2999(02)01987-8. [DOI] [PubMed] [Google Scholar]
- 49.Ahn HS, Choi JS, Choi BH, Kim MJ, Rhie DJ, et al. Inhibition of the cloned delayed rectifier K+ channels, Kv1.5 and Kv3.1, by riluzole. Neuroscience. 2005;133:1007–1019. doi: 10.1016/j.neuroscience.2005.03.041. [DOI] [PubMed] [Google Scholar]
- 50.Xu L, Enyeart JA, Enyeart JJ. Neuroprotective agent riluzole dramatically slows inactivation of Kv1.4 potassium channels by a voltage-dependent oxidative mechanism. J Pharmacol Exp Ther. 2001;299:227–237. [PubMed] [Google Scholar]
- 51.Huang CS, Song JH, Nagata K, Yeh JZ, Narahashi T. Effects of the neuroprotective agent riluzole on the high voltage-activated calcium channels of rat dorsal root ganglion neurons. J Pharmacol Exp Ther. 1997;282:1280–1290. [PubMed] [Google Scholar]
- 52.Siniscalchi A, Bonci A, Mercuri NB, Bernardi G. Effects of riluzole on rat cortical neurones: an in vitro electrophysiological study. Br J Pharmacol. 1997;120:225–230. doi: 10.1038/sj.bjp.0700905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stefani A, Spadoni F, Bernardi G. Differential inhibition by riluzole, lamotrigine, and phenytoin of sodium and calcium currents in cortical neurons: implications for neuroprotective strategies. Exp Neurol. 1997;147:115–122. doi: 10.1006/exnr.1997.6554. [DOI] [PubMed] [Google Scholar]
- 54.Umemiya M, Berger AJ. Inhibition by riluzole of glycinergic postsynaptic currents in rat hypoglossal motoneurones. Br J Pharmacol. 1995;116:3227–3230. doi: 10.1111/j.1476-5381.1995.tb15128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mohammadi B, Krampfl K, Moschref H, Dengler R, Bufler J. Interaction of the neuroprotective drug riluzole with GABA(A) and glycine receptor channels. Eur J Pharmacol. 2001;415:135–140. doi: 10.1016/s0014-2999(01)00847-0. [DOI] [PubMed] [Google Scholar]
- 56.He Y, Benz A, Fu T, Wang M, Covey DF, et al. Neuroprotective agent riluzole potentiates postsynaptic GABA(A) receptor function. Neuropharmacology. 2002;42:199–209. doi: 10.1016/s0028-3908(01)00175-7. [DOI] [PubMed] [Google Scholar]
- 57.Noh KM, Hwang JY, Shin HC, Koh JY. A novel neuroprotective mechanism of riluzole: direct inhibition of protein kinase C. Neurobiol Dis. 2000;7:375–383. doi: 10.1006/nbdi.2000.0297. [DOI] [PubMed] [Google Scholar]
- 58.Lips J, de Haan P, Bodewits P, Vanicky I, Dzoljic M, et al. Neuroprotective effects of riluzole and ketamine during transient spinal cord ischemia in the rabbit. Anesthesiology. 2000;93:1303–1311. doi: 10.1097/00000542-200011000-00025. [DOI] [PubMed] [Google Scholar]
- 59.McAdoo DJ, Hughes MG, Nie L, Shah B, Clifton C, et al. The effect of glutamate receptor blockers on glutamate release following spinal cord injury. Lack of evidence for an ongoing feedback cascade of damage --> glutamate release --> damage --> glutamate release --> etc. Brain Res. 2005;1038:92–99. doi: 10.1016/j.brainres.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 60.Mantz J, Chéramy A, Thierry AM, Glowinski J, Desmonts JM. Anesthetic properties of riluzole (54274 RP), a new inhibitor of glutamate neurotransmission. Anesthesiology. 1992;76:844–848. doi: 10.1097/00000542-199205000-00023. [DOI] [PubMed] [Google Scholar]
- 61.Kitzman PH. Effectiveness of riluzole in suppressing spasticity in the spinal cord injured rat. Neurosci Lett. 2009;455:150–153. doi: 10.1016/j.neulet.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 62.Bryan J, Aguilar-Bryan L. Sulfonylurea receptors: ABC transporters that regulate ATP-sensitive K(+) channels. Biochim Biophys Acta. 1999;1461:285–303. doi: 10.1016/s0005-2736(99)00164-9. [DOI] [PubMed] [Google Scholar]
- 63.Simard JM, Yurovsky V, Tsymbalyuk N, Melnichenko L, Ivanova S, et al. Protective effect of delayed treatment with low-dose glibenclamide in three models of ischemic stroke. Stroke. 2009;40:604–609. doi: 10.1161/STROKEAHA.108.522409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barthel W, Markwardt F. Aggregation of blood platelets by adrenaline and its uptake. Biochem Pharmacol. 1975;24:1903–1904. doi: 10.1016/0006-2952(75)90415-3. [DOI] [PubMed] [Google Scholar]
- 65.Simard JM, Woo SK, Bhatta S, Gerzanich V. Drugs acting on SUR1 to treat CNS ischemia and trauma. Curr Opin Pharmacol. 2008;8:42–49. doi: 10.1016/j.coph.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bessadok A, Garcia E, Jacquet H, Martin S, Garrigues A, et al. Recognition of sulfonylurea receptor (ABCC8/9) ligands by the multidrug resistance transporter P-glycoprotein (ABCB1): functional similarities based on common structural features between two multispecific ABC proteins. J Biol Chem. 2011;286:3552–3569. doi: 10.1074/jbc.M110.155200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mishra MK, Ray D, Barik BB. Microcapsules and transdermal patch: a comparative approach for improved delivery of antidiabetic drug. AAPS PharmSciTech. 2009;10:928–934. doi: 10.1208/s12249-009-9289-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tosun C, Koltz MT, Kurland DB, Ijaz H, Gurakar M, et al. The Protective Effect of Glibenclamide in a Model of Hemorrhagic Encephalopathy of Prematurity. Brain Sci. 2013;3:215–238. doi: 10.3390/brainsci3010215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rossignol S, Schwab M, Schwartz M, Fehlings MG. Spinal cord injury: time to move? J Neurosci. 2007;27:11782–11792. doi: 10.1523/JNEUROSCI.3444-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]