

Abstract
MicroRNAs (miRNAs) are a class of small noncoding RNAs that mediate posttranscriptional gene silencing. Mitochondrial fission participates in the induction of apoptosis. It remains largely unknown whether miRNAs can regulate mitochondrial fission. Reactive oxygen species and doxorubicin could induce mitochondrial fission and apoptosis in cardiomyocytes. Concomitantly, mitofusin 1 (Mfn1) was downregulated, whereas miRNA 140 (miR-140) was upregulated upon apoptotic stimulation. We investigated whether Mfn1 and miR-140 play a functional role in mitochondrial fission and apoptosis. Ectopic expression of Mfn1 attenuated mitochondrial fission and apoptosis. Knockdown of miR-140 inhibited mitochondrial fission. Our results further revealed that knockdown of miR-140 was able to reduce myocardial infarct sizes in an animal model. We observed that miR-140 could suppress the expression of Mfn1, and it exerted its effect on mitochondrial fission and apoptosis through targeting Mfn1. Our data revealed that mitochondrial fission occurs in cardiomyocytes and can be counteracted by Mfn1. However, the function of Mfn1 is negatively regulated by miR-140. Our present work suggests that Mfn1 and miR-140 are integrated into the program of cardiomyocyte apoptosis.
INTRODUCTION
MicroRNAs (miRNAs) are ∼22 nucleotides long and act as negative regulators of gene expression by inhibiting mRNA translation or promoting mRNA degradation (1). The function of miRNAs is to coordinately regulate target genes encoding proteins and their functions. Strikingly, a single miRNA is even able to modulate complex physiological or disease phenotypes by regulating entire functional networks (2–4). miRNAs can regulate cardiac function, including the conductance of electrical signals, heart muscle contraction, heart growth, and morphogenesis. In particular, miRNAs are involved in the pathogenesis of cardiac diseases, and manipulation of miRNAs can be developed for therapeutic targets (5–7).
Apoptosis can occur in the myocardium under a variety of pathological conditions (8, 9). For example, myocyte apoptosis is increased in myocardium from patients with myocardial infarction and heart failure and from experimental models of hypertrophy and heart failure (8, 10–14). Growing evidence has shown that miRNAs are involved in the regulation of cardiomyocyte apoptosis. For example, overexpression of miRNA 320 (miR-320) enhances apoptosis in cardiomyocytes, whereas knockdown of miR-320 can attenuate cell death (15).
A characteristic of cardiomyocytes is that they are enriched in mitochondria. As an organ supporting the energy needed for blood flow, the heart has perpetually high energy demands. Due to the limited ability for substrate storage, heart function stringently depends on ATP-generating pathways (16). The highly abundant existence of functional mitochondria in cardiomyocytes is essential for producing enough ATP for normal cardiac contractile function (17). However, it has been well documented that mitochondria contain a variety of proapoptotic factors that are able to initiate apoptosis. Mitochondria constantly undergo fusion and fission, which are necessary for the maintenance of organelle fidelity (18–21). Recently, accumulating evidence has shown that abnormal mitochondrial fusion and fission participate in the regulation of apoptosis (22–24). Mitochondrial fusion is able to inhibit apoptosis, whereas mitochondrial fission is involved in the initiation of apoptosis (25–28). It remains largely unknown as to whether miRNAs are involved in the program of mitochondrial fission.
Mitofusin 1 (Mfn1) is involved in the regulation of mitochondrial fusion (29). It can prevent mitochondrial fission in neurons (30). Our recent work has found that Mfn1 is able to inhibit mitochondrial fission and apoptosis in cardiomyocytes (31). However, Mfn1 expression is downregulated during apoptosis. It is not yet clear as to how Mfn1 is downregulated by apoptotic stimulation.
Our present work reveals that miR-140 can regulate mitochondrial fission and apoptotic programs. It can suppress Mfn1 expression and exert its effect through targeting Mfn1. Our data provide novel evidence demonstrating that the mitochondrial fission program can be regulated by miRNA.
MATERIALS AND METHODS
Cell cultures and treatment.
Monolayer cultures of neonatal rat cardiac cells were prepared as we described previously, with modifications (32). In brief, hearts from 1-day-old Wistar rats were dissected, minced, and placed into phosphate-buffered saline (PBS). The tissue was digested at 37°C in a HEPES-buffered saline solution (20 mM HEPES-NaOH [pH 7.35], 116 mM NaCl, 5 mM KCl, 1 mM NaH2PO4, 5 mM l-glucose, 0.4 mM MgSO4, 0.14 mg/ml collagenase, and 30 mg/ml pancreatin). After centrifugation, cells were resuspended in Dulbecco's modified Eagle medium–F-12 medium (Gibco) containing 5% heat-inactivated horse serum, 100 μM ascorbate, 1 μg/ml insulin, 1 μg/ml transferrin, 10 ng/ml selenium, 100 U/ml penicillin, and 100 μg/ml streptomycin. The dissociated cells were preplated at 37°C for 1 h to eliminate fibroblasts and then cultured for 48 h in a medium containing 0.1 mmol/liter bromodeoxyuridine to prevent proliferation of nonmyocytes. More than 95% of cells were cardiomyocytes, as detected by immunostaining with the anti-α-sarcomeric actin antibody.
Cardiomyocytes were treated with 100 μmol/liter hydrogen peroxide plus 0.1 mM ferrous sulfate for the indicated hours and further cultured in normal culture medium without hydrogen peroxide and ferrous sulfate, as we described previously (33). A total of 1.0 μmol/liter doxorubicin (Sigma, St. Louis, MO) was employed. Neonate rat cardiomyocytes and cardiac fibroblasts were isolated from 1-day-old Wistar rats as described previously (34).
Adenoviral infection.
The adenoviral β-galactosidase construct was described previously (35). Viruses were amplified in HEK293 cells and purified on a CsCl gradient. Cells were infected with the viruses at a multiplicity of infection (MOI) of 50 for 90 min. After washing with PBS, the culture medium was added, and cells were cultured until the indicated times.
Mitochondrial staining and immunofluorescence.
Mitochondrial staining was carried as we and others have described previously, with modifications (20, 36). Briefly, cells were plated onto coverslips coated with 0.01% poly-l-lysine. After treatment, they were stained for 20 min with 0.02 μM MitoTracker red CMXRos (Molecular Probes). Assessment and quantification of mitochondrial morphology were performed as described previously (20, 36). Briefly, the extent of mitochondrial fission was analyzed on a cell-to-cell basis. A punctiform mitochondrial phenotype was scored as fragmented mitochondrion when at least 90% of its tubular mitochondria were disintegrated. At least 200 randomly selected cells in multiple fields were assessed. Mitochondrial fission (percentage of cells) was calculated as the percentage of fragmented mitochondria to the total number of cells, which were randomly selected and scored.
Immunofluorescence was performed as we have described previously (37). Mitochondria and the samples were imaged by using a laser scanning confocal microscope (Zeiss LSM 510 Meta).
Fluorescent labeling of mitochondria.
For fluorescent labeling of mitochondria, cells were transfected with pAcGFP1-mito, which encodes a fusion of a mitochondrial targeting sequence derived from the precursor of subunit VIII of cytochrome c oxidase and the green fluorescent protein (GFP) (Clontech). Mitochondria were imaged by using a laser scanning confocal microscope (Zeiss LSM510 Meta).
DNA fragmentation and apoptosis assays.
DNA fragmentation was monitored by using the Cell Death Detection ELISA kit (Roche), as we described previously (38). Briefly, the antihistone monoclonal antibody was added to 96-well enzyme-linked immunosorbent assay (ELISA) plates and incubated overnight at 4°C. After recoating and three rinses, the cytoplasmic fractions were added and incubated for 90 min at room temperature. After washing three times, bound nucleosomes were detected by the addition of anti-DNA peroxidase monoclonal antibody and reacted for 90 min at room temperature. After the addition of the substrate, the optical density was read with an ELISA reader at 405 nm. For apoptosis analysis, the terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) kit (Clontech) was used according to the kit's instructions. A total of 150 to 200 cells were counted in 20 to 25 random fields for each group.
Preparations of subcellular fractions.
Subcellular fractions were prepared as described previously, with modifications (39). In brief, cells were washed twice with PBS, and the pellet was suspended in 0.2 ml of ice-cold buffer (20 mM potassium-HEPES [pH 7.8], 5 mM potassium acetate, 0.5 mM MgCl2, and 0.5 mM dithiothreitol [DTT]) containing a protease inhibitor cocktail. The cells were homogenized by 12 strokes in a Dounce homogenizer. The homogenates were centrifuged at 750 × g for 5 min at 4°C to collect nuclei. The supernatants were centrifuged at 10,000 × g for 15 min at 4°C to collect the heavy membrane (HM) pellet. The resulting supernatants were centrifuged at 100,000 × g for 1 h at 4°C. The final supernatants are referred to as cytosolic fractions.
Immunoblot analysis.
Immunoblotting was carried out as we described previously (35). Cells were lysed for 1 h at 4°C in a lysis buffer (20 mM Tris [pH 7.5], 2 mM EDTA, 3 mM EGTA, 2 mM DTT, 250 mM sucrose, 0.1 mM phenylmethylsulfonyl fluoride, 1% Triton X-100) containing a protease inhibitor cocktail (Sigma, St. Louis, MO). We harvested myocardial samples according to procedures described previously (40–42). In brief, ischemic/reperfused myocardial tissue was homogenized in ice-cold lysis buffer by using a homogenizer and lysed for 1 h at 4°C. The homogenates were centrifuged for 5 min at 10,000 × g at 4°C. Supernatants were collected. Samples were subjected to 12% SDS-PAGE and transferred onto nitrocellulose membranes. Equal protein loading was controlled by Ponceau red (Sigma, St. Louis, MO) staining of membranes. Blots were probed by using primary antibodies, followed by horseradish peroxidase-conjugated secondary antibodies. Anti-endonuclease G antibody, anti-Mfn1 antibody, anti-Mfn2 antibody, and anti-proliferating cell nuclear antigen (anti-PCNA) antibody were obtained from Santa Cruz Biotechnology. The antiactin antibody was obtained from Chemicon. The anti-cytochrome oxidase subunit V (anti-COX) antibody was obtained from Invitrogen. Antibodies to Drp1 and OPA1 were obtained from BD Biosciences. The anti-platelet-derived growth factor receptor alpha (anti-PDGFα) antibody was obtained from Cell Signaling Technology. Antigen-antibody complexes were visualized by enhanced chemiluminescence.
Adenoviral constructions.
Adult rat heart mRNA was prepared by using a Qiagen mRNA Minipreparation kit. First-strand cDNA synthesis was carried out by using a first-strand cDNA synthesis kit (Amersham Pharmacia Biotech) followed by PCR. The upstream primer for Mfn1 was 5′-ATGGCAGAAACGGTATCTCC-3′, and the downstream primer was 5′-TTAGGATTCTCCACTGCTCG-3′. The upstream primer for PDGFRα was 5′-ATGGGGACCTCCCAGGCCTT-3′, and the downstream primer was 5′-TTACAGGAAGCTGTCCTCCA-3′. To produce a mutated 3′ untranslated region (UTR) for Mfn1, the mutations (wild-type 3′ UTR, 5′-AACCACU-3′; mutated 3′ UTR, 5′-ATTATGU-3′ [mutations are underlined]) were generated by using a QuikChange II XL site-directed mutagenesis kit (Stratagene). Mfn1 or PDGFRα was finally cloned in to the Adeno-X expression system (Clontech) according to the manufacturer's instructions.
Constructions of adenoviral miR-140.
miR-140 was synthesized by PCR using rat genomic DNA as the template. The upstream primer was 5′-TGAAAGAACCCAGAGCAATG-3′; the downstream primer was 5′-TCAACTGTCACCCACCCAAC-3′. The PCR fragment was finally cloned into the Adeno-X expression system (Clontech) according to the manufacturer's instructions.
Lentiviral Mfn1 shRNA.
Lentiviral particles expressing small hairpin RNA (shRNA) for rat Mfn1 were purchased from Santa Cruz (catalog number sc-270320-V). Control shRNA lentiviral particles (catalog number sc-108080) were used as a negative control, which encodes a scrambled shRNA sequence.
Construction of parkin siRNA.
Parkin small interfering RNA (siRNA) was constructed by using the pSilencer adeno 1.0-CMV vector (Ambion) according to the manufacturer's instructions. The parkin siRNA sense sequence is 5′-TTCCAAACCGGATGAGTGG-3′; the antisense sequence is 5′-CCACTCATCCGGTTTGGAA-3′. The scramble parkin siRNA sense sequence is 5′-GCTCATCGAGCTAGTAGAG-3′; the scramble antisense sequence is 5′-CTCTACTAGCTCGATGAGC-3′.
The specificity of siRNA was confirmed by comparison with all other sequences in GenBank using Nucleotide BLAST. There was no homology to other known rat DNA sequences.
Preparation of the luciferase construct for the Mfn1 mRNA 3′ UTR.
The rat Mfn1 mRNA 3′ UTR was amplified by PCR. The forward primer was 5′-CCTATGCCTTGCGGAGATTG-3′. The reverse primer was 5′-TTCACATCAGTTTTAATGGG-3′. To produce a mutated 3′ UTR, the mutations (wild-type 3′ UTR, 5′-AACCACU-3′; mutated 3′ UTR, 5′-ATTATGU-3′ [mutated residues are underlined]) were generated by using the QuikChange II XL site-directed mutagenesis kit (Stratagene). The constructs were sequence verified. Wild-type and mutated 3′ UTRs were subcloned into the pGL3 vector (Promega) immediately downstream of the stop codon of the luciferase gene.
Luciferase assay.
A luciferase assay was performed as we described previously (43). In brief, cells were cotransfected with the plasmid constructs, 150 ng/well of pGL3-Mfn1-3′UTR and 300 ng/well of miR-140, by using Lipofectamine 2000 (Invitrogen). At 48 h after transfection, cells were lysed, and luciferase activity was measured by using a dual-luciferase reporter assay kit (Promega) according to the manufacturer's instructions.
Cell transfection.
The anti-miR for miR-140 designed to inhibit endogenous miR-140 expression and the anti-miR negative control (anti-miR-NC) were obtained from Ambion. Cells were transfected with anti-miR or anti-miR-NC at 50 nM by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Cardiomyocytes were transfected with the plasmid construct pAcGFP1-mito, encoding mitochondrion-targeted GFP, by using an Effectene transfection kit (Qiagen) according to the manufacturer's instructions.
Quantitative real-time PCR.
TaqMan microRNA assay kits were employed to analyze the levels of miR-140, miR-21, and miR-499 according to the manufacturer's instructions (Applied Biosystems). The results of quantitative real-time PCR (qRT-PCR) were normalized to those of U6.
Anoxia.
Anoxia was achieved as described previously (35). Briefly, cells were placed into an anoxic chamber with a water-saturated atmosphere composed of 5% CO2 and 95% N2.
Anti-miR delivery, ischemia-reperfusion, infarct size determination, and determination of apoptotic cardiomyocytes in the myocardium.
All animal experiments were performed according to protocols approved by the Animal Care Committee, Institute of Zoology, Chinese Academy of Sciences. We obtained C57BL/6 male mice from the Institute of Laboratory Animal Science of the Chinese Academy of Medical Sciences. Chemically modified antisense RNA oligonucleotides complementary to the mature miR-140 were used to inhibit endogenous miR-140 activity in vivo. The anti-miR-140 sequence is 5′-CUACCAUAGGGUAAAACCACUG-3′ (GenePharma Co. Ltd.). The anti-miR-NC sequence is 5′-CAGUACUUUUGUGUAGUACAA-3′ (GenePharma Co. Ltd.). All the bases were 2′-O-methyl modified. Anti-miR-140 and anti-miR-NC oligonucleotides were injected at 80 mg kg− of body weight day− through the tail vein for three consecutive days. We harvested the hearts 1 day after the last injection for measurement of the expression of miR-140. We exposed the hearts of mice to ischemia-reperfusion (I/R) injury 24 h after the last injection of anti-miR or anti-miR-NC. To overexpress miR-140 in the myocardium, we performed intracoronary delivery of adenoviruses as described previously (44). Mice were anesthetized by intraperitoneal injection of a mixture of ketamine (100 mg/kg) and xylazine (5 mg/kg) and fixed in the supine position on a warm pad to keep the body temperature constant. The chest was entered through a small left anterior thoracotomy, the pericardial sac was then removed, and adenoviral miR-140 at an MOI of 2 × 1010 was injected with a catheter from the apex of the left ventricle into the aortic root while the aorta and pulmonary arteries were cross-clamped. The clamp was maintained for 20 s when the heart pumped against a closed system.
We carried out standard cardiac ischemia-reperfusion on mice as we described previously (45). In brief, we exposed the mice to left chest thoracotomy and occluded the left anterior descending coronary artery (LAD) with a slipknot at the inferior border of the left auricle to induce cardiac ischemia. We loosened the ligation to reperfuse the heart after ischemia. The sham group experienced the same procedure except that the suture around the LAD was left untied.
We measured the myocardial infarct sizes as we described previously (45), at 24 h of reperfusion. The areas of infarction (INF), area at risk (AAR), and left ventricular (LV) area were measured by using computer-assisted planimetry (NIH Image 1.57) in a blind manner.
We detected apoptotic cardiomyocytes with TUNEL staining according to the manufacturer's instructions (Roche). We labeled cardiomyocytes with antibody to α-actinin (catalog number A7811; Sigma) and total nuclei with 4′,6-diamidino-2-phenylindole (DAPI). We counted the apoptotic cells according to protocols described previously (46–48). An investigator blind to the treatment quantified 25 random fields from three slides of each block. Data are expressed as TUNEL-positive cells as a percentage of total cells counted.
To knock down Mfn1 in vivo, we performed intracoronary delivery of lentiviral particles expressing small hairpin RNA (shRNA) for Mfn1 or lentiviral control shRNA as described previously (44). We exposed the mice to 45 min of ischemia and 3 h of reperfusion for immunoblot and apoptosis analyses and 24 h of reperfusion for myocardial infarction measurements, as described previously (45).
Electron microscopy.
We analyzed the myocardium ultrastructure to quantify mitochondrial fission in vivo, as we and others have described previously (45, 49).
Statistical analysis.
Paired data were evaluated by Student's t test. One-way analysis of variance (ANOVA) was used for multiple comparisons. A P value of <0.05 was considered significant.
RESULTS
Cardiomyocytes undergo mitochondrial fission upon apoptotic stimulation.
Cardiomyocyte apoptosis is a characteristic of a variety of cardiac diseases. To understand the molecular mechanism by which apoptosis is initiated in cardiomyocytes, we detected whether mitochondrial fission is a component of cardiomyocyte apoptosis. Hydrogen peroxide treatment led to the occurrence of mitochondrial fission, as revealed by morphological alterations. The reticular morphology of mitochondria becomes fragmented upon treatment with hydrogen peroxide (Fig. 1A). A time-dependent increase in the number of cells with mitochondrial fission was observed (Fig. 1B). Concomitantly, apoptosis was observed (Fig. 1C). We also detected that doxorubicin induced mitochondrial fission (Fig. 1D) and apoptosis (Fig. 1E) in cardiomyocytes. We also observed similar mitochondrial morphological changes in cardiomyocytes monitored with pAcGFP1-mito (Fig. 1F). Thus, it appears that mitochondrial fission occurs upon apoptotic stimulation in cardiomyocytes.
FIG 1.

Mitochondria undergo fission in cardiomyocytes upon apoptotic stimulation. (A) Representative images of mitochondria in cardiomyocytes without hydrogen peroxide (control) or with hydrogen peroxide (H2O2). Neonatal rat cardiomyocytes were treated with 100 μmol/liter hydrogen peroxide. Two hours after H2O2 treatment, cells were stained with 0.02 μM MitoTracker red for 20 min to visualize mitochondrial morphology. Red, MitoTracker red; blue, DAPI. Bar, 10 μm. (B) Quantification of mitochondrial fission. Cardiomyocytes were treated and stained as described for panel A. *, P < 0.05 compared with 0 h (control). (C) Apoptotic cardiac myocytes treated as described for panel A were quantified, and at the indicated time points, cells were stained for apoptosis by a TUNEL assay. *, P < 0.05 compared with 0 h (control). (D and E) Mitochondrial morphology and apoptosis of cardiomyocytes treated with doxorubicin (Dox). *, P < 0.05 compared with 0 h (control). (F) Mitochondrial morphology in cardiomyocytes transfected with pAcGFP1-mito and then exposed to H2O2. Green, pAcGFP1-mito; blue, DAPI. Scale bar, 10 μm. Data are expressed as means ± standard errors of the means of three independent experiments.
Mfn1 downregulation and miR-140 upregulation occur simultaneously in apoptosis.
Mfn1 participates in the regulation of the mitochondrial network (29). We detected the expression levels of Mfn1 and observed that it was downregulated upon hydrogen peroxide treatment (Fig. 2A). Doxorubicin treatment also resulted in a reduction of Mfn1 levels (Fig. 2B). miRNA participates in apoptosis by suppressing gene expression (7, 50). We detected miR-140 distribution by separating cardiomyocytes from fibroblasts and found that miR-140 is mainly expressed in cardiomyocytes (Fig. 2C). The decrease in Mfn1 expression levels led us to consider whether it was related to miRNA regulation. We analyzed the 3′ UTR of Mfn1 mRNA by using the bioinformatics program targetscan and found that Mfn1 is a potential target of miR-140 (Fig. 2D). Subsequently, we analyzed the expression level of miR-140 during apoptosis. miR-140 levels were elevated upon hydrogen peroxide treatment (Fig. 2E) as well as doxorubicin treatment (Fig. 2F). The effects were attenuated by the administration of the antioxidant N-acetylcysteine (NAC) (Fig. 2G and H). These results suggest that Mfn1 and miR-140 are the targets of apoptotic stimulation in cardiomyocytes.
FIG 2.
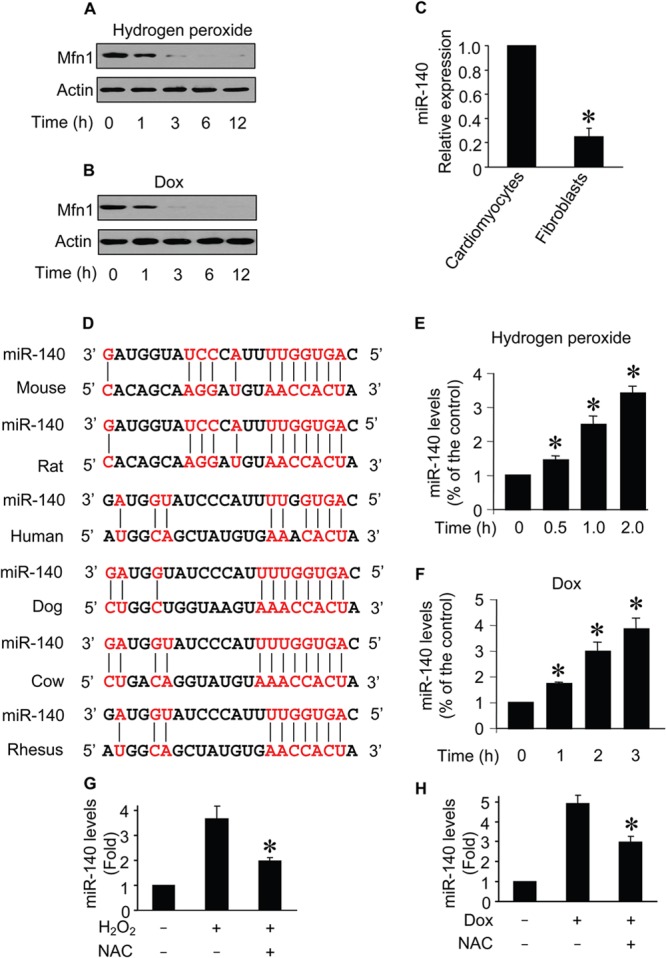
Mfn1 downregulation and miR-140 upregulation occur simultaneously in apoptosis. (A and B) Immunoblots of Mfn1 proteins from cardiomyocytes exposed to H2O2 (A) or doxorubicin (B). The number of hours after treatment is indicated. (C) Comparison of expression profiles of miR-140 in cardiomyocytes and cardiac fibroblasts. (D) Putative miR-140 binding sites (potential complementary residues are shown in red). (E and F) miR-140 levels from cardiomyocytes upon H2O2 (E) or doxorubicin (F) treatment. The number of hours after treatment is indicated. *, P < 0.05 compared with the control. (G and H) Cells were pretreated with 5 mM NAC and then exposed to 100 μmol/liter H2O2 (G) or 1.0 μmol/liter doxorubicin (H). Cells were harvested for the analysis of miR-140 by qRT-PCR, and the data were normalized to those for U6. *, P < 0.05. Data are expressed as the means ± standard errors of the means of three independent experiments.
miR-140 suppresses Mfn1 expression.
We tested whether miR-140 is related to Mfn1 expression. First, we attempted to understand whether endogenous miR-140 participates in the regulation of Mfn1 expression. To this end, the anti-miR of miR-140 (anti-miR-140) was employed. Administration of anti-miR-140 but not its negative control (anti-miR-NC) reduced miR-140 levels upon hydrogen peroxide treatment (Fig. 3A) and doxorubicin treatment (Fig. 3B). Concomitantly, anti-miR-140 administration attenuated the Mfn1 reduction induced by hydrogen peroxide (Fig. 3C) and doxorubicin (Fig. 3D). Second, we tested whether exogenous miR-140 is able to regulate Mfn1 expression. Ectopic expression of miR-140 led to a reduction of Mfn1 expression levels (Fig. 3E and F). However, anti-miR-140 administration had no effects on the levels of Mfn2, OPA1, and Drp1 upon treatment with hydrogen peroxide (Fig. 3G) and doxorubicin (Fig. 3H). Knockdown of parkin did not affect Mfn1 reduction induced by hydrogen peroxide (Fig. 3I). Taken together, these results show that Mfn1 can be a target of miR-140.
FIG 3.

miR-140 suppresses Mfn1 expression. (A and B) miR-140 levels from cardiomyocytes transfected with miR-140 anti-miR or the anti-miR control (anti-miR-NC) and then exposed to H2O2 (A) or doxorubicin (B). *, P < 0.05. (C and D) Immunoblots of Mfn1 proteins from cardiomyocytes treated as described above for panels A and B. (E and F) Immunoblots of Mfn1 proteins and miR-140 levels from cardiomyocytes infected with adenoviral miR-140 or the control (miR-con). *, P < 0.05. Data represent three separate experiments. (G and H) Immunoblots of Mfn2, Drp1, and OPA1 proteins from cardiomyocytes treated as described above for panels A and B. Results are representative of three independent experiments. (I) Immunoblot analysis of Mfn1. Cardiomyocytes were infected with adenoviral small interfering RNA (siRNA) constructs targeting parkin or its scrambled form (sc) and then exposed to H2O2. Data are expressed as means ± standard errors of the means of three independent experiments.
miR-140 inhibits Mfn1 expression through targeting the 3′ UTR.
To understand whether Mfn1 can be directly or indirectly regulated by miR-140, we tested whether miR-140 regulates Mfn1 dependent on the 3′ UTR of Mfn1 mRNA. We constructed the 3′ UTR of Mfn1 mRNA with the luciferase reporter vector (Fig. 4A), and miR-140 inhibited the translational activity of the wild-type 3′ UTR of Mfn1 mRNA. In contrast, the introduction of mutations into the 3′ UTR of Mfn1 mRNA resulted in a failure of miR-140 to influence the translational activity of the Mfn1 3′ UTR mRNA. The effect of miR-140 on the translational activity of the wild-type 3′ UTR of Mfn1 mRNA was inhibited by anti-miR-140 (Fig. 4B). We further analyzed the protein levels of Mfn1 in response to miR-140 stimulation. Mfn1 whose mRNA had the wild-type 3′ UTR but not that whose mRNA lacked the 3′ UTR was suppressed by miR-140 (Fig. 4C). The introduction of mutations into the 3′ UTR of Mfn1 mRNA led to the inability of miR-140 to inhibit Mfn1 expression (Fig. 4D). Thus, miR-140 represses Mfn1 expression dependent on the 3′ UTR of Mfn1 mRNA.
FIG 4.
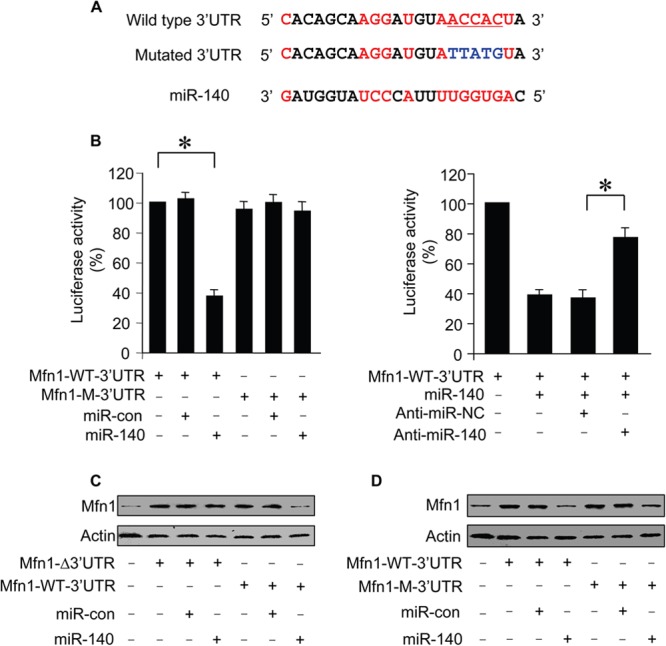
miR-140 inhibits Mfn1 expression through targeting the 3′ UTR. (A) Luciferase constructs of the Mfn1 mRNA 3′ UTR. (B) Luciferase activity measured from HEK293 cells transfected with the wild-type (Mfn1-WT-3′UTR) or mutated (Mfn1-M-3′UTR) Mfn1 mRNA 3′ UTR luciferase constructs, along with the expression plasmid for miR-140 or miR-con. Luciferase activity was measured from cardiomyocytes transfected with a luciferase construct containing the wild-type Mfn1 mRNA 3′ UTR, miR-140 anti-miR (AN), or the negative-control anti-miR (NC), along with the expression plasmid for miR-140. *, P < 0.05. (C) Immunoblots of Mfn1 proteins from cardiomyocytes infected with adenoviral Mfn1 whose mRNA had the wild-type 3′ UTR (Mfn1-WT-3′UTR) or lacked the 3′ UTR (Mfn1-Δ3′UTR), along with adenoviral miR-140 or miR-con. Results are representative of three independent experiments. (D) Immunoblots of Mfn1 proteins from cardiomyocytes infected with adenoviral Mfn1 whose mRNA had the wild-type 3′ UTR (Mfn1-WT-3′UTR) or the mutated 3′ UTR (Mfn1-M-3′UTR), along with adenoviral miR-140 or miR-con. Results are representative of three independent experiments. Data are expressed as means ± standard errors of the means of three independent experiments.
Both Mfn1 and miR-140 are able to regulate mitochondrial fission in cardiomyocytes.
We asked whether Mfn1 and miR-140 play a functional role in mitochondrial fission and apoptosis. Ectopic expression of Mfn1 prevents cells from undergoing mitochondrial fission and apoptosis induced by hydrogen peroxide (Fig. 5A). Doxorubicin-induced mitochondrial fission and apoptosis were also suppressed by Mfn1 (Fig. 5B). Because miR-140 was upregulated during apoptosis, as shown in Fig. 2, we therefore tested whether the knockdown of miR-140 was able to influence mitochondrial fission and apoptosis. Administration of the miR-140 anti-miR led to a reduction of mitochondrial fission and apoptosis (Fig. 5C to E). Mfn1 expression was monitored by immunofluorescence (Fig. 5F). Treatment of cardiomyocytes with adenoviral miR-140 at a high titer but not at a low dose led to mitochondrial fission (Fig. 5G). Ectopic expression of miR-140 sensitized cells to undergo mitochondrial fission and apoptosis (Fig. 5H). These data suggest that Mfn1 and miR-140 participate in the programs of mitochondrial fission and apoptosis.
FIG 5.

Mfn1 and miR-140 are able to regulate mitochondrial fission in cardiomyocytes. (A and B) Analysis of mitochondrial fission and apoptosis from cardiomyocytes infected with adenoviruses harboring the Mfn1 or β-galactosidase (β-gal) construct and then exposed to H2O2 (A) or doxorubicin (B) treatment. (A) Mitochondrial fission was analyzed 6 h after treatment. Apoptosis was analyzed 12 h after treatment by a TUNEL assay. (B) Mitochondrial fission was analyzed 5 h after treatment. Apoptosis was analyzed 15 h after treatment by a TUNEL assay. *, P < 0.05. (C) Effect of knockdown miR-140 on anoxia-induced apoptosis and mitochondrial fission. Cardiomyocytes were transfected with anti-miR-140 or the negative control (anti-miR-NC) and then exposed to anoxia. Data represent three separate experiments. *, P < 0.05. (D) Mitochondrial morphology in cardiomyocytes transfected with anti-miR of miR-140 or the negative control (anti-miR-NC) and then treated with H2O2. (E) Counting of cells with mitochondrial fission and apoptosis. Cardiomyocytes were treated as described for panel D. *, P < 0.05. (F) Analysis of Mfn1 expression by immunofluorescence. Cardiomyocytes were transfected with anti-miR of miR-140 or its negative control (NC) and then exposed to 100 μmol/liter H2O2. Immunofluorescence and TUNEL assays were performed after H2O2 treatment. Red, Mfn1 expression; green, TUNEL-positive cell; blue, DAPI staining. Bar, 10 mm. (G) Effect of miR-140 on mitochondria morphology and cell death in cardiomyocytes. Cardiomyocytes were infected with adenoviral miR-140 or the control (miR-con) at the indicated titers, and the percentage of cells with mitochondrial fission or undergoing cell death was determined. *, P < 0.05. (H) Counting of cardiomyocytes with mitochondrial fission and apoptotic cardiomyocytes infected with adenoviral miR-140 or the control (miR-con) and then treated with 20 μM H2O2. Data represent three separate experiments. *, P < 0.05 compared with the control. Data are expressed as the means ± standard errors of the means of three independent experiments.
Mfn1 and miR-140 are functionally related in the mitochondrial fission program.
The ability of miR-140 to regulate Mfn1 expression prompted us to test whether these two factors are functionally related to mitochondrial fission and apoptosis. We first studied whether Mfn1 function could be regulated by miR-140. Without the exogenous expression of miR-140, Mfn1 whose mRNA lacked the 3′ UTR or had the wild-type or mutated 3′ UTR antagonized mitochondrial fission and apoptosis (Fig. 6A). Second, we tested whether miR-140 exerted its effect dependent on Mfn1. The inhibitory effects of Mfn1 whose mRNA had the wild-type 3′ UTR on mitochondrial fission and apoptosis (Fig. 6B) were attenuated in the presence of miR-140. Furthermore, in response to treatment with a low dose of hydrogen peroxide, the knockdown of Mfn1 and ectopic expression of miR-140 have a synergistic effect on mitochondrial fission and apoptosis (Fig. 6C). Mfn1 downregulation reversed the effect of anti-miR-140 on mitochondrial fission and apoptosis (Fig. 6D). Thus, it appears that miR-140 exerts its effects through Mfn1. A miRNA may have multiple targets. To search for other potential downstream targets of miR-140 involved in apoptosis, we observed that platelet-derived growth factor receptor alpha (PDGFRα) expression was downregulated in cardiomyocytes upon hydrogen peroxide treatment (Fig. 6E). Knockdown of endogenous miR-140 regulated PDGFRα expression (Fig. 6F). PDGFRα inhibited cardiomyocyte apoptosis induced by oxidative stress (Fig. 6G).
FIG 6.

Mfn1 and miR-140 are functionally related in the mitochondrial fission program. (A) Counting of cardiomyocytes with mitochondrial fission and apoptosis. Cardiomyocytes were infected with adenoviral Mfn1 whose mRNA lacked the 3′ UTR (Mfn1-Δ3′UTR), had the wild-type 3′ UTR (Mfn1-WT-3′UTR), or had the mutated 3′ UTR (Mfn1-M-3′UTR). Twenty-four hours after infection, cells were treated with H2O2. *, P < 0.05 versus H2O2 alone. (B) Counting of mitochondrial fission and apoptosis. Cardiomyocytes were infected with adenoviral Mfn1 whose mRNA lacked a 3′ UTR (Mfn1-Δ3′UTR), had the wild-type 3′ UTR (Mfn1-WT-3′UTR), or had the mutated 3′ UTR (Mfn1-M-3′UTR), along with adenoviral miR-140, and then exposed to H2O2. *, P < 0.05. (C) Analysis of mitochondrial fission and apoptosis. Cardiomyocytes were infected with adenoviral miR-140, along with lentiviral Mfn1 small hairpin RNA (Mfn1-shRNA) or lentiviral control shRNA, and then exposed to 20 μmol/liter H2O2. *, P < 0.05. (D) Mfn1 downregulation reverses the effect of miR-140 knockdown on mitochondrial fission and cell death. Cardiomyocytes were infected with lentiviral Mfn1 shRNA or its control (control shRNA) and then transfected with anti-miR-140 or the negative control (anti-miR-NC). Twenty-four hours after transfection, cells were treated with H2O2. Mitochondrial fission was analyzed 6 h after treatment. Apoptosis was analyzed 24 h after treatment by a TUNEL assay. *, P < 0.05. (E) PDGFRα expression in cardiomyocytes upon treatment with H2O2. Cardiomyocytes were treated with 100 μmol/liter H2O2 and harvested at the indicated times for analysis of PDGFRα expression levels by immunoblotting. (F) Influence of miR-140 on PDGFRα expression. Cardiomyocytes were transfected with anti-miR-140 or anti-miR-NC and then treated with H2O2. The expression of PDGFRα was determined by immunoblotting. (G) Cardiomyocytes were infected with adenoviruses harboring PDGFRα or the β-galactosidase construct. Twenty-four hours after infection, cells were treated with H2O2. Apoptosis was analyzed 12 h after treatment by a TUNEL assay. *, P < 0.05. Data are expressed as means ± standard errors of the means of three independent experiments.
Knockdown of miR-140 attenuates myocardial infarction.
To further understand the pathophysiological significance of miR-140, we explored whether miR-140 is related to the pathogenesis of myocardial infarction. We employed anti-miR of miR-140 to test whether the knockdown of miR-140 can influence myocardial infarction. Ischemia induced an elevation in miR-140 expression levels. The administration of miR-140 anti-miR induced a reduction of miR-140 levels. Concomitantly, apoptosis was reduced (Fig. 7A), and the infarct sizes were attenuated upon knockdown of miR-140 (Fig. 7B). Mfn1 levels were reduced in mice treated with ischemia-reperfusion (I/R). However, knockdown of miR-140 attenuated the reduction of Mfn1 levels (Fig. 7C). Next, we examined the mitochondrial network in vivo upon ischemia-reperfusion. Ischemia-reperfusion led to mitochondrial fission, but this effect was attenuated by anti-miR-140 (Fig. 7D). Mfn1 downregulation reversed the effect of anti-miR-140 on I/R injury in vivo (Fig. 7E and Fig. 7F). These data suggest that miR-140 plays a role in the pathogenesis of myocardial infarction.
FIG 7.

Knockdown of miR-140 attenuates myocardial infarction. (A) miR-140 levels and apoptosis in ischemic hearts from mice treated with miR-140 anti-miR or its negative control (NC) and then exposed to ischemia-reperfusion (I/R). Representative images of ventricular myocardium sections are shown (n = 6 to 8). Green, TUNEL-positive myocyte nuclei; blue, DAPI-stained nuclei; red, cardiomyocytes labeled with antibody to α-actinin. Scale bar, 20 μm. *, P < 0.05 compared with anti-miR-NC plus I/R. (B) Infarct sizes and representative images of midventricular myocardial slices. AAR, area at risk; LV, left ventricular area; INF, infarct area. The ratios of AAR to LV, INF to AAR, and INF to LV are shown (n = 6 to 8). *, P < 0.05 compared with anti-miR-NC subjected to I/R. Scale bar, 2 mm. Data are expressed as means ± standard errors of the means. (C) Immunoblots of Mfn1 proteins. Results are representative of three independent experiments. (D) Mitochondrial morphology in ischemic hearts from mice treated as described for panel A. Representative electron microscopy images of mitochondria are shown (n = 6 to 8 mice per group). Scale bar, 2 μm. Arrows indicate fission mitochondria. *, P < 0.05. (E and F) Mfn1 downregulation reverses the effect of miR-140 knockdown on I/R injury. Mfn1 levels were measured by immunoblotting, and apoptosis was measured by TUNEL staining. Myocardial infarction was detected by Evans blue and 2,3,5-triphenyltetrazolium chloride (TTC) staining as described in Materials and Methods. AAR, area at risk; LV, left ventricular area; INF, infarct area. The ratios of AAR to LV, INF to AAR, and INF to LV are shown (n = 6 to 8 per group). *, P < 0.05. Data are expressed as means ± standard errors of the means.
miR-140 controls endonuclease G release through Mfn1.
Apoptosis is controlled by a complex interplay of apoptotic factors. We finally tested whether miR-140 can influence the downstream events of Mfn1 in mitochondrial fission and apoptosis. Apoptosis is characterized by the occurrence of DNA fragmentation. Mitochondria contain proapoptotic factors, including endonuclease G, which participates in causing DNA fragmentation (51, 52). Our previous work has shown that endonuclease G release from mitochondria can be controlled by Mfn1 (31). We tested whether this event is related to miR-140. Hydrogen peroxide induced endonuclease G release from mitochondria. Concomitantly, the levels of endonuclease G in nuclei were elevated. Mfn1 whose mRNA had the mutated 3′ UTR inhibited endonuclease G release from mitochondria more effectively (Fig. 8A). The function of endonuclease G is to cleave DNA. We thus tested whether DNA fragmentation can be influenced by miR-140. The Cell Death Detection ELISA, a method which can specifically detect histone-associated DNA fragmentation and thus is specific for apoptosis, was employed to analyze histone-associated DNA fragments. Mfn1 whose mRNA had the mutated 3′ UTR inhibited DNA fragmentation more effectively than that whose mRNA had the wild-type 3′ UTR in the presence of miR-140 (Fig. 8B). Thus, it appears that miR-140 controls the downstream events of Mfn1. Finally, knockdown of miR-140 (Fig. 8C and D) or Mfn-1 (Fig. 8E and F) did not affect the expression levels of miR-21 and miR-499 upon hydrogen peroxide treatment.
FIG 8.

miR-140 controls the downstream target of Mfn1. (A) Immunoblots of endonuclease G proteins in mitochondrion-enriched heavy membranes (HM) and nuclear fractions from cardiomyocytes. Cardiomyocytes were infected with adenoviral Mfn1 whose mRNA lacked the 3′ UTR (Mfn1-Δ3′UTR), had the wild-type 3′ UTR (Mfn1-WT-3′UTR), or had the mutated 3′ UTR (Mfn1-M-3′UTR), along with adenoviral miR-140, and then exposed to H2O2. Cytochrome oxidase subunit V (COX) served as a mitochondrial marker. Proliferating cell nuclear antigen (PCNA) served as a nuclear marker. (B) DNA fragmentation in cardiomyocytes treated as described for panel A. Twelve hours after treatment, DNA fragments were analyzed by the Cell Death Detection ELISA kit, which specifically monitors histone-associated DNA fragments. *, P < 0.05. (C to F) Quantification of miR-21 and miR-499 levels by qRT-PCR. Cardiomyocytes were transfected with anti-miR-140 or anti-miR-NC (C and D) or infected with lentiviral particles expressing small hairpin RNA (shRNA) for Mfn1 or lentiviral control shRNA (E and F) and treated with H2O2. *, P < 0.05 versus the control. Data are expressed as means ± standard errors of the means of three independent experiments.
DISCUSSION
Cardiomyocytes are enriched in mitochondria. However, growing evidence has shown that abnormal mitochondrial fission participates in the initiation of apoptosis. Hitherto, the molecular mechanism by which mitochondrial fission machinery is dysregulated has been poorly understood. More than 800 miRNAs have been identified in mammalian cells, and a large number of proteins control the mitochondrial fission program. It is necessary to characterize their relationship. Our present work reveals that Mfn1 is able to prevent mitochondrial fission. However, it is downregulated by apoptotic stimulation. We observed that miR-140 is upregulated and contributes to the suppression of Mfn1 expression during apoptosis. miR-140 can promote mitochondrial fission and apoptosis by targeting Mfn1. Our results reveal that miR-140 is able to facilitate the initiation of the mitochondrial fission program.
It is estimated that one-third of all mammalian mRNAs are regulated by miRNAs, indicating that miRNAs play critical roles in controlling gene expression (53). Apoptosis is essential for normal development and maintenance of tissue homeostasis. However, excessive apoptosis can occur in the myocardium and contributes to the pathogenesis of cardiac diseases. A variety of stimuli can trigger apoptosis in cardiomyocytes, such as reactive oxygen species (54, 55), hypoxia (56–58), and anthracyclines (59). Apoptosis is controlled by a complex interplay between pro- and antiapoptotic factors. Under physiological conditions, this interplay remains in equilibrium so that apoptosis is tightly controlled. The occurrence of apoptosis indicates that equilibrium is lost. Hitherto, few miRNAs have been demonstrated to be involved in controlling cardiomyocyte apoptosis. R-320 promotes apoptosis by targeting heat shock protein 20, which is known as a cardioprotective protein (15). Our recent work reveals that miR-30 family members are able to inhibit mitochondrial fission and apoptosis by suppressing the expression of p53 that can upregulate Drp1 (30). Thus, studies of miRNAs in cardiac apoptosis may reveal new insights into cardiac pathology.
Our present work illustrated for the first time that miR-140 levels are elevated in cardiomyocytes upon apoptotic stimulation and participate in the initiation of apoptosis. It has been demonstrated that miR-140 is abundantly expressed in cartilage in mouse embryos and zebrafish under physiological condition (60, 61), and it regulates cartilage development and homeostasis (62). In searching for the targets of miR-140, Adamts-5, a major cartilage matrix-degrading protease, has been found to be a target of miR-140 in regulating cartilage development and homeostasis (62). Histone deacetylases (HDACs) are generally transcriptional corepressors that modulate cell growth, differentiation, and apoptosis (63). HDAC4 has been identified to be a miR-140 target, although the functional role of this regulation remains to be elucidated (61). Our present study has elucidated that Mfn1 is regulated by miR-140 and is a target of miR-140 in the apoptotic cascades of cardiomyocytes.
Mfn1 and Mfn2 are guanosine triphosphatases (GTPases) that participate in mitochondrial fusion. It is of note that hearts express two isoforms of mitofusin, Mfn1 and Mfn2, but Mfn1 is expressed in the hearts at a higher level than Mfn2 (29). Our present study reveals that Mfn1 inhibits mitochondrial fission and consequent apoptosis. However, Mfn2 has been shown to be able to initiate apoptosis in cardiomyocytes (64). There have been several studies demonstrating that Mfn2 regulates a number of cellular processes that could not be related to its primary function as a mitochondrion-shaping protein (65). Our data show that Mfn1 is downregulated in response to apoptotic stimulation and that miR-140 contributes to the suppression of Mfn1. It would be interesting to elucidate whether Mfn1 is also regulated by other miRNAs in the apoptotic program.
Mfn1 regulates mitochondrial apoptotic pathways involved in a complex molecular mechanism. For example, Bcl-xL/Bax disturbs mitochondrial morphology by binding and inhibiting Mfn1 (66). Bak interacts with mitofusins, including Mfn1 (67, 68). The inhibition of Mfn1 by proapoptotic factors may lead to the permeabilization of the mitochondrial membranes, thereby causing the release of apoptotic factors from mitochondria into the cytosol. Indeed, endonuclease G is located to mitochondria under physiological conditions. However, it can be released from mitochondria and subsequently translocated to the nuclei, where it degrades DNA (51, 52). Our previous work has shown that Mfn1 is able to inhibit endonuclease G release (31). Our present work demonstrated for the first time that miR-140 can control the distributions of endonuclease G through targeting Mfn1.
Noticeably, mitochondrial fission and apoptosis can be separated (69). Our present work showed that fission is proportional to apoptosis in cardiomyocytes. One reason accounting for this phenomenon is that it is cell type and apoptotic stimuli dependent. We employed hydrogen peroxide and doxorubicin in our present work to induce apoptosis. Hydrogen peroxide has been shown to induce mitochondrial fission and apoptosis in cardiomyocytes (70, 71). Doxorubicin is able to cause the production of reactive oxygen species (72, 73). Thus, it appears that the mitochondrial network plays a functional role in the maintenance of cardiomyocyte integrity.
Our present work focused on Mfn1. The mitochondrial network is controlled by a variety of proteins. For example, knockdown of Drp1 can protect cells against doxorubicin-induced cell death (36). Our most recent study showed that knockdown of Drp1 can inhibit hydrogen peroxide-induced cell death (74). It would be interesting to delineate the relationship between Mfn1 and other fusion/fission proteins in the heart.
In summary, Mfn1 is a mitochondrial fusion protein. Our work reveals that miRNA is integrated into the mitochondrial network by controlling Mfn1. The axis of miR-140 and Mfn1 can be a therapeutic target for the development of interventional treatment of mitochondrion-related cardiac diseases.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (31010103911) and the NIH (5R01HL102202).
Footnotes
Published ahead of print 10 March 2014
REFERENCES
- 1.Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN. 2003. The nuclear RNase III Drosha initiates microRNA processing. Nature 425:415–419. 10.1038/nature01957 [DOI] [PubMed] [Google Scholar]
- 2.Valencia-Sanchez MA, Liu J, Hannon GJ, Parker R. 2006. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 20:515–524. 10.1101/gad.1399806 [DOI] [PubMed] [Google Scholar]
- 3.Bartel DP. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297. 10.1016/S0092-8674(04)00045-5 [DOI] [PubMed] [Google Scholar]
- 4.Ambros V. 2004. The functions of animal microRNAs. Nature 431:350–355. 10.1038/nature02871 [DOI] [PubMed] [Google Scholar]
- 5.Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, Gross C, Engelhardt S, Ertl G, Bauersachs J. 2007. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation 116:258–267. 10.1161/CIRCULATIONAHA.107.687947 [DOI] [PubMed] [Google Scholar]
- 6.Chien KR. 2007. Molecular medicine: microRNAs and the tell-tale heart. Nature 447:389–390. 10.1038/447389a [DOI] [PubMed] [Google Scholar]
- 7.Basson M. 2007. MicroRNAs loom large in the heart. Nat. Med. 13:541. 10.1038/nm0507-541 [DOI] [PubMed] [Google Scholar]
- 8.Crow MT, Mani K, Nam YJ, Kitsis RN. 2004. The mitochondrial death pathway and cardiac myocyte apoptosis. Circ. Res. 95:957–970. 10.1161/01.RES.0000148632.35500.d9 [DOI] [PubMed] [Google Scholar]
- 9.Reeve JL, Duffy AM, O'Brien T, Samali A. 2005. Don't lose heart—therapeutic value of apoptosis prevention in the treatment of cardiovascular disease. J. Cell. Mol. Med. 9:609–622. 10.1111/j.1582-4934.2005.tb00492.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fisher SA, Langille BL, Srivastava D. 2000. Apoptosis during cardiovascular development. Circ. Res. 87:856–864. 10.1161/01.RES.87.10.856 [DOI] [PubMed] [Google Scholar]
- 11.Poelmann RE, Molin D, Wisse LJ, Gittenberger-de Groot AC. 2000. Apoptosis in cardiac development. Cell Tissue Res. 301:43–52. 10.1007/s004410000227 [DOI] [PubMed] [Google Scholar]
- 12.Elsasser A, Suzuki K, Schaper J. 2000. Unresolved issues regarding the role of apoptosis in the pathogenesis of ischemic injury and heart failure. J. Mol. Cell. Cardiol. 32:711–724. 10.1006/jmcc.2000.1125 [DOI] [PubMed] [Google Scholar]
- 13.Hayakawa Y, Chandra M, Miao W, Shirani J, Brown JH, Dorn GW, II, Armstrong RC, Kitsis RN. 2003. Inhibition of cardiac myocyte apoptosis improves cardiac function and abolishes mortality in the peripartum cardiomyopathy of Galpha (q) transgenic mice. Circulation 108:3036–3041. 10.1161/01.CIR.0000101920.72665.58 [DOI] [PubMed] [Google Scholar]
- 14.Kang PM, Izumo S. 2000. Apoptosis and heart failure: a critical review of the literature. Circ. Res. 86:1107–1113. 10.1161/01.RES.86.11.1107 [DOI] [PubMed] [Google Scholar]
- 15.Ren XP, Wu J, Wang X, Sartor MA, Qian J, Jones K, Nicolaou P, Pritchard TJ, Fan GC. 2009. MicroRNA-320 is involved in the regulation of cardiac ischemia/reperfusion injury by targeting heat-shock protein 20. Circulation 119:2357–2366. 10.1161/CIRCULATIONAHA.108.814145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huss JM, Kelly DP. 2005. Mitochondrial energy metabolism in heart failure: a question of balance. J. Clin. Invest. 115:547–555. 10.1172/JCI200524405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin KM, Lin B, Lian IY, Mestril R, Scheffler IE, Dillmann WH. 2001. Combined and individual mitochondrial HSP60 and HSP10 expression in cardiac myocytes protects mitochondrial function and prevents apoptotic cell deaths induced by simulated ischemia-reoxygenation. Circulation 103:1787–1792. 10.1161/01.CIR.103.13.1787 [DOI] [PubMed] [Google Scholar]
- 18.Tanaka A, Youle RJ. 2008. A chemical inhibitor of DRP1 uncouples mitochondrial fission and apoptosis. Mol. Cell 29:409–410. 10.1016/j.molcel.2008.02.005 [DOI] [PubMed] [Google Scholar]
- 19.Yu T, Fox RJ, Burwell LS, Yoon Y. 2005. Regulation of mitochondrial fission and apoptosis by the mitochondrial outer membrane protein hFis1. J. Cell Sci. 118:4141–4151. 10.1242/jcs.02537 [DOI] [PubMed] [Google Scholar]
- 20.Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. 2001. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 1:515–525. 10.1016/S1534-5807(01)00055-7 [DOI] [PubMed] [Google Scholar]
- 21.Griffin EE, Graumann J, Chan DC. 2005. The WD40 protein Caf4p is a component of the mitochondrial fission machinery and recruits Dnm1p to mitochondria. J. Cell Biol. 170:237–248. 10.1083/jcb.200503148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ong S-B, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. 2010. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 121:2012–2022. 10.1161/CIRCULATIONAHA.109.906610 [DOI] [PubMed] [Google Scholar]
- 23.Ong S-B, Hall AR, Hausenloy DJ. 2013. Mitochondrial dynamics in cardiovascular health and disease. Antioxid. Redox Signal. 19:400–414. 10.1089/ars.2012.4777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim H, Scimia MC, Wilkinson D, Trelles RD, Wood MR, Bowtell D, Dillin A, Mercola M, Ronai ZA. 2011. Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol. Cell 44:532–544. 10.1016/j.molcel.2011.08.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J. 2008. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 14:193–204. 10.1016/j.devcel.2007.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC. 2003. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J. Cell Biol. 160:1115–1127. 10.1083/jcb.200212059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gomez-Lazaro M, Bonekamp NA, Galindo MF, Jordan J, Schrader M. 2008. 6-Hydroxydopamine (6-OHDA) induces Drp1-dependent mitochondrial fragmentation in SH-SY5Y cells. Free Radic. Biol. Med. 44:1960–1969. 10.1016/j.freeradbiomed.2008.03.009 [DOI] [PubMed] [Google Scholar]
- 28.Wasiak S, Zunino R, McBride HM. 2007. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J. Cell Biol. 177:439–450. 10.1083/jcb.200610042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Santel A, Frank S, Gaume B, Herrler M, Youle RJ, Fuller MT. 2003. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J. Cell Sci. 116:2763–2774. 10.1242/jcs.00479 [DOI] [PubMed] [Google Scholar]
- 30.Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Graber S, Kovacs I, Lee WD, Waggoner J, Cui J, White AD, Bossy B, Martinou JC, Youle RJ, Lipton SA, Ellisman MH, Perkins GA, Bossy-Wetzel E. 2006. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 25:3900–3911. 10.1038/sj.emboj.7601253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li J, Zhou J, Li Y, Qin D, Li P. 2010. Mitochondrial fission controls DNA fragmentation by regulating endonuclease G. Free Radic. Biol. Med. 49:622–631. 10.1016/j.freeradbiomed.2010.05.021 [DOI] [PubMed] [Google Scholar]
- 32.Lin Z, Murtaza I, Wang K, Jiao J, Gao J, Li PF. 2009. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc. Natl. Acad. Sci. U. S. A. 106:12103–12108. 10.1073/pnas.0811371106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.von Harsdorf R, Li PF, Dietz R. 1999. Signaling pathways in reactive oxygen species-induced cardiomyocyte apoptosis. Circulation 99:2934–2941. 10.1161/01.CIR.99.22.2934 [DOI] [PubMed] [Google Scholar]
- 34.De Windt LJ, Willemsen PH, Pöpping S, Van der Vusse GJ, Reneman RS, Van Bilsen M. 1997. Cloning and cellular distribution of a group II phospholipase A expressed in the heart. J. Mol. Cell. Cardiol. 29:2095–2106. 10.1006/jmcc.1997.0444 [DOI] [PubMed] [Google Scholar]
- 35.Li YZ, Lu DY, Tan WQ, Wang JX, Li PF. 2008. p53 initiates apoptosis by transcriptionally targeting the anti-apoptotic protein ARC. Mol. Cell. Biol. 28:564–574. 10.1128/MCB.00738-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang JX, Li Q, Li PF. 2009. Apoptosis repressor with caspase recruitment domain contributes to the chemotherapy resistance by abolishing mitochondrial fission mediated by dynamin-related protein-1. Cancer Res. 69:492–500. 10.1158/0008-5472.CAN-08-2962 [DOI] [PubMed] [Google Scholar]
- 37.Li PF, Li J, Müller EC, Otto A, Dietz R, von Harsdorf R. 2002. Phosphorylation by protein kinase CK2: a signaling switch for the caspase-inhibiting protein ARC. Mol. Cell 10:247–258. 10.1016/S1097-2765(02)00600-7 [DOI] [PubMed] [Google Scholar]
- 38.Li PF, Dietz R, von Harsdorf R. 1999. p53 regulates mitochondrial membrane potential through reactive oxygen species and induces cytochrome c-independent apoptosis blocked by Bcl-2. EMBO J. 18:6027–6036. 10.1093/emboj/18.21.6027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krude T, Jackman M, Pines J, Laskey RA. 1997. Cyclin/Cdk-dependent initiation of DNA replication in a human cell-free system. Cell 88:109–119. 10.1016/S0092-8674(00)81863-2 [DOI] [PubMed] [Google Scholar]
- 40.Bao W, Hu E, Tao L, Boyce R, Mirabile R, Thudium DT, Ma XL, Willette RN, Yue TL. 2004. Inhibition of Rho-kinase protects the heart against ischemia/reperfusion injury. Cardiovasc. Res. 61:548–558. 10.1016/j.cardiores.2003.12.004 [DOI] [PubMed] [Google Scholar]
- 41.Jiao XY, Gao E, Yuan Y, Wang Y, Lau WB, Koch W, Ma XL, Tao L. 2009. INO-4885 [5,10,15,20-tetra[N-(benzyl-4′-carboxylate)-2-pyridinium]-21H,23H-porphine iron(III) chloride], a peroxynitrite decomposition catalyst, protects the heart against reperfusion injury in mice. J. Pharmacol. Exp. Ther. 328:777–784. 10.1124/jpet.108.144352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tao L, Jiao X, Gao E, Lau WB, Yuan Y, Lopez B, Christopher T, RamachandraRao SP, Williams W, Southan G, Sharma K, Koch W, Ma XL. 2006. Nitrative inactivation of thioredoxin-1 and its role in postischemic myocardial apoptosis. Circulation 114:1395–1402. 10.1161/CIRCULATIONAHA.106.625061 [DOI] [PubMed] [Google Scholar]
- 43.Wang K, Li PF. 2010. Foxo3a regulates apoptosis by negatively targeting miR-21. J. Biol. Chem. 285:16958–16966. 10.1074/jbc.M109.093005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang K, Long B, Jiao JQ, Wang JX, Liu JP, Li Q, Li PF. 2012. miR-484 regulates mitochondrial network through targeting Fis1. Nat. Commun. 3:781. 10.1038/ncomms1770 [DOI] [PubMed] [Google Scholar]
- 45.Wang J-X, Jiao J-Q, Li Q, Long B, Wang K, Liu J-P, Li Y-R, Li P-F. 2011. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat. Med. 17:71–78. 10.1038/nm.2282 [DOI] [PubMed] [Google Scholar]
- 46.Tsutsumi YM, Horikawa YT, Jennings MM, Kidd MW, Niesman IR, Yokoyama U, Head BP, Hagiwara Y, Ishikawa Y, Miyanohara A, Patel PM, Insel PA, Patel HH, Roth DM. 2008. Cardiac-specific overexpression of caveolin-3 induces endogenous cardiac protection by mimicking ischemic preconditioning. Circulation 118:1979–1988. 10.1161/CIRCULATIONAHA.108.788331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oshima Y, Ouchi N, Sato K, Izumiya Y, Pimentel DR, Walsh K. 2008. Follistatin-like 1 is an Akt-regulated cardioprotective factor that is secreted by the heart. Circulation 117:3099–3108. 10.1161/CIRCULATIONAHA.108.767673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu HR, Tao L, Gao E, Lopez BL, Christopher TA, Willette RN, Ohlstein EH, Yue TL, Ma XL. 2004. Anti-apoptotic effects of rosiglitazone in hypercholesterolemic rabbits subjected to myocardial ischemia and reperfusion. Cardiovasc. Res. 62:135–144. 10.1016/j.cardiores.2003.12.027 [DOI] [PubMed] [Google Scholar]
- 49.Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C. 2007. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. U. S. A. 104:15560–15565. 10.1073/pnas.0705891104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. 2006. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc. Natl. Acad. Sci. U. S. A. 103:18255–18260. 10.1073/pnas.0608791103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li LY, Luo X, Wang X. 2001. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 412:95–99. 10.1038/35083620 [DOI] [PubMed] [Google Scholar]
- 52.Parrish J, Li L, Klotz K, Ledwich D, Wang X, Xue D. 2001. Mitochondrial endonuclease G is important for apoptosis in C. elegans. Nature 412:90–94. 10.1038/35083608 [DOI] [PubMed] [Google Scholar]
- 53.Lewis BP, Burge CB, Bartel DP. 2005. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120:15–20. 10.1016/j.cell.2004.12.035 [DOI] [PubMed] [Google Scholar]
- 54.Neuss M, Monticone R, Lundberg MS, Chesley AT, Fleck E, Crow MT. 2001. The apoptotic regulatory protein ARC (apoptosis repressor with caspase recruitment domain) prevents oxidant stress-mediated cell death by preserving mitochondrial function. J. Biol. Chem. 276:33915–33922. 10.1074/jbc.M104080200 [DOI] [PubMed] [Google Scholar]
- 55.van Empel VP, Bertrand AT, van der Nagel R, Kostin S, Doevendans PA, Crijns HJ, de Wit E, Sluiter W, Ackerman SL, De Windt LJ. 2005. Downregulation of apoptosis-inducing factor in harlequin mutant mice sensitizes the myocardium to oxidative stress-related cell death and pressure overload-induced decompensation. Circ. Res. 96:e92–e101. 10.1161/01.RES.0000172081.30327.28 [DOI] [PubMed] [Google Scholar]
- 56.Tanaka M, Ito H, Adachi S, Akimoto H, Nishikawa T, Kasajima T, Marumo F, Hiroe M. 1994. Hypoxia induces apoptosis with enhanced expression of Fas antigen messenger RNA in cultured neonatal rat cardiomyocytes. Circ. Res. 75:426–433. 10.1161/01.RES.75.3.426 [DOI] [PubMed] [Google Scholar]
- 57.Long X, Boluyt MO, Hipolito ML, Lundberg MS, Zheng JS, O'Neill L, Cirielli C, Lakatta EG, Crow MT. 1997. p53 and the hypoxia-induced apoptosis of cultured neonatal rat cardiac myocytes. J. Clin. Invest. 99:2635–2643. 10.1172/JCI119452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Webster KA, Discher DJ, Kaiser S, Hernandez O, Sato B, Bishopric NH. 1999. Hypoxia-activated apoptosis of cardiac myocytes requires reoxygenation or a pH shift and is independent of p53. J. Clin. Invest. 104:239–252. 10.1172/JCI5871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kitsis RN, Mann DL. 2005. Apoptosis and the heart: a decade of progress. J. Mol. Cell. Cardiol. 38:1–2. 10.1016/j.yjmcc.2004.11.008 [DOI] [PubMed] [Google Scholar]
- 60.Wienholds E, Kloosterman WP, Miska E, Alvarez-Saavedra E, Berezikov E, de Bruijn E, Horvitz HR, Kauppinen S, Plasterk RH. 2005. MicroRNA expression in zebrafish embryonic development. Science 309:310–311. 10.1126/science.1114519 [DOI] [PubMed] [Google Scholar]
- 61.Tuddenham L, Wheeler G, Ntounia-Fousara S, Waters J, Hajihosseini MK, Clark I, Dalmay T. 2006. The cartilage specific microRNA-140 targets histone deacetylase 4 in mouse cells. FEBS Lett. 580:4214–4217. 10.1016/j.febslet.2006.06.080 [DOI] [PubMed] [Google Scholar]
- 62.Miyaki S, Sato T, Inoue A, Otsuki S, Ito Y, Yokoyama S, Kato Y, Takemoto F, Nakasa T, Yamashita S, Takada S, Lotz MK, Ueno-Kudo H, Asahara H. 2010. MicroRNA-140 plays dual roles in both cartilage development and homeostasis. Genes Dev. 24:1173–1185. 10.1101/gad.1915510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang XJ, Gregoire S. 2005. Class II histone deacetylases: from sequence to function, regulation, and clinical implication. Mol. Cell. Biol. 25:2873–2884. 10.1128/MCB.25.8.2873-2884.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shen T, Zheng M, Cao C, Chen C, Tang J, Zhang W, Cheng H, Chen KH, Xiao RP. 2007. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J. Biol. Chem. 282:23354–23361. 10.1074/jbc.M702657200 [DOI] [PubMed] [Google Scholar]
- 65.de Brito OM, Scorrano L. 2009. Mitofusin-2 regulates mitochondrial and endoplasmic reticulum morphology and tethering: the role of Ras. Mitochondrion 9:222–226. 10.1016/j.mito.2009.02.005 [DOI] [PubMed] [Google Scholar]
- 66.Cleland MM, Norris KL, Karbowski M, Wang C, Suen DF, Jiao S, George NM, Luo X, Li Z, Youle RJ. 2011. Bcl-2 family interaction with the mitochondrial morphogenesis machinery. Cell Death Differ. 18:235–247. 10.1038/cdd.2010.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ. 2006. Role of Bax and Bak in mitochondrial morphogenesis. Nature 443:658–662. 10.1038/nature05111 [DOI] [PubMed] [Google Scholar]
- 68.Brooks C, Wei Q, Feng L, Dong G, Tao Y, Mei L, Xie ZJ, Dong Z. 2007. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc. Natl. Acad. Sci. U. S. A. 104:11649–11654. 10.1073/pnas.0703976104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martinou JC, Youle RJ. 2006. Which came first, the cytochrome c release or the mitochondrial fission? Cell Death Differ. 13:1291–1295. 10.1038/sj.cdd.4401985 [DOI] [PubMed] [Google Scholar]
- 70.Clerk A, Kemp TJ, Zoumpoulidou G, Sugden PH. 2007. Cardiac myocyte gene expression profiling during H2O2-induced apoptosis. Physiol. Genomics 29:118–127. 10.1152/physiolgenomics.00168.2006 [DOI] [PubMed] [Google Scholar]
- 71.Fan X, Hussien R, Brooks GA. 2010. H2O2-induced mitochondrial fragmentation in C2C12 myocytes. Free Radic. Biol. Med. 49:1646–1654. 10.1016/j.freeradbiomed.2010.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Danz ED, Skramsted J, Henry N, Bennett JA, Keller RS. 2009. Resveratrol prevents doxorubicin cardiotoxicity through mitochondrial stabilization and the Sirt1 pathway. Free Radic. Biol. Med. 46:1589–1597. 10.1016/j.freeradbiomed.2009.03.011 [DOI] [PubMed] [Google Scholar]
- 73.Niu J, Azfer A, Wang K, Wang X, Kolattukudy PE. 2009. Cardiac-targeted expression of soluble fas attenuates doxorubicin-induced cardiotoxicity in mice. J. Pharmacol. Exp. Ther. 328:740–748. 10.1124/jpet.108.146423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li J, Donath S, Li Y, Qin D, Prabhakar BS, Li P. 2010. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 6:e1000795. 10.1371/journal.pgen.1000795 [DOI] [PMC free article] [PubMed] [Google Scholar]