

Abstract
Aberrant activation of the ubiquitous transcription factor STAT3 is a major driver of solid tumor progression and pathological angiogenesis. STAT3 activity is regulated by numerous posttranslational modifications (PTMs), including Tyr705 phosphorylation, which is widely used as an indicator of canonical STAT3 function. Here, we report a noncanonical mechanism of STAT3 activation that occurs independently of Tyr705 phosphorylation. Using quantitative liquid chromatography-tandem mass spectrometry, we have discovered and characterized a novel STAT3 phosphoform that is simultaneously phosphorylated at Thr714 and Ser727 by glycogen synthase kinase 3α and -β (GSK-3α/β). Both Thr714 and Ser727 are required for STAT3-dependent gene induction in response to simultaneous activation of epidermal growth factor receptor (EGFR) and protease-activated receptor 1 (PAR-1) in endothelial cells. In this combinatorial signaling context, preventing formation of doubly phosphorylated STAT3 by depleting GSK-3α/β is sufficient to disrupt signal integration and inhibit STAT3-dependent gene expression. Levels of doubly phosphorylated STAT3 but not of Tyr705-phosphorylated STAT3 are remarkably elevated in clear-cell renal-cell carcinoma relative to adjacent normal tissue, suggesting that the GSK-3α/β–STAT3 pathway is active in the disease. Collectively, our results describe a functionally distinct, noncanonical STAT3 phosphoform that positively regulates target gene expression in a combinatorial signaling context and identify GSK-3α/β–STAT3 signaling as a potential therapeutic target in renal-cell carcinoma.
INTRODUCTION
The signal transducers and activators of transcription (STATs) are a family of seven transcription factors that regulate numerous physiological and pathophysiological processes, including immunity, angiogenesis, cellular survival, metastasis, and oncogenesis (1, 2). STAT3 is aberrantly activated in the vast majority of human cancers and is a downstream target of several oncogenic tyrosine kinases, including epidermal growth factor receptor (EGFR), JAKs, and Src family kinases (SFKs) (3–5). Consequently, much research has focused on understanding the role of STAT3 in malignancies, and studies are ongoing to determine the efficacy of STAT3 inhibition in treating human cancers (6, 7). It is therefore critical to identify and characterize novel mechanisms of STAT3 activation in order to elucidate unexplored opportunities to inhibit its function.
A wide range of stimuli, including growth factors, oncogenic kinases, and cytokines, can activate STAT3 (8). These stimuli modulate STAT3 function by regulating a diverse set of posttranslational modifications (PTMs), including tyrosine and serine phosphorylation, lysine acetylation, and lysine and arginine methylation (9–16). Activation of receptor and nonreceptor tyrosine kinases stimulates STAT3 Tyr705 phosphorylation to induce dimerization and increase STAT3 DNA binding activity (8, 13, 17, 18). Phosphorylation of Ser727 is mediated by various serine kinases (e.g., mitogen-activated protein kinases, cyclin-dependent kinases, and protein kinase Cs), and this modification increases STAT3 transcriptional activity by facilitating protein-protein interactions with transcriptional coactivators (8, 15, 19–22). Acetylation of several lysine residues, most notably Lys685, has also been reported to regulate STAT3 dimer formation and transcriptional activity (10–12, 23). STAT3 is methylated at Lys140 in response to interleukin 6 (IL-6), and this modification can inhibit or enhance STAT3-dependent transcription in a gene-specific manner (16). STAT3 has also been reported to be methylated at Arg31 by PRMT2 to negatively regulate leptin signaling (9).
The abundance and diversity of STAT3 PTMs suggest that numerous distinctly modified STAT3 forms (mod-forms) may be simultaneously present in a given cellular context. Indeed, there potentially exist 2n STAT3 mod-forms, where n is the number of modified STAT3 sites. As n increases or as the number of possible PTMs at a single site increases (e.g., acetylation or methylation of lysine), there is a corresponding exponential increase in the potential proteomic complexity of STAT3 mod-forms. This mechanism of proteomic expansion has been suggested to increase the functional repertoire of cellular proteins and is likely to confer signal integration potential on STAT3 (24).
We previously reported that STAT3 is a critical signal integrator downstream of coincident EGFR and protease-activated receptor 1 (PAR-1) signaling in vascular endothelial cells (EC) (25). In this context, glycogen synthase kinase 3α and -β (GSK-3α/β)-dependent phosphorylation of STAT3 Ser727 is required to trigger inducible expression of the transcription factor early growth response 1 (EGR1). Importantly, STAT3-dependent gene expression is triggered only when EGFR and PAR-1 are simultaneously activated, suggesting that the temporal information of coincident EGFR/PAR-1 activation is transduced via GSK-3α/β–STAT3 signaling. GSK-3α/β are multifunctional serine/threonine kinases that regulate substrates with multiple phosphorylation sites in a manner that often requires a “priming” phosphorylation (26). Recently, proteome-wide analyses have identified STAT3 Thr714 as a novel phosphorylation site (27–29), but the regulation and function of this modification have not been investigated.
In this report, we provide evidence that GSK-3α/β directly phosphorylate STAT3 to generate a STAT3 phosphoform that is simultaneously modified at Thr714 and Ser727. Both Thr714 and Ser727 phosphorylation are required for stimulus-dependent Mcl1 induction in a combinatorial signaling context, suggesting that the doubly modified phosphoform is the mediator of signal integration and gene induction during simultaneous EGFR/PAR-1 activation. Levels of doubly phosphorylated STAT3 are significantly elevated in renal tumors relative to matched normal tissue, suggesting that the GSK-3α/β–STAT3 signaling axis may be active in the disease. In summary, we provide a seminal example of temporal information encoding via multisite STAT3 phosphorylation in the context of combinatorial signaling.
MATERIALS AND METHODS
Cell culture, transfections, and treatments.
Human EC were isolated by trypsinization of umbilical veins as previously described (30). The EC were plated on fibronectin-coated cell culture dishes and maintained in MCDB–F-12 medium containing 15% fetal bovine serum (FBS), 0.009% heparin, and 0.015% endothelial cell growth supplement. All experiments were carried out using cells between the third and fifth passages. EC were transfected using Targefect reagents (Targeting Systems) according to the manufacturer's protocol. Unless otherwise indicated, EC were serum starved for 2 h prior to treatment with EGF (16 ng/ml), thrombin receptor agonist peptide (TRAP) (100 μM), or thrombin (5 U/ml). Small interfering RNAs (siRNAs) targeting STAT3 (s743), GSK-3α (s6237), and GSK-3β (s6241) were purchased from Life Technologies. STAT3 3′ untranslated region (UTR)-targeting siRNA was purchased from Dharmacon (D-003544-19-0005).
SDS-PAGE and Western blotting.
Total cell lysate from approximately 105 EC was resolved using Bis-Tris-buffered SDS-PAGE gels ranging from 8 to 12%, depending on the protein of interest. The gels were soaked in protein transfer buffer (48 mM Tris, 39 mM glycine, 20% methanol, 0.0375% SDS) and transferred to a polyvinylidene difluoride (PVDF) membrane using a Bio-Rad semidry transfer cell. After transfer, the PVDF membranes were washed briefly in TBST (150 mM NaCl, 50 mM Tris-HCl, pH 7.5, 0.1% Tween 20) and then blocked for 2 h in protein-free TBST blocking buffer (Pierce). After blocking, primary antibodies were diluted 1:1,000 in blocking buffer and incubated overnight at 4°C. The membranes were washed and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h, and the HRP signals were detected by chemiluminescence. Antibodies for the following targets were purchased from Cell Signaling Technology (catalog numbers are in parentheses): STAT3 (9139 and 4904), phosphorylated Tyr705 (pTyr705) STAT3 (9131, 9138, and 9145), Pro-Met-pSer-Pro (PMpSP) motif (2325), pThr-Pro motif (9391), GSK-3α/β (5676), GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (5174), and Mcl1 (5453). Anti-α-tubulin (T5168) antibody was purchased from Sigma-Aldrich. Anti-pSer727 STAT3 antibody (sc-136193) was purchased from Santa Cruz Biotechnology.
Immunoprecipitation.
EC were washed once with ice-cold phosphate-buffered saline (PBS) and lysed in RIPA buffer (50 mM Tris-HCl, pH 7.4, 1% NP-40, 150 mM NaCl, 0.1% SDS, 1 mM EDTA, 5 mM Na3VO4) supplemented with protease inhibitor cocktail (Roche) and phosphatase inhibitors (Roche). Lysis was allowed to proceed for 30 min at 4°C under gentle agitation. The lysates were cleared by high-speed centrifugation, and primary antibodies were added for overnight incubation. Antibody complexes were precipitated with protein A/G-agarose beads, washed three times with ice-cold RIPA buffer, and denatured with 2× Laemmli buffer for SDS-PAGE.
EGR1 promoter luciferase reporter and luciferase assay.
Luciferase reporter assays were performed using a 2.1-kb fragment of the EGR1 promoter, as previously described (25). Briefly, luciferase assays were performed by transfecting EC with EGR1 promoter reporter 24 h prior to treatment. The cells were lysed with passive lysis buffer (Promega), and luciferase activity was assayed using the luciferase assay system (Promega) according to the manufacturer's instructions.
Quantitative mass spectrometry.
After immunoprecipitation with an anti-STAT3 antibody (CST 9139), an ∼86-kDa band was cut from the Coomassie-stained gel, digested with trypsin, and analyzed by capillary column liquid chromatography-tandem mass spectrometry (LC–MS-MS) to identify phosphopeptides. The LC–MS-MS system was a Finnigan LTQ linear ion trap mass spectrometer and a LTQ-Orbitrap Elite system. The high-performance liquid chromatography (HPLC) column was a self-packed 9-cm by 75-μm (inside diameter [i.d.]) Phenomenex Jupiter C18 (LTQ) or a Dionex 15-cm by 75-μm (i.d.) Acclaim Pepmap C18 reverse-phase capillary column (Orbitrap). The digest was analyzed in both a survey manner and a targeted manner. The survey experiments were performed using the data-dependent multitask capability of the instrument, acquiring full-scan mass spectra to determine peptide molecular weights and product ion spectra to determine the amino acid sequence in successive instrument scans. These data were analyzed by using all collision-induced dissociation (CID) spectra collected in the experiment to search the human reference sequence database with the search program Mascot. The targeted experiments involved the analysis of specific STAT3 peptides, including the phosphorylated and unmodified forms of the Y705, S727, and T714 tryptic peptides. The chromatograms for these peptides were plotted based on known fragmentation patterns, and the peak areas of the chromatograms were used to determine the extent of phosphorylation.
Protein extraction from tumor tissue.
Tumor tissue was procured from fresh-frozen nephrectomy specimens, and an experienced kidney pathologist confirmed clear-cell renal-cell carcinoma (ccRCC) or papillary renal-cell carcinoma (pRCC) diagnosis by histology. Samples (∼100 mg) of tumor tissue were solubilized in RIPA buffer supplemented with protease and phosphatase inhibitors using a Dounce homogenizer, and the lysates were cleared once by high-speed centrifugation, tumbled end over end overnight, and cleared again by centrifugation, after which STAT3 was immunoprecipitated using an anti-STAT3 monoclonal antibody.
Development of polyclonal antiserum for STAT3 Thr714 phosphorylation.
Anti-STAT3 pThr714 polyclonal antiserum was generated by immunizing a rabbit with a phosphorylated peptide corresponding to phosphorylation at that site (KFICVpTPTTC).
In vitro kinase assays.
Recombinant STAT3 was purchased from Abcam (ab64310), and active GSK-3β was purchased from New England BioLabs (NEB P6040) and utilized according to the manufacturer's instructions. Briefly, STAT3 was incubated with GSK-3β in ATP-supplemented kinase buffer (20 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 5 mM dithiothreitol [DTT]) for 1 h at 30°C. For immune complex phosphorylation assays, recombinant wild-type (WT) STAT3, T714A STAT3, and S727A STAT3 were expressed in EC by transient transfection of STAT3 cDNAs. Prior to lysis, the EC were serum starved for 2 h to reduce STAT3 basal phosphorylation. The EC were lysed, and STAT3 was immunopurified using an anti-STAT3 monoclonal antibody and protein A/G beads. The beads were washed with RIPA buffer and resuspended in kinase buffer supplemented with ATP in the absence or presence of GSK-3β for 1 h at 30°C. The reactions were terminated with 2× SDS buffer and analyzed by SDS-PAGE and immunoblotting.
Chromatin immunoprecipitation.
Chromatin immunoprecipitations (ChIP) were performed using a commercially available kit according to the manufacturer's instructions (Millipore; 17-295). Human EC treated with EGF plus TRAP or IL-6 for 15 min were cross-linked by the addition of formaldehyde. The cross-linked EC were lysed in SDS lysis buffer and sonicated using a Sonicator 3000 (Misonix Inc., Farmingdale, NY). Chromatin-protein complexes were immunoprecipitated overnight with an anti-STAT3 antibody, and genomic DNA was purified using DNA PrepEase columns (Affymetrix). Purified genomic DNA was analyzed by quantitative real-time PCR using primers flanking the upstream GAS element in the EGR1 promoter (25): forward primer, 5′-CAGGAGGAGCCTTCCCTTCCCG-3′; reverse primer, 5′-CTGGGGCCGAACGCAACAG-3′. The data were normalized relative to the amount of input DNA for each sample and expressed as the fold change relative to the untreated control sample.
Statistical analyses.
Unless otherwise indicated, data are expressed as means and standard errors of the mean (SEM). Differences between groups were analyzed by analysis of variance (ANOVA). Bonferroni post hoc tests were performed to calculate multiplicity-adjusted P values and to evaluate pairwise differences between groups. Differences between tumors and normal tissue were evaluated by two-tailed paired t tests.
RESULTS
Multiple STAT3 phosphoforms are induced by combinatorial EGFR/PAR-1 signaling in endothelial cells.
We previously showed that GSK-3α/β participate in STAT3 activation downstream of simultaneous EGFR/PAR-1 signaling by positively regulating STAT3 Ser727 phosphorylation (Fig. 1A). STAT3 Thr714 is located in an evolutionarily conserved region that includes conservation of proline in the +1 position, raising the possibility that it is a direct substrate of GSK-3α/β. We therefore examined Thr714 and Ser727 phosphorylation in EC by quantitative mass spectrometry. STAT3 was immunoprecipitated from EC treated with EGF plus TRAP, a PAR-1 activator. Immunoprecipitates were resolved by SDS-PAGE, and ∼86-kDa protein bands were cut from the gel for LC–MS-MS analysis. We readily detected phosphorylation of STAT3 at Thr714 or Ser727 (Fig. 1B and C). We also identified STAT3 phosphopeptides that were phosphorylated at both Thr714 and Ser727 (Fig. 1D). Surprisingly, we failed to detect STAT3 Tyr705 phosphopeptides by mass spectrometry under these conditions (Fig. 1B).
FIG 1.

Identification of STAT3 phosphoforms in endothelial cells. (A) Coincident PAR-1 activation prevents EGF-induced inhibition of GSK-3α/β by AKT, thus allowing GSK-3α/β to positively regulate Ser727 phosphorylation (25). As proline-directed kinases, GSK-3α/β may directly phosphorylate both Thr714 and Ser727. (B) Serum-starved EC were treated with EGF plus TRAP for 15 min, after which STAT3 was immunoprecipitated, and STAT3 phosphorylation was quantified by selected reaction monitoring targeting the specific STAT3 peptides listed in the table. +, peptide identified; −, peptide not identified. (C) CID spectrum of the singly phosphorylated form of the T710-729 STAT3 peptide, FICVTPTTCSNTIDLPMoSPR. This spectrum is consistent with the presence of two different forms of the peptide. The y5 ion at 683.3 Da is consistent with phosphorylation at S727. The presence of an unmodified y15 ion at 1,705 Da and only a modified y16 ion at 1,886 Da indicates that a second isoform with modification at T714 is also present. (D) CID spectrum of the doubly phosphorylated form of the T710-729 STAT3 peptide, FICVTPTTCSNTIDLPMoPR. The identification of the C-terminal y5 ion at 683 Da is consistent with phosphorylation at S727. In addition, the mass difference between the y16 and y15 ions is 181 Da and is consistent with the second site of phosphorylation occurring at T714. Mo, oxidized form of methionine.
Differential induction of STAT3 phosphoforms by EGFR/PAR-1 signaling.
We next sought to characterize signaling mechanisms regulating the inducible formation of STAT3 phosphoforms. EC were stimulated with EGF plus TRAP for up to 1 h, STAT3 was immunoprecipitated and resolved by SDS-PAGE (Fig. 2A), and the kinetics with which STAT3 phosphoforms were induced were analyzed by LC–MS-MS. Singly phosphorylated Thr714 or Ser727 phosphoforms are induced as early as 5 min following EGF-plus-TRAP treatment and reach a maximum at ∼15 min posttreatment (Fig. 2B and C, red and blue). Levels of Thr714-phosphorylated STAT3 and Ser727-phosphorylated STAT3 begin to decrease after 15 min and stabilize at ∼50% of their maximum levels between 30 and 60 min after stimulation. Doubly phosphorylated STAT3 exists in much lower abundance than singly modified phosphoforms (Fig. 2B, green) but demonstrates the highest degree of stimulus-dependent inducibility (Fig. 2C). Further, the level of doubly phosphorylated STAT3 remains elevated for up to 30 min after stimulus exposure, after which it returns to ∼5-fold the basal level (Fig. 2C).
FIG 2.

Quantitative analysis of STAT3 phosphoform abundance following EGFR/PAR-1 activation. (A) STAT3 was immunoprecipitated (IP) from the pooled extracts and resolved by SDS-PAGE. An ∼86-kDa band corresponding to Coomassie-stained STAT3 was cut from the gel, digested with trypsin, and subjected to LC–MS-MS and selected reaction monitoring (SRM) to quantify specific STAT3 phosphoforms. This method allowed independently treated replicates to yield sufficient endogenous STAT3 for LC–MS-MS analysis. (B) Serum-starved EC were treated with EGF and TRAP for the indicated times, after which they were lysed in RIPA buffer, and STAT3 was immunoprecipitated for mass spectrometry analysis as described in Materials and Methods. Three independently treated 100-mm dishes were pooled and used for each point indicated on the graph. Percent phosphorylation was calculated as the abundance of a specific phosphoform divided by the abundance of all STAT3 tryptic peptides at that site. (C) Data from panel B presented as fold change relative to basal levels for each specific STAT3 phosphoform. Curve fit analyses were performed using a two-phase exponential model. (D) Coomassie-stained polyacrylamide gel of STAT3 immunoprecipitates after treatment with EGF (16 ng/ml), TRAP (100 μM), or both for 15 min. (E) Serum-starved EC treated with EGF, TRAP, or both for 15 min and processed for quantitative mass spectrometry of Thr714 phosphorylation as in panel B. (F) Mass spectrometry analysis of Ser727 phosphorylation as in panel B. (G) Mass spectrometry analysis of doubly phosphorylated STAT3 as in panel B. (H) The synergism ratios for individual STAT3 phosphoforms were calculated as follows: responseEGF + TRAP/(responseEGF + responseTRAP).
To determine the extent to which STAT3 phosphoforms are regulated by EGFR/PAR-1 cross talk, EC were treated with EGF, TRAP, or both for 15 min (Fig. 2D), and STAT3 phosphoforms were measured by LC–MS-MS. Singly phosphorylated Thr714 STAT3 was moderately induced by either EGF or TRAP alone (Fig. 2E), and combination treatment caused an almost perfectly additive increase in pThr714 STAT3 abundance (Fig. 2E and H). Singly phosphorylated Ser727 STAT3 was also moderately induced by either EGF or TRAP alone and was synergistically induced by combination treatment with EGF plus TRAP (Fig. 2F and H). Doubly phosphorylated STAT3 was undetectable under unstimulated conditions, and stimulation with either EGF or TRAP caused a minimal increase in the abundance of this phosphoform (Fig. 2G). Remarkably, combination treatment with EGF plus TRAP increased the levels of doubly phosphorylated STAT3 (Fig. 2G) in a manner that was strongly synergistic compared to the response with either EGF or TRAP alone (Fig. 2H).
EGF-plus-TRAP-induced STAT3 phosphorylation is distinct from IL-6-induced phosphorylation.
Next, we sought to validate our mass spectrometry results for Thr714, Ser727, and Tyr705 phosphorylation. STAT3 was immunoprecipitated from human umbilical vein endothelial cells (HUVEC), and various phosphospecific antibodies were used to assess the levels of distinct STAT3 phosphorylations. To investigate stimulus-inducible Thr714 phosphorylation by immunoblotting, a polyclonal STAT3 anti-pThr714 antibody was generated by immunizing a rabbit with a phosphopeptide corresponding to the STAT3 residues at that phosphosite. STAT3 Thr714 phosphorylation was synergistically induced in response to 15-min EGF-plus-TRAP treatment (Fig. 3A and B). Ser727 phosphorylation was also synergistically induced when STAT3 immunoprecipitates were probed with an anti-PMpSP antibody that is designed to recognize phosphorylated serine in the MAP kinase consensus motif corresponding to Ser727 (Fig. 3A and C). We previously showed that a commercially available pSer727 STAT3 antibody recognizes synergistically induced Ser727 phosphorylation in a manner consistent with mass spectrometry results after 5 min of stimulation with EGF plus TRAP (25). However, after 15 min of stimulation, the commercial antibody does not reflect the ∼8- to 10-fold induction of Ser727 phosphorylation that occurs (as indicated by mass spectrometry and PMpSP antibody), possibly due to the increased abundance of cooperating modifications near that site (Fig. 3A). Therefore, in subsequent experiments, we used immunoprecipitation and anti-PMpSP immunoblots to assess Ser727 phosphorylation.
FIG 3.
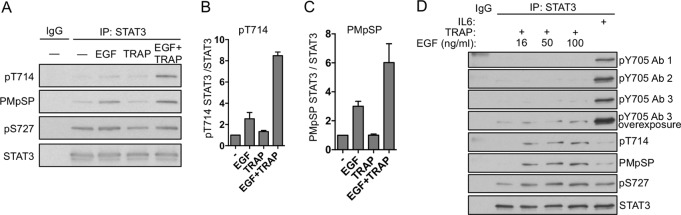
Immunoblot analysis of STAT3 phosphorylations. (A) Serum-starved EC were treated with EGF, TRAP, or both for 15 min, and STAT3 was immunoprecipitated and resolved by SDS-PAGE. STAT3 phosphorylations were assessed by immunoblotting. The anti-pThr714 antibody was generated by immunizing a rabbit with a phosphorylated peptide corresponding to phosphorylation at that site (KFICVpTPTTC). STAT3 Ser727 phosphorylation was detected using a Pro-Met-phospho-Ser-Pro motif antibody and a commercially available antibody from Santa Cruz Biotechnology. (B) Densitometry analysis of Thr714 phosphorylation from 3 independent experiments performed as for panel A. (C) Densitometry analysis of Ser727 phosphorylation from 3 independent experiments performed as for panel A. (D) Serum-starved EC were treated with EGF at the indicated concentrations in the presence of TRAP for 15 min. The EC were lysed with RIPA buffer, and STAT3 was immunoprecipitated and resolved by SDS-PAGE. Three antibodies (Ab) were used to assess Tyr705 phosphorylation. Ab 1 is Cell Signaling Technology (CST) 9131, Ab 2 is CST 9145, and Ab 3 is CST 9138. The immunoblot with CST 9138 was deliberately overexposed to film to detect basal STAT3 Tyr705 phosphorylation. Thr714 and Ser727 phosphorylation was assessed as for panel A. IL-6 (10 ng/ml) was included as a positive control for Tyr705 phosphorylation. The error bars indicate SEM.
We next sought to standardize immunoblotting methods for detecting multiple STAT3 phosphorylations. Our mass spectrometry results suggested that phosphorylation of STAT3 Tyr705 is low or absent in EC. To validate this result, we performed a dose-response experiment using increasing levels of EGF in the presence of TRAP and analyzed Tyr705 phosphorylation by immunoblotting using three commercially available antibodies (Fig. 3D). Tyr705 phosphorylation was strongly induced by IL-6 but not by EGF plus TRAP, even at high EGF concentrations. Basal Tyr705 phosphorylation was detectable upon overexposure of the film, suggesting that this modification occurs basally at very low abundance in EC. A commercially available pSer727 antibody did not detect a strong EGF-plus-TRAP-induced phosphorylation at Ser727, suggesting that there is a loss of affinity of the antibody for phosphorylated STAT3 at this time point (Fig. 3D). EGF-plus-TRAP stimulation increased Thr714 phosphorylation and PMpSP phosphorylation (Ser727) of STAT3 at all concentrations (Fig. 3D).
GSK-3α/β phosphorylate STAT3 to generate a doubly modified phosphoform.
We previously reported that GSK-3α/β positively regulated STAT3 Ser727 phosphorylation and STAT3-dependent gene expression in EC (25). Therefore, we investigated the extent to which GSK-3α/β regulate the stimulus-inducible generation of specific STAT3 phosphoforms downstream of EGFR/PAR-1 signaling. Depletion of GSK-3α/β by RNA interference (RNAi) caused a 90% decrease in the levels of inducible doubly phosphorylated STAT3 while causing only a partial reduction in single pThr714 or pSer727 STAT3 (Fig. 4A). This result was verified by immunoblotting STAT3 immunoprecipitates using a STAT3 pThr714 antibody (Fig. 4B) and by anti-pThr-Pro and Pro-Met-pSer-Pro motif antibodies to detect the levels of pThr714 and pSer727, respectively (Fig. 4C and D). In all cases, GSK-3α/β depletion caused a reduction in total pThr714 and pSer727, demonstrating that GSK-3α/β positively regulate STAT3 Ser/Thr phosphorylation. Moreover, this result demonstrates that there are both GSK-3α/β-dependent and GSK-3α/β-independent components of STAT3 Thr714 and Ser727 phosphorylation, and GSK-3α/β are required for generating doubly phosphorylated STAT3 in this context. GSK-3β efficiently phosphorylated STAT3 in vitro at a pThr-Pro site (Fig. 4E). To test the ability of GSK-3β to phosphorylate STAT3 at Thr714 and Ser727, we immunopurified WT STAT3, S727A STAT3, and T714A STAT3 for use as in vitro GSK-3β substrates. GSK-3β efficiently phosphorylated both Thr714 and Ser727 (Fig. 4F). Mutation of Ser727 to alanine dramatically reduced the ability of GSK-3β to phosphorylate Thr714. In contrast, mutation of Thr714 to alanine did not affect Ser727 phosphorylation by GSK-3β (Fig. 4F), suggesting that Ser727 may regulate priming or processive phosphorylation of STAT3 Thr714 by GSK-3α/β.
FIG 4.

GSK-3α/β directly phosphorylate STAT3 at Thr714 and Ser727. (A) GSK-3α/β were depleted from EC by RNAi; 48 h after siRNA transfection, the EC were stimulated with EGF plus TRAP for 15 min, and STAT3 phosphoform abundance was analyzed by mass spectrometry. Red bars, STAT3 phosphorylated at Ser727; blue bars, STAT3 phosphorylated at Thr714; green bars, STAT3 phosphorylated at both Thr714 and Ser727. (B) GSK-3α/β were depleted from EC by RNAi, and the effect on EGF-plus-TRAP-induced STAT3 Thr714 phosphorylation was analyzed by immunoblotting of STAT3 immunoprecipitates using anti-STAT3 pThr714 polyclonal antisera. (C) GSK-3α/β were depleted from EC by RNAi, and the effect on EGF-plus-TRAP-induced STAT3 Thr714 phosphorylation was analyzed by immunoblotting of STAT3 immunoprecipitates using anti-Thr-Pro and anti-Pro-Met-pSer-Pro motif antibodies. The amino acid sequence under the immunoblot represents STAT3 710 to 729, with the sites targeted for immunodetection indicated. (D) Densitometry of 3 independent immunoblots performed as for panel C. The error bars indicate SEM. (E) In vitro kinase assay using recombinant GSK-3β (New England BioLabs) and STAT3 (Abcam) substrate was performed for 1 h at 30°C, and threonine phosphorylation was detected using a pThr-Pro motif antibody. (F) Endogenous STAT3 was depleted by RNAi targeting the 3′ UTR of STAT3, and recombinant WT STAT3, S727A STAT3, or T714A STAT3 was immunopurified from serum-starved EC and used as an in vitro substrate for GSK-3β. The amino acid sequence under the immunoblot represents STAT3 710 to 729, with the sites targeted for immunodetection indicated.
STAT3 phosphorylation at Thr714 by GSK-3α/β positively regulates target gene expression.
We previously showed that simultaneous EGFR/PAR-1 activation causes synergistic induction of EGR1 in a manner that requires STAT3 Ser727 phosphorylation (25). To identify established STAT3 targets that are regulated by EGFR/PAR-1 signaling, we examined Bcl2 family members that are known to require STAT3 Ser727 phosphorylation, including Mcl1 (31). Indeed, we found that EGF plus TRAP maximally induced Mcl1 at 2 h posttreatment (Fig. 5A), and this induction was synergistic compared to treatment with either EGF or TRAP alone (Fig. 5B and C). Depletion of STAT3 by RNAi completely inhibited synergistic induction of Mcl1 (Fig. 5D), demonstrating that STAT3 is critical for EGFR/PAR-1 cross talk, consistent with our previously published results for EGR1 induction (25). Inducible expression of Mcl1 was reduced ∼50% when GSK-3α/β were depleted by RNAi, consistent with our hypothesis that GSK-3α/β positively regulate STAT3 activity (Fig. 5E and F). Further, WT STAT3, but not T714A, S727A, or T714A/S727A STAT3 mutants, rescued EGF-plus-TRAP-induced Mcl1 expression when endogenous STAT3 was depleted by RNAi (Fig. 5G and H). In contrast, a STAT3 Y705F mutant lacking the canonical phosphorylation site rescued Mcl1 induction to an extent similar to that of WT STAT3, suggesting that combinatorial activation of STAT3 by EGFR/PAR-1 is Tyr705 independent (Fig. 5I to J).
FIG 5.

STAT3 Thr714 and Ser727 phosphorylation positively regulate target gene expression. (A) Serum-starved EC were treated with EGF plus TRAP over a time course of up to 8 h, and inducible Mcl1 expression was analyzed by immunoblotting. (B) Serum-starved EC were treated for 2 h with EGF, TRAP, or both, and Mcl1 expression was analyzed by immunoblotting. (C) Densitometry analysis of 3 independent immunoblots performed as for panel B. (D) STAT3 was depleted by RNAi; EC were treated with EGF, TRAP, or both for 2 h; and Mcl1 expression was analyzed by immunoblotting. (E and F) GSK-3α/β were depleted from EC by RNAi, and the effect on EGF-plus-TRAP-dependent Mcl1 induction was analyzed by immunoblotting (E) and densitometry (F) of 4 independent experiments. (G and H) Endogenous STAT3 was depleted by RNAi with siRNA targeting the 3′ UTR; STAT3 levels were reconstituted by transient expression of recombinant WT, T714A, S727A, or T714A/S727A STAT3; and the effect on Mcl1 expression was analyzed by immunoblotting (G) and densitometry (H) of 6 independent experiments for GFP/control, GFP/siSTAT3, WT/siSTAT3, and T714A/siSTAT3 and 3 experiments for S727A and T714A/S727A STAT3. (I) Experiment performed as for panel G using a STAT3 mutant in which Tyr705 is mutated to phenylalanine. (H and J) Densitometry values of 3 independent experiments performed as for panel I. *, P < 0.05; **, P < 0.01. The error bars indicate SEM.
Simultaneous EGFR/PAR-1 signaling induces STAT3 binding to a distal GAS element in the EGR1 promoter.
We next sought to compare combinatorial activation of STAT3 by EGF plus TRAP to canonical activation of STAT3 by gamma interferon (IFN-γ) or IL-6. Simultaneous EGFR/PAR-1 activation strongly induced STAT3 Thr-Pro and Thr714 phosphorylation at 15 min (Fig. 6A). A PMpSP motif antibody (Ser727) detected phosphorylated STAT3 Ser727 in a manner largely consistent with data from mass spectrometry experiments (Fig. 6A). Neither IL-6 nor IFN-γ induced a significant degree of STAT3 threonine phosphorylation. IL-6 and IFN-γ, but not EGF plus TRAP, strongly induced STAT3 Tyr705 phosphorylation, suggesting that STAT3-dependent gene expression can be triggered by EGF plus TRAP in the absence of Tyr705 phosphorylation. Further, we previously showed that an upstream GAS element in the EGR1 promoter was critical for EGF-plus-TRAP-induced promoter activation (25). EGF plus TRAP, but not IL-6, induced STAT3 binding to this upstream promoter region (Fig. 6B). Additionally, when EC were cotransfected with an EGR1 promoter reporter, EGR1 promoter activation was significantly reduced by cotransfection with T714A STAT3 relative to WT STAT3 (Fig. 6C). Coupled with our previous results, these data demonstrate that both Thr714 and Ser727 are required for maximal EGR1 promoter activation (25). Overexpression of a phosphomimetic T714D STAT3 mutant was not sufficient to increase basal EGR1 promoter activity relative to WT STAT3 (Fig. 6D), suggesting that other modifications and signaling pathways are required. To test the requirement for Tyr705 in EGR1 promoter activation, we overexpressed WT STAT3 or Y705F STAT3 in STAT3-depleted EC and measured EGR1 promoter activity in response to EGF plus TRAP. EGF-plus-TRAP-induced promoter activation was similar for WT STAT3 and Y705F STAT3 (Fig. 6E).
FIG 6.

Combinatorial control of STAT3 and EGR1 promoter activation. (A) Serum-starved EC were treated with EGF plus TRAP, IFN-γ (10 ng/ml), or IL-6 (10 ng/ml) for 15 min, after which STAT3 was immunoprecipitated and phosphorylations were analyzed by immunoblotting. (B) EC were stimulated for 15 min with EGF plus TRAP or IL-6, after which STAT3 was immunoprecipitated from cross-linked cell extracts. DNA was isolated from the immunoprecipitates, and the EGR1 promoter was PCR amplified using forward and reverse primers flanking the upstream GAS element of the EGR1 promoter. Binding of STAT3 to the EGR1 promoter was verified by DNA sequencing of PCR amplicons. (C) EC were cotransfected with an EGR1 promoter reporter and WT STAT3 or T714A STAT3. EGF-plus-TRAP-induced luciferase activity was measured after 4 h of treatment using a luminometer. Endogenous STAT3 was depleted by siRNA targeting the 3′ UTR of STAT3 under all conditions. RLU, relative light units. (D) EC were cotransfected with an EGR1 promoter reporter and WT STAT3 or T714D STAT3. Basal luciferase activity was measured after 8 h of serum starvation using a luminometer. (E) EC were cotransfected with an EGR1 promoter reporter and WT STAT3 or Y705F STAT3. EGF-plus-TRAP-induced luciferase activity was measured after 4 h of treatment using a luminometer. *, P < 0.05; **, P < 0.01; ***, P < 0.001; N.S., not significant. The error bars indicate SEM.
Doubly phosphorylated STAT3 is elevated in human renal tumors.
Preclinical studies of RCC have validated both STAT3 and GSK-3α/β as targetable molecules for the therapeutic inhibition of RCC progression (32–35). Additionally, aberrant nuclear accumulation of GSK-3β has been reported to occur in >90% of human RCC cases. Based on these studies and our discovery of a direct link between GSK-3α/β and STAT3 Thr714 phosphorylation, we hypothesized that doubly phosphorylated STAT3 may be elevated in renal tumors. We analyzed STAT3 PTMs in a cohort of 4 ccRCC and 4 pRCC patients to determine the relative degrees of STAT3 Thr714 phosphorylation in these cases. Thr714 phosphorylation was detectable in all patients by immunoblotting but was remarkably elevated in 2/4 ccRCC cases and 1/4 pRCC cases (Fig. 7A). STAT3 Ser727 phosphorylation (anti-PMpSP) was elevated in 4/4 ccRCC cases and 1/4 pRCC cases. STAT3 pTyr705 was elevated in 4/4 ccRCC patients and 1/4 pRCC patients. The same cases were analyzed by LC–MS-MS to measure the abundance of STAT3 phosphoforms (Fig. 7B to E). STAT3 phosphoform abundance was generally elevated in ccRCC versus pRCC, and levels of pSer727 and pTyr705 were significantly elevated in ccRCC (Fig. 7C and E).
FIG 7.

Detection and abundance of STAT3 phosphoforms in renal-cell carcinoma. (A) Human renal-tumor samples (clear cell and papillary) were isolated from tissue after nephrectomy. The tumor tissue was solubilized in RIPA buffer and cleared by high-speed centrifugation, and STAT3 was immunoprecipitated overnight. Half of the immunoprecipitate was analyzed by immunoblotting to confirm the presence of STAT3 and its phosphorylations. Recombinant (Rec.) STAT3 was included as a nonphosphorylated reference. (B to E) The remaining immunoprecipitates were processed by LC–MS-MS to measure the abundances of STAT3 phosphoforms in the tumors. C1 and C2 indicate the clear-cell renal-cell carcinoma case from panel A with elevated STAT3 phosphorylation. Horizontal lines indicate the median values of the samples. (F to I) Ten cases of ccRCC and patient-matched normal tissue were examined by microscopy to confirm the diagnosis and to estimate the number of tumor nuclei per field. Tumor tissue and matched tissue controls were solubilized as for panel A, and STAT3 was immunoprecipitated to analyze phosphoform abundance by LC–MS-MS. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (J to M) An experienced kidney pathologist estimated the percentage of tumor nuclei per field. Pearson correlation coefficients were calculated based on the percentage of tumor nuclei (x axis) versus phosphoform abundance (y axis). Pearson r values and two-tailed P values for correlation with tumor nuclei were as follows: pThr714, r = 0.37, P = 0.29; pSer727, r = −0.42, P = 0.23; double pSTAT3, r = 0.44, P = 0.20; pTyr705, r = −0.38, P = 0.28.
Based on these results, we procured 10 additional cases of ccRCC with matched normal tissue to determine the extent to which STAT3 phosphoform abundances differed in tumor tissue relative to adjacent normal tissue. Singly phosphorylated STAT3 at Thr714 or Ser727 was significantly elevated in tumor tissue relative to adjacent normal tissue (Fig. 7F and G). Doubly phosphorylated STAT3 was also significantly more abundant in tumor tissue, indicating that GSK-3α/β–STAT3 signaling may be active in ccRCC (Fig. 7H). In contrast, pTyr705 STAT3 displayed a high degree of variability and was not significantly different between normal and tumor tissue (Fig. 7I). To examine the relative contribution of cancer cells versus stromal cells to tumor-associated STAT3 phosphorylation, we examined the extent to which STAT3 phosphoforms correlated with the percentage of tumor nuclei in ccRCC cases. pThr714 and doubly phosphorylated STAT3 exhibited a positive correlative trend versus the percentage of tumor nuclei in each sample, but this relationship was not statistically significant (Fig. 7J and L). Neither pSer727 nor pTyr705 correlated with the percentage of tumor nuclei (Fig. 7K and M). These data clearly demonstrate that Thr714- and Ser727-phosphorylated STAT3 are elevated in renal-tumor tissue, suggesting that GSK-3α/β–STAT3 signaling is a novel targetable signaling axis in ccRCC.
DISCUSSION
The transcriptional activity of STAT3 is modulated by reversible PTMs that regulate dimerization, DNA binding, and protein-protein interactions. Activated STAT3, in turn, transcriptionally activates genes that promote growth and survival and is thought to be a major driver of solid tumor progression. We now report that STAT3 Thr714 is phosphorylated, in addition to Ser727, in response to EGF signaling during coincident GPCR activation, and both of these modifications are critical for STAT3-dependent expression of Mcl1 and EGR1 (Fig. 5G to H). Therefore, the data presented here strongly suggest that EGR1 and Mcl1 induction is driven by a low-abundance but dynamically regulated STAT3 phosphoform that is simultaneously modified at Thr714 and Ser727, likely in the absence of Tyr705 phosphorylation (Fig. 2B and C, 3D, and 6A). Further, our data demonstrate that GSK-3α/β exclusively mediate the generation of doubly phosphorylated STAT3 to induce Mcl1 and EGR1 during coincident EGFR/PAR-1 activation (Fig. 4A and F). The additive response of pThr714 STAT3 to EGF plus TRAP (Fig. 2H) and the observation that T714A does not affect Ser727 phosphorylation (Fig. 4F) suggest that there is little to no conversion of pThr714 STAT3 to doubly phosphorylated STAT3 in this context (Fig. 8). We therefore propose that doubly phosphorylated STAT3 mediates a noncanonical mechanism of STAT3 activation that is triggered specifically by combinatorial EGFR/PAR-1 signaling and is aberrantly regulated in human renal tumors (Fig. 8).
FIG 8.
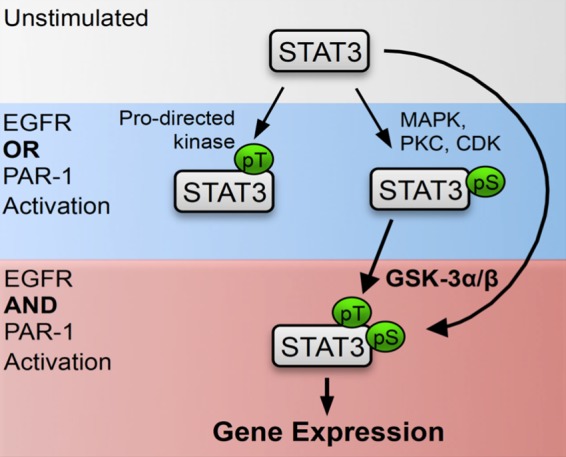
GSK-3α/β regulate a novel mechanism of STAT3 activation via direct Thr714/Ser727 phosphorylation. The model shows the inducible flux of STAT3 phosphoforms stimulated with different combinations of EGF plus TRAP. Simultaneous EGFR/PAR-1 activation is required to drive significant formation of doubly phosphorylated STAT3 and STAT3-dependent gene expression in this context. Furthermore, GSK-3α/β are required for nearly all of the doubly phosphorylated STAT3 generated following EGFR/PAR-1 activation (red area). During individual activation of EGFR or PAR-1, or in the absence of GSK-3α/β, STAT3 is inducibly phosphorylated at Thr714 or Ser727 (blue area). Numerous Ser727 kinases are known, but additional proline-directed Thr714 kinases that catalyze single phosphorylation of Thr714 in the absence of GSK-3α/β and Ser727 phosphorylation likely exist.
STAT3 Thr714 phosphorylation adds to the high frequency of PTMs within a relatively small region of the STAT3 COOH terminus. Between Lys679 and Ser727, researchers have identified acetylation of Lys679, Lys685, Lys707, and Lys709 (10–12, 23) and phosphorylation at Tyr705, Thr714, and Ser727 (13, 15). The mechanisms by which canonical STAT3 modifications (pTyr705 and pSer727) regulate STAT3 function are relatively clear. However, the exact mechanisms by which lysine acetylation/methylation and threonine phosphorylation regulate transcriptional activity of STAT3 are not well understood, and more work is required to understand the structural basis for promoter-specific functions of individual STAT3 PTMs. In any event, the diversity and abundance of STAT3 PTMs that is now reported, coupled with the established functions of unphosphorylated STAT3 (U-STAT3) (36–38), demonstrate that Tyr705 phosphorylation is neither necessary nor sufficient for STAT3 activation in a number of signaling contexts (36, 37, 39–42). Further, STAT1/3 phosphorylations at Tyr701/705 and Ser727 is often mediated by the same kinases, as the sequences surrounding these residues are highly similar. However, the amino acids surrounding Ser708 in STAT1 and Thr714 in STAT3 are unique, suggesting a possible basis for the differential activation of STAT1/3 via regulation of distinct proximal kinases. In support of this notion, others have shown that IKKε phosphorylates STAT1 Ser708 to regulate antiviral immunity (43, 44), and we now present evidence that STAT3 Thr714 is mediated by GSK-3α/β to positively regulate Mcl1 and EGR1 expression.
It is known that STAT3 can dimerize in the absence of Tyr705 phosphorylation (38). Furthermore, recent studies using both atomic force microscopy and X-ray crystallography have directly visualized U-STAT3 binding to GAS elements as an unphosphorylated dimer (37, 45). Our results suggest that STAT3 binds to an upstream GAS element in the EGR1 promoter in the absence of Tyr705 phosphorylation. Interestingly, others have shown that oncostatin M induces STAT3 Tyr705 phosphorylation and STAT3 binding to the EGR1 promoter within 30 min of stimulation (46). However, the same study also showed that IFN-γ strongly induces STAT3 Tyr705 phosphorylation but does not elicit STAT3 binding to the EGR1 promoter, consistent with our results for IL-6 (Fig. 6A and B). Although direct promoter binding of Tyr705-phosphorylated STAT3 was not tested in that study, our results raise the possibility that canonical and noncanonical mechanisms of STAT3 activation converge on common GAS elements to regulate transcription. This is in agreement with structural studies that suggest U-STAT3 and Tyr705-phosphorylated STAT3 bind to the same DNA elements (45, 47).
The identification of doubly phosphorylated STAT3 in human RCC indicates that the GSK-3α/β–STAT3 axis may be active in a subset of human tumors. The multikinase inhibitors sorafenib and sunitinib are approved for use in RCC patients, and it is thought that their major mechanism of action is through the inhibition of angiogenesis (48, 49). Sorafenib and sunitinib effectively prolong progression-free survival (50, 51), but resistance to these drugs invariably limits their efficacy, and mechanisms of resistance are poorly defined (52). It was recently reported that sorafenib treatment increased GSK-3β activity in RCC cells and that combination therapy with sorafenib plus GSK-3α/β inhibitors synergistically inhibited xenograft tumor growth in a mouse model of RCC (35). Additionally, aberrant nuclear accumulation of GSK-3β is reported to occur in >90% of RCC cases (34), and an immunohistochemistry analysis of renal tumors showed that tyrosine-phosphorylated nuclear STAT3 is present in approximately 60% of ccRCC and pRCC cases (53). Importantly, a STAT3 inhibitor reduced tumor-associated angiogenesis in a mouse model of RCC, and sunitinib-induced RCC cell apoptosis is thought to be mediated in part via inactivation of STAT3 (32, 33).
The previous studies of STAT3 in RCC emphasized the role of pTyr705 as an indicator of STAT3 activation in renal tumors. Our results suggest that Tyr705 phosphorylation is not significantly elevated in ccRCC, and Thr714 and Ser727 phosphorylation may be reliable predictors of GSK-3α/β–STAT3 signaling in this disease. Similar results for STAT3 have been reported in peripheral blood cells of patients with chronic lymphocytic leukemia (CLL). Hazan-Halevy et al. reported that STAT3 is constitutively phosphorylated at Ser727 but not Tyr705 in the disease (42). Furthermore, Ser727-phosphorylated STAT3 that was isolated from peripheral blood cells of CLL patients bound DNA in the absence of Tyr705 phosphorylation (42). STAT3 also facilitates Ras-dependent malignant transformation in a Tyr705-independent manner by localizing to mitochondria and regulating the activity of the electron transport chain (54). Yet another study used a phosphomimetic STAT3 mutant in which Ser727 was replaced with glutamate (S727E) to show that Ser727 phosphorylation increases prostate cancer cell tumorigenicity and invasive capacity (41). Importantly, a STAT3 double mutant (Y705F/S727E) behaved similarly to the S727E condition, suggesting that the tumorigenic effects of STAT3 Ser727 phosphorylation are Tyr705 independent in a prostate cancer cell line (41). Our own results demonstrate that singly phosphorylated Ser727 STAT3 is more abundant than doubly phosphorylated Thr714/Ser727 STAT3 in EC and ccRCC. One possible explanation for this difference is that Ser727-phosphorylated STAT3 exists in higher abundance due to its dual role as a transcriptional regulator and modulator of oxidative respiration.
Our results describe a novel mechanism of signal integration in which combinatorial EGFR/PAR-1 signaling regulates a GSK-3α/β–STAT3 signaling axis. Direct multisite phosphorylation of STAT3 by GSK-3α/β, therefore, serves as the molecular conduit through which temporal information of coincident receptor activation is transduced to regulate gene expression in a strict combinatorial manner. This is the first report that Thr714 phosphorylation regulates STAT3 function, and the data here reveal a new opportunity for the therapeutic inhibition of STAT3 transcriptional activity downstream of an oncogenic receptor tyrosine kinase.
ACKNOWLEDGMENTS
We thank Chad Braley and Lisa Dechert for cell culture assistance. We thank Earl Poptic and the hybridoma core of the Lerner Research Institute for assistance with polyclonal antibody production.
This study was supported by grant HL29582 from the National Institutes of Health (P. E. DiCorleto). Umbilical vein endothelial cells were harvested from human umbilical cords collected from the Birthing Services Department at the Cleveland Clinic (Hillcrest Hospital) and the Perinatal Clinical Research Center (PCRC) at the Cleveland MetroHealth Hospital. The PCRC has been supported by a grant (UL1TR000439) awarded to the Clinical and Translational Science Collaborative of Cleveland by the National Center for Advancing Translational Sciences component of the NIH and NIH Roadmap for Medical Research. The Orbitrap Elite instrument was purchased via an NIH shared instrument grant (1S10RR031537-01).
We have no conflicts of interest.
Footnotes
Published ahead of print 10 March 2014
REFERENCES
- 1.Stark GR, Darnell JE. 2012. The JAK-STAT pathway at twenty. Immunity 36:503–514. 10.1016/j.immuni.2012.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu H, Pardoll D, Jove R. 2009. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer. 9:798–809. 10.1038/nrc2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levy DE, Inghirami G. 2006. STAT3: a multifaceted oncogene. Proc. Natl. Acad. Sci. U. S. A. 103:10151–10152. 10.1073/pnas.0604042103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aznar S, Valerón PF, del Rincon SV, Pérez LF, Perona R, Lacal JC. 2001. Simultaneous tyrosine and serine phosphorylation of STAT3 transcription factor is involved in Rho A GTPase oncogenic transformation. Mol. Biol. Cell 12:3282–3294. 10.1091/mbc.12.10.3282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim LC, Song L, Haura EB. 2009. Src kinases as therapeutic targets for cancer. Nat. Rev. Clin. Oncol. 6:587–595. 10.1038/nrclinonc.2009.129 [DOI] [PubMed] [Google Scholar]
- 6.Sen M, Joyce S, Panahandeh M, Li C, Thomas SM, Maxwell J, Wang L, Gooding WE, Johnson DE, Grandis JR. 2012. Targeting Stat3 abrogates EGFR inhibitor resistance in cancer. Clin. Cancer Res. 18:4986–4996. 10.1158/1078-0432.CCR-12-0792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sen M, Thomas SM, Kim S, Yeh JI, Ferris RL, Johnson JT, Duvvuri U, Lee J, Sahu N, Joyce S, Freilino ML, Shi H, Li C, Ly D, Rapireddy S, Etter JP, Li P-K, Wang L, Chiosea S, Seethala RR, Gooding WE, Chen X, Kaminski N, Pandit K, Johnson DE, Grandis JR. 2012. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: implications for cancer therapy. Cancer Discov. 2:694–705. 10.1158/2159-8290.CD-12-0191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aggarwal BB, Kunnumakkara AB, Harikumar KB, Gupta SR, Tharakan ST, Koca C, Dey S, Sung B. 2009. Signal transducer and activator of transcription-3, inflammation, and cancer: how intimate is the relationship? Ann. N. Y. Acad. Sci. 1171:59–76. 10.1111/j.1749-6632.2009.04911.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iwasaki H, Kovacic JC, Olive M, Beers JK, Yoshimoto T, Crook MF, Tonelli LH, Nabel EG. 2010. Disruption of protein arginine N-methyltransferase 2 regulates leptin signaling and produces leanness in vivo through loss of STAT3 methylation. Circ. Res. 107:992–1001. 10.1161/CIRCRESAHA.110.225326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan Z-L, Guan Y-J, Chatterjee D, Chin YE. 2005. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 307:269–273. 10.1126/science.1105166 [DOI] [PubMed] [Google Scholar]
- 11.Sestito R, Madonna S, Scarponi C, Cianfarani F, Failla CM, Cavani A, Girolomoni G, Albanesi C. 2011. STAT3-dependent effects of IL-22 in human keratinocytes are counterregulated by sirtuin 1 through a direct inhibition of STAT3 acetylation. FASEB J. 25:916–927. 10.1096/fj.10-172288 [DOI] [PubMed] [Google Scholar]
- 12.Nie Y, Erion DM, Yuan Z, Dietrich M, Shulman GI, Horvath TL, Gao Q. 2009. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat. Cell Biol. 11:492–500. 10.1038/ncb1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong Z, Wen Z, Darnell JE. 1994. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 264:95–98. 10.1126/science.8140422 [DOI] [PubMed] [Google Scholar]
- 14.Wen Z, Darnell JE. 1997. Mapping of Stat3 serine phosphorylation to a single residue (727) and evidence that serine phosphorylation has no influence on DNA binding of Stat1 and Stat3. Nucleic Acids Res. 25:2062–2067. 10.1093/nar/25.11.2062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wen Z, Zhong Z, Darnell JE. 1995. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 82:241–250. 10.1016/0092-8674(95)90311-9 [DOI] [PubMed] [Google Scholar]
- 16.Yang J, Huang J, Dasgupta M, Sears N, Miyagi M, Wang B, Chance MR, Chen X, Du Y, Wang Y, An L, Wang Q, Lu T, Zhang X, Wang Z, Stark GR. 2010. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. U. S. A. 107:21499–21504. 10.1073/pnas.1016147107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song L, Turkson J, Karras JG, Jove R, Haura EB. 2003. Activation of Stat3 by receptor tyrosine kinases and cytokines regulates survival in human non-small cell carcinoma cells. Oncogene 22:4150–4165. 10.1038/sj.onc.1206479 [DOI] [PubMed] [Google Scholar]
- 18.Guschin D, Rogers N, Briscoe J, Witthuhn B, Watling D, Horn F, Pellegrini S, Yasukawa K, Heinrich P, Stark GR. 1995. A major role for the protein tyrosine kinase JAK1 in the JAK/STAT signal transduction pathway in response to interleukin-6. EMBO J. 14:1421–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schuringa JJ, Schepers H, Vellenga E, Kruijer W. 2001. Ser727-dependent transcriptional activation by association of p300 with STAT3 upon IL-6 stimulation. FEBS Lett. 495:71–76. 10.1016/S0014-5793(01)02354-7 [DOI] [PubMed] [Google Scholar]
- 20.Shi X, Zhang H, Paddon H, Lee G, Cao X, Pelech S. 2006. Phosphorylation of STAT3 serine-727 by cyclin-dependent kinase 1 is critical for nocodazole-induced mitotic arrest. Biochemistry 45:5857–5867. 10.1021/bi052490j [DOI] [PubMed] [Google Scholar]
- 21.Chung J, Uchida E, Grammer TC, Blenis J. 1997. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol. Cell. Biol. 17:6508–6516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aziz MH, Manoharan HT, Church DR, Dreckschmidt NE, Zhong W, Oberley TD, Wilding G, Verma AK. 2007. Protein kinase C epsilon interacts with signal transducers and activators of transcription 3 (Stat3), phosphorylates Stat3 Ser727, and regulates its constitutive activation in prostate cancer. Cancer Res. 67:8828–8838. 10.1158/0008-5472.CAN-07-1604 [DOI] [PubMed] [Google Scholar]
- 23.Lee J-L, Wang M-J, Chen J-Y. 2009. Acetylation and activation of STAT3 mediated by nuclear translocation of CD44. J. Cell Biol. 185:949–957. 10.1083/jcb.200812060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prabakaran S, Lippens G, Steen H, Gunawardena J. 2012. Post-translational modification: nature's escape from genetic imprisonment and the basis for dynamic information encoding. Wiley Interdiscip. Rev. Syst. Biol. Med. 4:565–583. 10.1002/wsbm.1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waitkus MS, Chandrasekharan UM, Willard B, Haque SJ, DiCorleto PE. 2013. STAT3-mediated coincidence detection regulates noncanonical immediate early gene induction. J. Biol. Chem. 288:11988–12003. 10.1074/jbc.M112.428516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sutherland C. 2011. What are the bona fide GSK3 substrates? Int. J. Alzheimers Dis. 2011:505607. 10.4061/2011/505607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dephoure N, Zhou C, Villén J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. 2008. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U. S. A. 105:10762–10767. 10.1073/pnas.0805139105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daub H, Olsen JV, Bairlein M, Gnad F, Oppermann FS, Körner R, Greff Z, Kéri G, Stemmann O, Mann M. 2008. Kinase-selective enrichment enables quantitative phosphoproteomics of the kinome across the cell cycle. Mol. Cell 31:438–448. 10.1016/j.molcel.2008.07.007 [DOI] [PubMed] [Google Scholar]
- 29.Hoffert JD, Pisitkun T, Wang G, Shen R-F, Knepper MA. 2006. Quantitative phosphoproteomics of vasopressin-sensitive renal cells: regulation of aquaporin-2 phosphorylation at two sites. Proc. Natl. Acad. Sci. U. S. A. 103:7159–7164. 10.1073/pnas.0600895103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DiCorleto PE, de la Motte CA. 1989. Thrombin causes increased monocytic-cell adhesion to endothelial cells through a protein kinase C-dependent pathway. Biochem. J. 264:71–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu H, Ma Y, Cole SM, Zander C, Chen K-H, Karras J, Pope RM. 2003. Serine phosphorylation of STAT3 is essential for Mcl-1 expression and macrophage survival. Blood 102:344–352. 10.1182/blood-2002-11-3396 [DOI] [PubMed] [Google Scholar]
- 32.Horiguchi A, Asano T, Kuroda K, Sato A, Asakuma J, Ito K, Hayakawa M, Sumitomo M. 2010. STAT3 inhibitor WP1066 as a novel therapeutic agent for renal cell carcinoma. Br. J. Cancer 102:1592–1599. 10.1038/sj.bjc.6605691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. 2009. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 69:2506–2513. 10.1158/0008-5472.CAN-08-4323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bilim V, Ougolkov A, Yuuki K, Naito S, Kawazoe H, Muto A, Oya M, Billadeau D, Motoyama T, Tomita Y. 2009. Glycogen synthase kinase-3: a new therapeutic target in renal cell carcinoma. Br. J. Cancer 101:2005–2014. 10.1038/sj.bjc.6605437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawazoe H, Bilim VN, Ugolkov AV, Yuuki K, Naito S, Nagaoka A, Kato T, Tomita Y. 2012. GSK-3 inhibition in vitro and in vivo enhances antitumor effect of sorafenib in renal cell carcinoma (RCC). Biochem. Biophys. Res. Commun. 423:490–495. 10.1016/j.bbrc.2012.05.147 [DOI] [PubMed] [Google Scholar]
- 36.Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. 2007. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 21:1396–1408. 10.1101/gad.1553707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Timofeeva OA, Chasovskikh S, Lonskaya I, Tarasova NI, Khavrutskii L, Tarasov SG, Zhang X, Korostyshevskiy VR, Cheema A, Zhang L, Dakshanamurthy S, Brown ML, Dritschilo A. 2012. Mechanisms of unphosphorylated STAT3 transcription factor binding to DNA. J. Biol. Chem. 287:14192–14200. 10.1074/jbc.M111.323899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Braunstein J, Brutsaert S, Olson R, Schindler C. 2003. STATs dimerize in the absence of phosphorylation. J. Biol. Chem. 278:34133–34140. 10.1074/jbc.M304531200 [DOI] [PubMed] [Google Scholar]
- 39.Yang J, Stark GR. 2008. Roles of unphosphorylated STATs in signaling. Cell Res. 18:443–451. 10.1038/cr.2008.41 [DOI] [PubMed] [Google Scholar]
- 40.Yang J, Chatterjee-Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, Stark GR. 2005. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 65:939–947 [PubMed] [Google Scholar]
- 41.Qin HR, Kim H-J, Kim J-Y, Hurt EM, Klarmann GJ, Kawasaki BT, Duhagon Serrat MA, Farrar WL. 2008. Activation of signal transducer and activator of transcription 3 through a phosphomimetic serine 727 promotes prostate tumorigenesis independent of tyrosine 705 phosphorylation. Cancer Res. 68:7736–7741. 10.1158/0008-5472.CAN-08-1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hazan-Halevy I, Harris D, Liu Z, Liu J, Li P, Chen X, Shanker S, Ferrajoli A, Keating MJ, Estrov Z. 2010. STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood 115:2852–2863. 10.1182/blood-2009-10-230060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ng S-L, Friedman BA, Schmid S, Gertz J, Myers RM, Tenoever BR, Maniatis T. 2011. IκB kinase epsilon (IKK(epsilon)) regulates the balance between type I and type II interferon responses. Proc. Natl. Acad. Sci. U. S. A. 108:21170–21175. 10.1073/pnas.1119137109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tenoever BR, Ng S-L, Chua MA, McWhirter SM, García-Sastre A, Maniatis T. 2007. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science 315:1274–1278. 10.1126/science.1136567 [DOI] [PubMed] [Google Scholar]
- 45.Nkansah E, Shah R, Collie GW, Parkinson GN, Palmer J, Rahman KM, Bui TT, Drake AF, Husby J, Neidle S, Zinzalla G, Thurston DE, Wilderspin AF. 2013. Observation of unphosphorylated STAT3 core protein binding to target dsDNA by PEMSA and X-ray crystallography. FEBS Lett. 587:833–839. 10.1016/j.febslet.2013.01.065 [DOI] [PubMed] [Google Scholar]
- 46.Schiavone D, Avalle L, Dewilde S, Poli V. 2011. The immediate early genes Fos and Egr1 become STAT1 transcriptional targets in the absence of STAT3. FEBS Lett. 585:2455–2460. 10.1016/j.febslet.2011.06.020 [DOI] [PubMed] [Google Scholar]
- 47.Becker S, Groner B, Mu CW. 1998. Three-dimensional structure of the Stat3 NL homodimer bound to DNA 394:145–151 [DOI] [PubMed] [Google Scholar]
- 48.Tamaskar I, Garcia JA, Elson P, Wood L, Mekhail T, Dreicer R, Rini BI, Bukowski RM. 2008. Antitumor effects of sunitinib or sorafenib in patients with metastatic renal cell carcinoma who received prior antiangiogenic therapy. J. Urol. 179:81–86. 10.1016/j.juro.2007.08.127 [DOI] [PubMed] [Google Scholar]
- 49.Chang YS, Adnane J, Trail PA, Levy J, Henderson A, Xue D, Bortolon E, Ichetovkin M, Chen C, McNabola A, Wilkie D, Carter CA, Taylor IC, Lynch M, Wilhelm S. 2007. Sorafenib (BAY 43-9006) inhibits tumor growth and vascularization and induces tumor apoptosis and hypoxia in RCC xenograft models. Cancer Chemother. Pharmacol. 59:561–574. 10.1007/s00280-006-0393-4 [DOI] [PubMed] [Google Scholar]
- 50.Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, Rolland F, Demkow T, Hutson TE, Gore M, Freeman S, Schwartz B, Shan M, Simantov R, Bukowski RM. 2007. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 356:125–134. 10.1056/NEJMoa060655 [DOI] [PubMed] [Google Scholar]
- 51.Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST, Chen I, Bycott PW, Baum CM, Figlin RA. 2007. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 356:115–124. 10.1056/NEJMoa065044 [DOI] [PubMed] [Google Scholar]
- 52.Najjar YG, Rini BI. 2012. Novel agents in renal carcinoma: a reality check. Ther. Adv. Med. Oncol. 4:183–194. 10.1177/1758834012443725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo C, Yang G, Khun K, Kong X, Levy D, Lee P, Melamed J. 2009. Activation of Stat3 in renal tumors. Am. J. Transl. Res. 1:283–290 [PMC free article] [PubMed] [Google Scholar]
- 54.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. 2009. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 324:1713–1716. 10.1126/science.1171721 [DOI] [PMC free article] [PubMed] [Google Scholar]