

Abstract
cGMP and cAMP-dependent protein kinases (PKG and PKA) are closely related homologs, and the cyclic nucleotide specificity of each kinase is crucial for keeping the two signaling pathways segregated, but the molecular mechanism of cyclic nucleotide selectivity is unknown. Here we report that the PKG Iβ C-terminal cyclic nucleotide binding domain (CNB-B) is highly selective for cGMP binding, and have solved crystal structures of CNB-B with and without bound cGMP. These structures, combined with a comprehensive mutagenic analysis, allowed us to identify Leu296 and Arg297 as key residues which mediate cGMP selectivity. In addition, by comparing the cGMP bound and unbound structures, we observed large conformational changes in the C-terminal helices in response to cGMP binding, which were stabilized by recruitment of Tyr351 as a “capping residue” for cGMP. The observed rearrangements of the C-terminal helices provide a mechanical insight into release of the catalytic domain and kinase activation.
PKG and PKA are homologous kinases in the protein kinase A, G and C (AGC) family that mediate pathway-specific cellular responses through the phosphorylation of unique substrates and down-stream effectors, often regulating opposing physiological responses – for example, in cardiac tissue, cAMP has been shown to cause positive inotropy, while cGMP has been shown to cause negative inotropy (Beavo and Brunton, 2002; Francis and Corbin, 1999; Pearce et al., 2010; Rehmann et al., 2007; Schlossmann and Hofmann, 2005). While PKG and PKA signaling specificity is mediated in part through subcellular compartmentalization and protein-protein interactions (Francis et al., 2010), specific binding of each cyclic nucleotide is crucial for keeping the two signaling pathways segregated at the molecular level. At this time, the molecular mechanism of cyclic nucleotide selectivity is poorly understood.
PKG is a central down-stream mediator of the nitric oxide (NO)-cGMP signaling pathway and regulates key physiological processes such as vasodilation, inhibition of platelet aggregation, nociception, and smooth muscle tone (Francis et al., 2010). Genetic ablation of PKG in mice results in phenotypes that reflect the necessity of the kinase in vivo. For example, PKG I knockout mice have a hypertensive phenotype, dysfunctional smooth muscle contraction, and die at an early age due to loss of intestinal peristalsis (Pfeifer et al., 1998). Furthermore, a recent study identified a mutation in human PKG I that causes Thoracic Aortic Aneurysms and Dissections (TAAD), demonstrating the importance of proper cGMP/PKG I signaling to normal aortic function (Guo et al., 2013). These observations, combined with the fact that many existing therapeutics act indirectly on PKG by elevating cellular cGMP levels, has generated interest in targeting PKG directly with PKG-specific activators (Schlossmann and Hofmann, 2005). However, this approach is hampered by the lack of structural information on PKG and an incomplete mechanistic understanding of how PKG is specifically activated by cGMP.
PKG I can be divided into N-terminal regulatory (R)- and C-terminal catalytic (C)-domains. PKG I has two splice variants, PKG Iα and PKG Iβ, which differ in their first ~100 amino acids, giving each isoform unique dimerization and autoinhibitory domains, conferring differences in cyclic nucleotide binding (CNB) affinity, and modulating the ability of cGMP to induce kinase activation (Ruth et al., 1991). These are followed by the two cyclic nucleotide binding (CNB) domains (CNB-A and CNB-B) that bind cGMP with different affinities and which make up the rest of the regulatory domain (Francis and Corbin, 1999) (Figure 1A). The CNB domain is a regulatory module which is highly conserved in diverse proteins such as PKA, the catabolite activator protein (CAP), cyclic nucleotide-gated (CNG) and hyperpolarization-activated cyclic nucleotide-gated (HCN) channels, and exchange proteins activated by cAMP (EPAC). The domain consists of approximately 120 residues forming an eight-stranded β-barrel with a variable number of α-helices (Berman et al., 2005; Rehmann et al., 2007). The CNB site is located within the β-barrel with the cyclic phosphate making multiple contacts with residues at the base of the barrel in a structure referred to as the Phosphate Binding Cassette (PBC), which consists of β-strand 6, a short helix followed by a loop, and β-strand 7. In the absence of cGMP, the R-domain binds and inhibits the C-domain. Binding of cGMP to both sites in the R-domain is required for full kinase activation, and cGMP-induced conformational changes in the R-domain release and activate the C-domain (Alverdi et al., 2008; Rehmann et al., 2007; Smith et al., 2000; Wall et al., 2003). However, the underlying molecular mechanism that allows cGMP to drive this conformational change is unknown. Here we report for the first time high resolution structures of the C-terminal cyclic nucleotide binding domain (CNB-B) of PKG I with and without cGMP. These structures reveal the mechanism for cGMP-selectivity in PKG I and allow us to propose a mechanism for cGMP-selective activation.
Figure 1. Domain organization and overall structure of the PKG Iβ (219-369):cGMP complex.

(A) Domain organization of PKG Iβ with CNB-B highlighted. AI: autoinhibitor sequence. (B) Affinity measurements of PKG Iβ (219-369) for cGMP and cAMP The construct has a 240-fold selectivity for cGMP. Competition FP curves are shown in red and competition SPR curves are shown in black. Error bars denote standard error of measurement. (C) Overall structure of the PKG Iβ 219-369:cGMP complex with the secondary structure elements labeled. The phosphate binding cassette (PBC) is colored yellow, the αB and αC helices red, the N-terminal helices teal, and cGMP is colored by atom type. The N- and C-termini are labeled with their corresponding residue number seen in the final model. The disordered β4-β5 loop in the PKG Iβ (219-369):cGMP complex is shown as a dotted line. All structure images were generated using PyMOL (Delano Scientific).
Results
PKG I CNB-B is responsible for cGMP selectivity
We previously found that PKG I CNB-A bound both cGMP and cAMP with relatively high affinity (12 ± 1.6 vs. 27 ± 3.6 nM, respectively). While crystal structures of the N-terminal CNB (CNB-A) of PKG Iβ and tandem CNBs (CNB-A/B) of PKG Iα are available (Kim et al., 2011; Osborne et al 2011), only the former was solved with bound cGMP, revealing specific contacts between residues in the CNB-A β5 strand and cGMP Despite these unique contacts, the fact that CNB-A of PKG Iβ shows poor cyclic nucleotide selectivity indicates that other domains are required to mediate cGMP specificity (Kim et al., 2011).
Therefore, we decided to test whether the PKG I CNB-B domain (PKG Iβ residues 219-369) bound cGMP selectively. Using competition fluorescence polarization (FP), we found that the PKG I CNB-B domain binds cGMP with an EC50 of 215nM whereas it binds cAMP with an EC50 of 52μM (Figure 1B and Table S1). Thus, affinity for cGMP over cAMP is approximately 240-fold higher in the CNB-B domain, and from this result we conclude that CNB-B represents the minimal domain required for cGMP selectivity in PKG I.
Strand β5 and the αC-helix provide novel interactions for cGMP
To understand the structural basis for cGMP selectivity seen in CNB-B, we solved a crystal structure of CNB-B bound to cGMP at 1.65Å resolution using iodide single wavelength anomalous dispersion (SAD) for phase determination (Figure 1C and Table 1). The final model includes residues 223-351 with the large portion of the C-terminus disordered (residues 352-369). The structure of the CNB-B:cGMP complex clearly shows electron density for cGMP in the syn configuration at the PBC (Figure S1). The interactions between CNB-B and the ribose and the cyclic phosphate of cGMP are virtually identical to those seen in the CNB-A:cGMP complex (Figure S2) (Kim et al., 2011). In addition, Thr317 forms hydrogen bonds with guanine that mirror those seen between cGMP and Thr193. However, other interactions with the guanine moiety are distinct in CNB-B. The CNB-B:cGMP complex reveals that Leu296 and Arg297 on the β5 strand provide a unique docking site for cGMP (Figure 2A). Leu296 interacts with the guanine through Van der Waals (VDW) interactions whereas Arg297 interacts through two hydrogen bonds. The unique side chain orientation of Arg297 aligns its guanidinium group with the guanine ring, placing its amine and protonated Nε within hydrogen bonding distance of the C6 carbonyl and unprotonated N7 nitrogen of cGMP, respectively (Figure S3). Notably, although they are consecutive residues, the unusual backbone geometry at this regions enables both side chains of Leu296 and Arg297 to point towards the binding pocket and interact with cGMP.
Table 1.
Data and refinement statistics.
| Apo | cGMP bound High-resolution | cGMP-Bound (Iodine SAD) | |
|---|---|---|---|
| Data collection | |||
| Wavelength (Å) | 0.97931 | 0.97931 | 1.54178 |
| Space group | P3121 | P41212 | P41212 |
| Cell dimensions | |||
| a,b,c (Å) | 78.8,78.8,147.6 | 47.2, 47.2,101.9 | 47.9,47.9,104.1 |
| α,β,γ (°) | 90,90,120 | 90,90,90 | 90,90,90 |
| Resolution (Å) | 36.9-2.00 | 23.8-1.65 | 15.0-1.80 |
| Rsym or Rmerge | 11.4(45.1)* | 7.7(47.1) | 6.0(21.4) |
| I/σI | 10.2(3.4) | 20.1(5.5) | 32.2/(2.1) |
| Completeness (%) | 99.4(96.4) | 99.6(100) | 91.2(35.7) |
| Redundancy | 4.8(4.4) | 13.5(13.8) | 7.2(5.9) |
| Refinement | |||
| Resolution (Å) | 36.9 – 2.00 | 23.8 – 1.65 | 15.0 – 1.80 |
| No. reflections | 36516 | 14492 | 19090 |
| Rwork/Rfree† | 18.82/22.69 | 19.74/24.47 | 18.9/24.4 |
| No. atoms | |||
| Proteins | 2934 | 985 | 985 |
| Ligand/ion | 14 | 46 | 45 |
| Water | 343 | 68 | 56 |
| B-factors | |||
| Protein | 46.424 | 28.464 | 37.069 |
| Ligand/ion | 83.734 | 32.253 | 49.169 |
| Water | 45.140 | 38.431 | 37.139 |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.006 | 0.006 | 0.007 |
| Bond angles (°) | 0.980 | 1.093 | 1.149 |
Highest resolution shell is shown in parenthesis.
5.0% of the observed intensities was excluded from refinement for cross validation purposes.
Figure 2. cGMP binding pocket of CNB-B and its comparison with the PKA:cAMP complex.
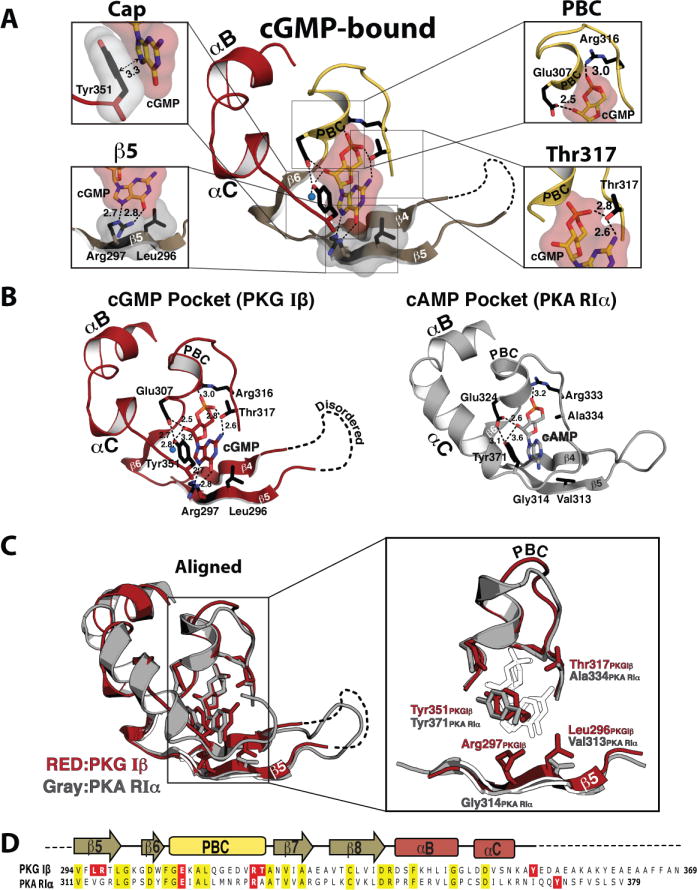
(A) Detailed interactions between cGMP and CNB-B. Zoomed in views for each cGMP binding site are shown on either side. The individual cGMP interacting residues are shown, with the following color theme: side chain carbon, black; oxygen, red; nitrogen, blue. The cyclic nucleotide, β5 and capping residues are shown with transparent surface. Contacts are shown as dotted lines with their distances (Å) (B) Comparison between the PKG Iβ cGMP binding pocket and PKA RIα cAMP pocket. The cGMP pocket of PKG Iβ in red is shown on the left panel and the cAMP binding pocket of PKA RIα (PDB code: 1RGS) in gray is shown on the right. Key cyclic nucleotide binding residues are shown as sticks. An ordered water molecule is shown as blue sphere. (C) Superposition of two structures shown in B are shown on the left, with a zoomed in view on the right (cyclic nucleotides are showm as white sticks). (D) Sequence Alignments of CNB-B from PKG Iβ and PKA RIα. Conserved residues are shaded in yellow and cyclic nucleotide interacting residues through their side chains are shaded in red for both proteins.
In our previous crystal structure of the CNB-A:cGMP complex, the side chain of Leu172, which is in an analogous position to Leu296, interacts with cGMP in a similar manner (Kim et al., 2011). However, Cys173, corresponding to Arg297 in CNB-B, does not form hydrogen bonds and only shows VDW interaction with the guanine moiety (Figure S2). The absence of these contacts may at least partially explain why CNB-A is not selective for cGMP. Moreover, in the β5 strand of PKA RIα, Val313 and Gly314 reside in analogous positions to Leu296 and Arg297 of PKG Iβ and do not interact with cAMP (Figures 2B and 2C). Similarly in the β5 strand of PKA RIIβ, Ile339 and Ala440 reside in analogous positions to Leu296 and Arg297 of PKGIβ and form only van der Waals interactions with cAMP (Diller et al., 2001). These structural differences provide evidence that support the role of Arg297 in mediating cGMP selectivity.
In addition to the novel interactions at the β5-strand, the CNB-B:cGMP complex structure shows that Tyr351 in the C-helix interacts with cGMP through a π stacking interaction (Figure 2A). This finding is consistent with our previous hydrogen/deuterium (H/D) exchange data which showed cGMP induced slowing of H/D exchange around Tyr351 (Lee et al., 2011). The phenol group of Tyr351 interacts with one side of the guanine moiety, sandwiching it against Leu296 (Figure 2A). Unlike the continuous helix seen in PKA RIα, the αC helix shows only one helical turn followed by a short loop (Figures 2B and 2C) (Su et al., 1995). Despite low sequence and structural similarity at this region, superimposing PKA and PKG structures shows that Tyr351 of PKG Iβ overlaps with Tyr371 of RIα, and that these tyrosines act as “capping residues” for the cyclic nucleotide binding pockets (Figures 2C and 2D).
Mutagenesis of contact residues results in reduced cGMP selectivity and activation
To test the contribution of Leu296, Arg297, Thr317 and Tyr351 to cGMP selectivity, we mutated them one at a time to alanine and measured cGMP and cAMP affinities of the mutants. Mutating any of these residues dramatically reduced cGMP affinity with relatively little change in cAMP affinity (Table S1). Next, we mutated these residues in full-length PKG Iβ (residues 5-686) and measured their effect on kinase activation using a microfluidic mobility-shift assay. Mutating any of these residues except Thr317 increases activation constants (Ka) for cGMP (Table S2). Consistent with these observations, a previous study measuring the contribution of Thr317 to cGMP-dependent activation reported only a minor increase in Ka upon replacing this residue with an alanine (Reed et al., 1996). In contrast, the roles of Leu296, Arg297 and Tyr351 in PKG I activation have not been previously appreciated, and our data show that these residues are important not only for the cGMP binding, but also for kinase activation.
Binding of cGMP induces large conformational changes in the CNB-B helices
To investigate the structural changes upon cGMP binding, we obtained a crystal structure of CNB-B in the absence of cGMP. The apo structure was solved by molecular replacement (MR) at 2.00 Å resolution using the CNB-B:cGMP complex as a MR model. The apo structure contains three molecules per unit cell (Figure S4). Independent of the different packing environments, the overall structures of all three molecules in the unit cell are very similar with rmsd values of less than 1 Å for at least 80 Cα atoms. Only one out of three molecules in the unit cell showed most of the helical subdomains, which were stabilized by crystal contacts (Figure S4C), indicating that the helical domains in the isolated CNB-B are highly dynamic in the absence of cGMP The final model for the ordered molecule contained residues 217-365. Comparing the apo structure to the cGMP-bound structure shows that the β-barrel regions do not vary whereas the helical subdomains undergo major conformational changes (Figure 3 and Figure S5). The structural changes occur in three distinct regions of CNB-B: the PBC, the αB-helix, and the αC-helix. In the absence of cGMP, the PBC is in a open conformation tilting away from the rest of the β barrel. The αB-helix interacts with the αA-helix mainly through hydrophobic interactions formed between amphipathic helices running parallel to each other. Upon cGMP binding, the αB-helix and PBC, coupled through a hydrophobic interaction between Leu310 and Phe336, tilt towards the β barrel in a concerted motion (Figure 3A). The closer proximity of the αB-helix to the binding pocket then allows the flexible αC-helix to close onto the binding pocket, resulting in capping of cGMP by Tyr351.
Figure 3. Structural comparison between the apo- and cGMP bound PKG Iβ (219-369).

(A) Regions mediating cGMP interactions are shown. The disordered β4-β5 loop and disordered portion of the αC helix are indicated with a dotted line. The Cα atoms of the Gly-Gly hinge residues (Gly341 and Gly342) are shown as balls. Leu310, Phe336, Tyr351 and cGMP are shown as sticks with transparent surface. (B) The apo (gray) and PKG Iβ 219-369:cGMP (red) structures aligned at the β barrel region. An enhanced view of the binding pocket highlighting the β5 and capping residue interactions with cGMP (black sticks) is shown on the right. In the apo structure, the side chain of Arg297 is extended in a conformation partially occupying the empty cGMP binding pocket.
Out of all these movements, the most drastic change occurs at the αC-helix. For the molecule in the apo structure with ordered helical subdomains, the αC-helix is more ordered compared to the cGMP-bound structure, showing five additional helical turns (Figure 3A). Tyr351 is on the second turn of the αC-helix, and not at the end of a loop as seen in the cGMP-bound complex. Due to these structural changes, the capping residues are over 20 Å apart between the bound and unbound structures (Figure 3B). Gly341 and Gly342 located between the αB- and αC-helices seem to form a flexible “Gly-Gly hinge” enabling the large conformational flexibility (Figure 3A). This Gly-Gly hinge explains why the overall position of the αB- and αC- helices are dramatically different between the apo and cGMP bound structures.
The Gly-Gly hinge Allows Conformational Flexibility of the αB- and αC-helices
Next, we compared our structures with the crystal structure of PKG Iα CNB-A/B reported by Osborne et al. (Osborne et al., 2011). Excluding dramatic conformational differences at the αC-helix, their structure of PKG Iα CNB-A/B closely resembles our apo structure, which is unsurprising since its CNB-B also lacked cGMP (Figure S6). In their structure, the αC helix (corresponding to residues 347-371 in PKG Iβ) is completely removed from the rest of CNB-B and forms an approximately 90° angle compared to our apo CNB-B. Furthermore, the capping residues are over 18 Å apart between the two structures. Since both PKG I isoforms have the same amino acid sequence at this region, these differences could be explained by the flexible Gly-Gly hinge, which enables multiple conformations of the αC-helix that can be stabilized in different conformations by alternative crystal packing. Specifically, PKG Iα CNB-A/B formed a symmetry related dimer through the hydrophobic contacts (referred to as a “knob-nest” assembly) between the αC-helix of one protomer and CNB-B of the other, and these contacts likely stabilized the open conformation of the αC-helix. However, although the CNB-B domain of PKG Iβ contains the corresponding residues that form these contacts, our apo structure shows no such “knob-nest” assembly, as residues 366-369 corresponding to the knob are disordered (Figure S4).
To investigate the structural changes associated with cGMP binding in solution, we used NMR to obtain secondary structure probabilities of the αB and αC helices. In the presence of cGMP, the secondary structural probabilities agreed well with the crystal structure, indicating that crystal packing minimally influences their conformation (Figure S7). Additionally, residues 352-369, which were disordered in the crystal structure, show a low probability of being an α-helix in solution. In the absence of cGMP, NMR showed very little α-helix preference in these C-terminal residues. This finding corresponds to the fact that two of the molecules in the unit cell of the apo structure showed no electron density for this region, and the one molecule that was structured was stabilized by crystal contacts; again, indicating that this region is more dynamic in the absence of cGMP. Furthermore, dilution of the sample resulted in no significant change in 1H,15N-HSQC spectra, suggesting no change in oligomerization state between these two concentrations (Figure S8A). Finally, we tested whether PKG Iβ 92-369, a construct containing CNB-A and CNB-B that aligns closely with PKG Iα CNB-A/B, migrates as a dimer on a size exclusion column (Figure S8B and C). The elution profile of PKG Iβ 92-369 is consistent with a monomeric 48 kDa species, suggesting that the interchain interface between αC helices described by Osborne et al. is a crystal contact unlikely to occur in solution. Taken together, these results demonstrate that αB and αC helices of CNB-B are more flexible without cGMP in solution and support the observation derived from our crystal structures that the αB and αC helices close onto the binding pocket upon cGMP binding.
Discussion
The biochemical and structural data presented here clearly demonstrate the high cGMP selectivity of CNB-B in PKG I and reveal specific amino acid contacts that explain its high selectivity. In light of the finding that CNB-A is not selective for cGMP (Kim et al., 2011), our measurements demonstrating 240-fold selectivity for cGMP in CNB-B, combined with our data demonstrating that mutations that disrupt CNB-B's interaction with cGMP significantly decrease the ability of cGMP to activate full-length PKG I, suggest that CNB-B is not only a minimal construct selective for cGMP but is also responsible for conferring cGMP selectivity to full-length PKG I. While its overall fold and interaction with cGMP are similar to CNB-A, the structures of CNB-B reveal unique Leu296 and Arg297 residues on the β5 strand that explain the high selectivity for cGMP. Specifically, while Cys173 at the analogous position on the β5 strand of CNB-A makes a weak VDW interaction with cGMP, the side chain of Arg297 forms two strong hydrogen bonds with the guanine moiety. Additionally, because mutating Arg297 to alanine partially improves affinity for cAMP, we hypothesize that Arg297 on the β5 strand acts as a filter for cAMP by weakening CNB-B's affinity for cAMP. Consistent with this hypothesis, a conserved glycine is located at the analogous position in the PKA RI subunit. Taken together, these results demonstrate that the high selectivity of full-length PKG I is achieved in part by the placement of Arg297 in CNB-B, which forms specific interactions with cGMP while reducing affinity for cAMP.
More generally, our results allow us to propose the role of CNB-B in mediating cGMP-specific signaling. Studies on intracellular levels of cGMP and cAMP in intact cells reported that, while they are in constant flux, the highest concentrations range between 2-5μM for cGMP or cAMP (Ponsioen et al., 2004; Trivedi and Kramer, 1998). Previous reports in the literature have observed that, at these concentrations, cross-talk between the cGMP and cAMP pathways can occur through binding of cGMP to PKA or binding of cAMP to PKG, resulting in at least partial activation of these kinases and affecting physiological outcome (Cornwell et al., 1994; Jiang et al., 1992; Komalavilas and Lincoln, 1996). However, since these levels of cyclic nucleotide are far below CNB-B's EC50 for cAMP (52μM), but well above its EC50 for cGMP (215nM), full activation of PKG I likely occurs only in response to cGMP. Thus, CNB-B plays a “checkpoint” role by limiting cAMP mediated activation of PKG I. Consistent with this hypothesis, biochemical evidence and Small-Angle X-ray Scattering (SAXS) data suggested that binding of cGMP to CNB-B is required for the full activation of PKG I (Francis and Corbin, 1999; Smith et al., 2000; Wall et al., 2003). Although cross-talk between the cGMP and cAMP pathways can affect physiological outcome, the observation that cGMP and cAMP can mediate drastically different physiological responses in specific environments, such as negative and positive inotropy in cardiac tissue, suggest a need for CNB-B in PKG to filter out cAMP and limit cross-talk between these two signaling pathways, at least in certain tissues (Huang et al., 1999; Pellegrino et al., 2009). Thus, our data provides key insights for developing PKG-selective activators in the rational design of anti-hypertensive therapeutics.
Additionally, our structures show for the first time that Tyr351 provides a capping interaction for cGMP in PKG I. This aromatic residue at the αC helix undergoes a π stacking interaction with the bound cyclic nucleotide and a similar interaction is found in PKA CNB domains (Su et al., 1995; Wu et al., 2004). Specifically, comparison of our structure of PKG I CNB-B with the structures of PKA RIα CNB-B domain shows that Tyr351 of PKG Iβ structurally aligns with Tyr371 of RIα. Because it forms a hydrophobic interaction with cyclic nucleotides seen also in PKA subunits, we hypothesized that Tyr351 does not contribute to cGMP selectivity but instead stabilizes the activated conformation through the capping interaction. Indeed, comparison of the cGMP-bound and apo structures of CNB-B revealed dramatic conformational changes in the αC-helix, enabled by the flexibility of the Gly-Gly hinge and by the capping interaction (Figure 4). Furthermore, mutating Tyr351 to alanine reduces binding affinity and increases Ka values for both cGMP and cAMP, confirming the role of Tyr351 in both cyclic nucleotide binding and kinase activation. Thus, we hypothesize that this capping interaction provides an allosteric mechanism of activation by drastically changing the conformation of the αC-helix upon cGMP binding.
Figure 4. Role of CNB-B in activation and stepwise model for cGMP binding.

(A) Role of cGMP contact residues in kinase activation. Ka values were measured using a microfluidic mobility shift assay, and error bars denote standard error of measurement. (B) Stepwise model for cGMP binding and kinase activation. (1) cGMP binds to the pocket, interacting with the PBC and the β5 strand. (2) This interaction causes the PBC to tilt towards the core of CNB-B, causing the αB and αC helices to compact against the binding pocket. (3) The capping of cGMP by Tyr351. (4) The capping interaction stabilizes the activated conformation of CNB-B, dislodging the C-domain and activating the enzyme.
Our findings describing the dynamic nature of the αC-helix, and its rearrangement upon cyclic nucleotide binding, are consistent with studies on other CNB-containing proteins. As previously mentioned, structures of the R-subunit of PKG's closest homologue, PKA, have demonstrated that conformational changes occur in the αC-helix upon cAMP binding, and these changes provide an allosteric mechanism of activation (Su et al., 1995; Wu et al., 2004). Furthermore, recent FRET studies on more distantly related proteins that contain CNB domains, such as the HCN and CNG channels, have shown that cyclic nucleotide binding causes conformational rearrangement of the αC-helices in their CNB domains, favoring the opening of the channels (Puljung and Zagotta, 2013; Taraska et al., 2009). Another atypical example comes from recent structures of CNB homology domains in the ether-a-go-go (EAG) channel, which does not bind cyclic nucleotides but instead responds to changes in voltage when a short β-strand at the end of the αC-helix occupies the binding pocket and acts as an “intrinsic ligand”(Brelidze et al., 2012). Mutating residues on the αC-helix that occupy the binding pocket affects the ability of EAG to respond to change in electrical potential, suggesting that the positioning of the αC-helix regulates EAG's activity, even in the absence of cyclic nucleotide (Marques-Carvalho et al., 2012). Taken together, these examples suggest that while the αC-helix in CNB domains has evolved in diverse ways, repositioning of the αC-helix is a general mechanism used by CNB domains to regulate protein activity.
Consistent with these observations, the data presented here suggest a role for the αC-helix in CNB-B for PKG I activation (Figure 4). In the absence of cGMP, the R-domain latches onto the C-domain and keeps it inactive. Because the αC-helix interconnects the two domains, we hypothesize that the R-C interaction immobilizes the αC-helix leading to an ordered helical state similar to what we observed in the apo structure. Upon cGMP binding, the guanine moiety recruits the capping residue, Tyr351, rearranging the αB and αC-helices and causing disordering of the αC-helix as seen in our CNB-B:cGMP complex structure. SAXS experiments on PKG Iβ (53-686) showed that cGMP binding to CNB-B is required for both elongation and activation (Wall et al., 2003). Thus, the conformational change in the αB and αC-helices upon cGMP binding may contribute to release of the C-domain in response to cGMP binding, resulting in kinase activation. However, the exact activation mechanism awaits more detailed structural studies on the global architecture of PKG I.
Experimental Procedures
Protein Expression and Purification
Sequences encoding wild-type and mutant PKG Iβ 219-369 were cloned into pQTEV from a template containing full-length PKG Iβ (Genbank: ABQ59040.1) (Bussow et al., 2005). The proteins were expressed by transforming the construct into BL21(DE3) E. coli, growing the cells at 37°C to an OD600nm of 0.6, and inducing the cells with 0.5 mM IPTG (Isopropyl β-D-1-thiogalactopyranoside). The cells were grown for an additional 18 hours at 20°C. Cells were resuspended in 50 mM potassium phosphate pH 7.0, 150 mM NaCl, 1 mM β-mercaptoethanol and lysed using a cell disruptor (Constant Systems). His-tagged PKG Iβ 219-369 was purified with BioRad IMAC resin on a BioRad Profinia purification system. The protein was eluted with cell suspension buffer containing 250 mM imidazole. To remove the His-tag, the sample was incubated with TEV protease in a 50:1 molar ratio at 4°C overnight and loaded onto BioRad IMAC resin. The tagless samples were collected from flow-through fractions. A final buffer exchange into 25 mM Tris pH 7.5, 150 mM NaCl, and 1 mM TCEP was performed with a Hiload 16/60 Superdex 75 column (GE Healthcare).
The sequence encoding wild-type PKGIβ 92–369 was similarly cloned into pQTEV but was transformed into the ΔAcya TP2000 mutant strain of E. coli to ensure no contamination by cAMP (Brickman et al., 1973). Purification was performed using the same protocol as for PKG Iβ 219-369.
Crystallization, Data Collection, Phasing, Model Building and Refinement
To obtain crystals of the CNB-B:cGMP complex, the protein sample was pre-incubated with 5 mM equivalent cGMP and concentrated to 50 mg/mL using a 10 kDa cutoff Amicon Ultra (Millipore). Crystals were obtained using the hanging drop method. 2.5 μL of protein solution was mixed with 2 μL of reservoir solution containing 1.6 M tri-sodium citrate at pH 6.5, and 0.5 μL of 1 M sodium iodide. After one week incubation at 22°C, drops produced bipyramidal crystals belonging to a P41212 space group that diffracted to 1.65 Å resolution. Apo crystals were obtained at a much higher concentration of 106 mg/mL at 4°C in a crystallizing solution containing 25% (w/v) PEG 1500 and 0.1 M SPG Buffer pH 5.5. The apo crystal belongs to a P3121 space group and diffracted to 2.0 Å resolution. All protein concentrations were measured using a Bradford assay, and all crystals were cryoprotected with Paratone-N before freezing.
Initial diffraction experiments on the CNB-B:cGMP crystals were performed on a Rigaku FRE-SuperBright with a CuKα anode (1.54 Å wavelength). Data was collected with 0.5° oscillation angles and 30 second exposures, and diffraction was observed at 1.8 Å resolution. The data were processed using HKL-2000 (Minor W et al., 2006). Thirteen initial iodide sites were located using Phenix Autosol (Terwilliger et al., 2009) with a figure of merit equal to 0.53. An initial model was obtained using Autobuild (Langer et al., 2008) and refined using Phenix.Refine (Afonine et al., 2012) against a 1.65 Å data set collected at the Advanced Photon Source (Argonne, IL). The final model was manually built using Coot (Cowtan and Emsley, 2004).
Diffraction experiments on the apo crystals were performed at the Advanced Photon Source. The structure was solved by molecular replacement using AutoMR and a truncated version of the cGMP-bound structure of CNB-B as a search model. The structure was built and refined as previously described.
Fluorescence polarization
The direct fluorescence polarization (FP) assay was performed following the procedure from Moll et al (Moll et al., 2006). Measurements were performed in 150 mM NaCl, 20 mM MOPS plus 0.005% (w/v) CHAPS pH 7.0 using the Fusion ™a-FP microtiter plate reader at room temperature in a 384 well microtiterplate (Perkin Elmer, Optiplate, black). The protein concentration was varied while the concentration of 8-Fluo-cAMP/8-Fluo-cGMP (Biolog Life Science Institute (Bremen, Germany)) was fixed at 1 - 5 nM. The FP signal was detected for 2 seconds at Ex 485 nm and Em 535 nm with a PMT Voltage of 1,100. Data were analyzed with GraphPad Prism 5.03 (GraphPad Software, San Diego, CA) by plotting the polarization signal in mPol against the logarithm of the protein concentration. The KD values were calculated from sigmoidal dose-response curves.
In FP competition experiments the protein concentration and the concentration of 8-Fluo-cGMP were fixed at a polarization signal of 50% of the maximum value obtained from direct FP measurements. The protein was incubated with 8-Fluo-cGMP and varying concentrations of unlabelled cGMP or cAMP (Biolog Life Science Institute (Bremen, Germany)). FP signals were detected as indicated above. Data were analyzed with GraphPad Prism 5.03 by plotting the polarization signal in mPol against the logarithm of the cyclic nucleotide concentration. The EC50 values were calculated from sigmoidal dose-response curves.
Surface Plasmon Resonance (SPR)
Protein/nucleotide interactions were monitored as solution competition experiments at 25°C following the procedure from Moll et al. using a Biacore T100 instrument (Biacore GE Healthcare) (Moll et al., 2006). 8-AHT-cGMP (Biolog Life Science Institute (Bremen, Germany) was covalently coupled to a CM5 chip (research grade, Biacore GE Healthcare) using standard amine coupling (Johnsson et al., 1991). Binding experiments were performed in 20 mM MOPS, 100 mM NaCl pH 7.0, plus 0.005 % (v/v) surfactant P20 at 25°C. Proteins were preincubated with varying concentrations of unlabelled cAMP or cGMP and the mixture subsequently injected over the nucleotide surfaces. Association was monitored for 150 - 300 sec, and the residual binding signal was collected at the end of the association phase and plotted with GraphPad Prism 5.03 against the logarithm of the protein concentration. EC50 values were calculated from sigmoidal dose-response curves.
Microfluidic mobility shift assay
HEK293T cells were grown to 80% confluency in complete medium and transfected with FLAG-tagged wild-type and mutant PKG Iβ constructs using Lipofectamine2000. After transfection, cells were lysed with 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100 with protease inhibitors. Protein samples were purified by loading the lysate onto Agarose-FLAG-M2 beads (Sigma-Aldrich), washing with 1× TBS, and eluting with 1× TBS containing 5 mg/mL 3×FLAG peptide (Sigma-Aldrich).
Kinase activity was determined using a microfluidic mobility-shift assay on a Caliper DeskTop Profiler (Caliper Life Sciences, PerkinElmer). Human PKG Iβ was incubated for 2 hr at 25 °C in a 384 well assay plate (Corning®, low volume, non-binding surface) in 20 μL buffer (20 mM MOPS pH 7.0, 150 mM NaCl, 0.1 mg/mL BSA, 1 mM DTT, 0.05% L-31, 10 μM FITC-Kemptide, 990 μM Kemptide, 1 mM ATP, 10 mM MgCl2) and various concentrations of cGMP or cAMP (3 nM - 5 mM), respectively. Reaction mixtures without cyclic nucleotide were used as controls. For electrophoretic separation of substrate and product a ProfilerPro™ LabChip (4-sipper mode; Caliper Life Sciences, PerkinElmer) was used under the following conditions: downstream voltage -150 V, upstream voltage -1,800 V with a screening pressure of -1.7 psi. Substrate conversion was plotted against the logarithmic cyclic nucleotide concentration and activation constants (Ka) were calculated from sigmoidal dose-response curves employing GraphPad Prism 5.03.
NMR Analysis
The PKG Iβ 219-369 construct was expressed with an N-terminal poly-L-histidine tag, in E. coli strain BL21(DE3). The cells were grown at 37°C in isotopically enriched minimal media supplemented with trace metals, D-biotin and thiamine-HCl. Expression was induced with 0.4 mM IPTG at an OD600nm of 1.0, and the cells were further incubated for 16 hours at 20 °C before being harvested by centrifugation.
The harvested cells were lysed using a homogenizer, and cell debris was subsequently removed by centrifugation (20,000×g for one hour). For this process, the cells were resuspended in 50 mM Tris buffer pH 7.6 with 150 mM NaCl, 1 mM β-mercaptoethanol and 0.2 mM AEBSF. An initial purification of the PKG construct was then performed using Ni2+–Sepharose resin in a 5-mL gravity column. After passing the cell lysate through the column, the column was rinsed with lysis buffer, followed by 50 mM Tris buffer pH 7.6 with 500 mM NaCl, 20 mM imidazole and 1 mM β-mercaptoethanol. The protein was then eluted from the column using 50 mM Tris buffer pH 7.6 with 50 mM NaCl, 300 mM imidazole and 1 mM β-mercaptoethanol.
The collected gravity column eluant was dialyzed in 50 mM Tris, 100 mM NaCl, 1 mM β-mercaptoethanol pH 7.6, and cleaved with TEV protease for 48 hours to remove the N-terminal poly-L-histidine tag from the PKG construct. The tag and TEV protease were removed by passing the sample through a Ni2+–Sepharose gravity column, followed by dialysis buffer. The flow-through and rinse were concentrated and loaded onto HiLoad Superdex 75, equilibrated 50 mM Tris, 100 mM NaCl, 1 mM DTT, 0.2%(w/v) NaN3 pH 7.0. An apo sample for NMR analysis was prepared by concentrating an aliquot of the purified protein solution to ∼300 μM, and adding 5% (v/v) D2O. A cGMP-bound sample was prepared similarly, with the addition of 2 mM cGMP (Sigma-Aldrich) to the protein solution.
Two-dimensional 1H,15N-HSQC and three-dimensional triple-resonance NMR spectra were acquired for apo and cGMP-bound samples of PKG Iβ 219-369. All NMR spectra were acquired at 306 K with a Bruker Avance 700-MHz NMR spectrometer equipped with a 5 mm TCI cryoprobe. The spectra were processed with NMRPipe (Delaglio et al., 1995) and analyzed using Sparky (Goddard and Kneller, 2006). Automated assignment and secondary structure probability calculations were performed using the PINE-NMR server, and assignments were confirmed by manual examination of the spectra with Sparky (Bahrami et al., 2009).
Size Exclusion Chromatography
SEC experiments to measure the molecular weight of PKG Iβ 92-369 were performed on a Superdex 75 10/300 GL column (GE Healthcare) at 4°C using 25mM Tris pH 7.5, 150mM NaCl, and 1mM DTT as running buffer. Elution of PKGIβ 92-369 was compared against the following standards: conalbumin, 75 kDa; ovalbumin, 44kDa; carbonic anhydrase, 29 kDa; ribonuclease A, 13.7 kDa; aprotinin, 6.5 kDa.
Supplementary Material
Acknowledgments
We thank K. Sippel, M. Zhou, C. Peters, C.J. Lim, A. Koyfman and S.S. Taylor for critical reading of the manuscript. We also thank E. Franz (University of Kassel) for expert technical assistance, S.R. Wasserman (Eli Lilly Beamline, APS, Argonne, IL) for his assistance with data collection, and S. Badal and F. Danesh (BCM) for their assistance with HEK293T culture. C.K. is funded by NIH grant R01 GM090161 and a BCM seed grant. G.Y.H. is supported by the Houston Area Molecular Biophysics Program, National Institute of General Medical Science (NIGMS) T32GM008280. G.M. is funded by the Canadian Institute of Health Research (CIHR). F.W.H is supported in part by European Union grant “Affinomics” (Contract 222635) and the Federal Ministry of Education and Research Project “NoPain” (FKZ0316177F). The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health, National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Author Contributions: G.Y.H., J.J.K., and C.K. designed the experiments required for addressing the questions in this study. G.Y.H., J.J.K., C.Z., and E.M. designed the constructs and purification protocols. G.Y.H., J.J.K., and C.Z. identified and optimized crystallization conditions. G.Y.H., A.S.R., J.W.P, and B.S. collected the diffraction data. G.Y.H. and A.S.R. solved the structures and refined the crystallographic models. R.L., D.B., and F.W.H. designed the fluorescence polarization protocol, while R.L. and G.Y.H. generated the KD/EC50 measurements. R.L., D.B., and F.W.H. designed the surface plasmon resonance protocol, while R.L. performed the EC50 measurements. D.E.C. designed and built the full-length PKG wild type and mutant constructs, and GYH. and D.E.C. cultured HEK293T and performed transfections, while R.L., D.B., and F.W.H. measured activation constants. B.V., R.S., and GM. designed and performed NMR experiments. G.Y.H., J.J.K., and C.K. wrote the bulk of the manuscript and created the figures. Atomic coordinates and structure factors of the PKG Iβ (219-369):cGMP complex and apo structures have been deposited in the Protein Data Bank (www.pdb.org) under accession numbers 4KU7 and 4KU8. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart P, Adams PD. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alverdi V, Mazon H, Versluis C, Hemrika W, Esposito G, van den Heuvel R, Scholten A, Heck AJ. cGMP-binding prepares PKG for substrate binding by disclosing the C-terminal domain. Journal of molecular biology. 2008;375:1380–1393. doi: 10.1016/j.jmb.2007.11.053. [DOI] [PubMed] [Google Scholar]
- Bahrami A, Assadi AH, Markley JL, Eghbalnia HR. Probabilistic interaction network of evidence algorithm and its application to complete labeling of peak lists from protein NMR spectroscopy. PLoS Comput Biol. 2009;5:e1000307. doi: 10.1371/journal.pcbi.1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beavo JA, Brunton LL. Cyclic nucleotide research -- still expanding after half a century. Nat Rev Mol Cell Biol. 2002;3:710–718. doi: 10.1038/nrm911. [DOI] [PubMed] [Google Scholar]
- Berman HM, Ten Eyck LF, Goodsell DS, Haste NM, Kornev A, Taylor SS. The cAMP binding domain: an ancient signaling module. Proc Natl Acad Sci U S A. 2005;102:45–50. doi: 10.1073/pnas.0408579102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brelidze TI, Carlson AE, Sankaran B, Zagotta WN. Structure of the carboxy-terminal region of a KCNH channel. Nature. 2012;481:530–533. doi: 10.1038/nature10735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickman E, Soll L, Beckwith J. Genetic characterization of mutations which affect catabolite-sensitive operons in Escherichia coli, including deletions of the gene for adenyl cyclase. J Bacteriol. 1973;116:582–587. doi: 10.1128/jb.116.2.582-587.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussow K, Scheich C, Sievert V, Harttig U, Schultz J, Simon B, Bork P, Lehrach H, Heinemann U. Structural genomics of human proteins-target selection and generation of a public catalogue of expression clones. Microb Cell Fact. 2005;4 doi: 10.1186/1475-2859-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornwell TL, Arnold E, Boerth NJ, Lincoln TM. Inhibition of smooth muscle cell growth by nitric oxide and activation of cAMP-dependent protein kinase by cGMP. Am J Physiol. 1994;267:C1405–1413. doi: 10.1152/ajpcell.1994.267.5.C1405. [DOI] [PubMed] [Google Scholar]
- Cowtan K, Emsley P. COOT:model building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Diller TC, Madhusudan, Xuong NH, Taylor SS. Molecular basis for regulatory subunit diversity in cAMP-dependent protein kinase: crystal structure of the type II beta regulatory subunit. Structure. 2001;9:73–82. doi: 10.1016/s0969-2126(00)00556-6. [DOI] [PubMed] [Google Scholar]
- Francis SH, Busch JL, Corbin JD, Sibley D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev. 2010;62:525–563. doi: 10.1124/pr.110.002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis SH, Corbin JD. Cyclic nucleotide-dependent protein kinases: intracellular receptors for cAMP and cGMP action. Crit Rev Clin Lab Sci. 1999;36:275–328. doi: 10.1080/10408369991239213. [DOI] [PubMed] [Google Scholar]
- Goddard TD, Kneller DG. SPARKY - NMR Assignment and Integration Software. University of California; San Francisco: 2006. [Google Scholar]
- Guo DC, Regalado E, Casteel DE, Santos-Cortez RL, Gong L, Kim JJ, Dyack S, Horne SG, Chang G, Jondeau G, et al. Recurrent Gain-of-Function Mutation in PRKG1 Causes Thoracic Aortic Aneurysms and Acute Aortic Dissections. Am J Hum Genet. 2013 doi: 10.1016/j.ajhg.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang MH, Knight PR, 3rd, Izzo JL., Jr Ca2+-induced Ca2+ release involved in positive inotropic effect mediated by CGRP in ventricular myocytes. Am J Physiol. 1999;276:R259–264. doi: 10.1152/ajpregu.1999.276.1.R259. [DOI] [PubMed] [Google Scholar]
- Jiang H, Colbran JL, Francis SH, Corbin JD. Direct evidence for cross-activation of cGMP-dependent protein kinase by cAMP in pig coronary arteries. J Biol Chem. 1992;267:1015–1019. [PubMed] [Google Scholar]
- Johnsson B, Lofas S, Lindquist G. Immobilization of proteins to a carboxymethyldextran-modified gold surface for biospecific interaction analysis in surface plasmon resonance sensors. Anal Biochem. 1991;198:268–277. doi: 10.1016/0003-2697(91)90424-r. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Casteel DE, Huang G, Kwon TH, Ren RK, Zwart P, Headd JJ, Brown NG, Chow DC, Palzkill T, Kim C. Co-crystal structures of PKG Ibeta (92-227) with cGMP and cAMP reveal the molecular details of cyclic-nucleotide binding. PLoS One. 2011;6:e18413. doi: 10.1371/journal.pone.0018413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komalavilas P, Lincoln TM. Phosphorylation of the inositol 1,4,5-trisphosphate receptor. Cyclic GMP-dependent protein kinase mediates cAMP and cGMP dependent phosphorylation in the intact rat aorta. J Biol Chem. 1996;271:21933–21938. doi: 10.1074/jbc.271.36.21933. [DOI] [PubMed] [Google Scholar]
- Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nature protocols. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Li S, Liu T, Hsu S, Kim C, Woods VL, Jr, Casteel DE. The amino terminus of cGMP-dependent protein kinase Ibeta increases the dynamics of the protein's cGMP-binding pockets. International journal of mass spectrometry. 2011;302:44–52. doi: 10.1016/j.ijms.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Carvalho MJ, Sahoo N, Muskett FW, Vieira-Pires RS, Gabant G, Cadene M, Schonherr R, Morais-Cabral JH. Structural, biochemical, and functional characterization of the cyclic nucleotide binding homology domain from the mouse EAG1 potassium channel. Journal of molecular biology. 2012;423:34–46. doi: 10.1016/j.jmb.2012.06.025. [DOI] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, C M. HKL-3000: the integration of data reduction and structure solution-from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- Moll D, Prinz A, Gesellchen F, Drewianka S, Zimmermann B, Herberg FW. Biomolecular interaction analysis in functional proteomics. Journal of neural transmission. 2006;113:1015–1032. doi: 10.1007/s00702-006-0515-5. [DOI] [PubMed] [Google Scholar]
- Osborne B, Wu J, McFarland C, NIckl C, Sankaran B, Casteel DE, Woods VJ, Kornev A, Taylor S, Dostmann WR. Crystal structure of cGMP-dependent protein kinase reveals novel site of interchain communication. Structure. 2011;19:1317–1327. doi: 10.1016/j.str.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- Pellegrino D, Shiva S, Angelone T, Gladwin MT, Tota B. Nitrite exerts potent negative inotropy in the isolated heart via eNOS-independent nitric oxide generation and cGMP-PKG pathway activation. Biochim Biophys Acta. 2009;1787:818–827. doi: 10.1016/j.bbabio.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Pfeifer A, Klatt P, Massberg S, Ny L, Sausbier M, Hirneiss C, Wang GX, Korth M, Aszodi A, Andersson KE, et al. Defective smooth muscle regulation in cGMP kinase I-deficient mice. EMBO J. 1998;17:3045–3051. doi: 10.1093/emboj/17.11.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponsioen B, Zhao J, Riedl J, Zwartkruis F, van der Krogt G, Zaccolo M, Moolenaar WH, Bos JL, Jalink K. Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO reports. 2004;5:1176–1180. doi: 10.1038/sj.embor.7400290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puljung MC, Zagotta WN. A Secondary Structural Transition in the C-helix Promotes Gating of Cyclic Nucleotide-regulated Ion Channels. J Biol Chem. 2013;288:12944–12956. doi: 10.1074/jbc.M113.464123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed RB, Sandberg M, Jahnsen T, Lohmann SM, Francis SH, Corbin JD. Fast and slow cyclic nucleotide-dissociation sites in cAMP-dependent protein kinase are transposed in type Ibeta cGMP-dependent protein kinase. J Biol Chem. 1996;271:17570–17575. doi: 10.1074/jbc.271.29.17570. [DOI] [PubMed] [Google Scholar]
- Rehmann H, Wittinghofer A, Bos JL. Capturing cyclic nucleotides in action: snapshots from crystallographic studies. Nat Rev Mol Cell Biol. 2007;8:63–73. doi: 10.1038/nrm2082. [DOI] [PubMed] [Google Scholar]
- Ruth P, Landgraf W, Keilbach A, May B, Egleme C, Hofmann F. The activation of expressed cGMP-dependent protein kinase isozymes I alpha and I beta is determined by the different amino-termini. Eur J Biochem. 1991;202:1339–1344. doi: 10.1111/j.1432-1033.1991.tb16509.x. [DOI] [PubMed] [Google Scholar]
- Schlossmann J, Hofmann F. cGMP-dependent protein kinases in drug discovery. Drug Discov Today. 2005;10:627–634. doi: 10.1016/S1359-6446(05)03406-9. [DOI] [PubMed] [Google Scholar]
- Smith JA, Reed RB, Francis SH, Grimes K, Corbin JD. Distinguishing the roles of the two different cGMP-binding sites for modulating phosphorylation of exogenous substrate (heterophosphorylation) and autophosphorylation of cGMP-dependent protein kinase. The Journal of biological chemistry. 2000;275:154–158. doi: 10.1074/jbc.275.1.154. [DOI] [PubMed] [Google Scholar]
- Su Y, Dostmann WR, Herberg FW, Durick K, Xuong NH, Ten Eyck L, Taylor SS, Varughese KI. Regulatory subunit of protein kinase A: structure of deletion mutant with cAMP binding domains. Science. 1995;269:807–813. doi: 10.1126/science.7638597. [DOI] [PubMed] [Google Scholar]
- Taraska JW, Puljung MC, Olivier NB, Flynn GE, Zagotta WN. Mapping the structure and conformational movements of proteins with transition metal ion FRET. Nat Methods. 2009;6:532–537. doi: 10.1038/nmeth.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Adams PD, Read RJ, McCoy AJ, Moriarty NW, Grosse-Kunstleve RW, Afonine PV, Zwart P, Hung LW. Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr D Biol Crystallogr. 2009;65:582–601. doi: 10.1107/S0907444909012098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi B, Kramer RH. Real-time patch-cram detection of intracellular cGMP reveals long-term suppression of responses to NO and muscarinic agonists. Neuron. 1998;21:895–906. doi: 10.1016/s0896-6273(00)80604-2. [DOI] [PubMed] [Google Scholar]
- Wall ME, Francis SH, Corbin JD, Grimes K, Richie-Jannetta R, Kotera J, Macdonald BA, Gibson RR, Trewhella J. Mechanisms associated with cGMP binding and activation of cGMP-dependent protein kinase. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:2380–2385. doi: 10.1073/pnas.0534892100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Brown S, Xuong NH, Taylor SS. RIalpha subunit of PKA: a cAMP-free structure reveals a hydrophobic capping mechanism for docking cAMP into site B. Structure. 2004;12:1057–1065. doi: 10.1016/j.str.2004.03.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.