

Abstract
Embryonic stem (ES) cells express pluripotency-associated genes and repress differentiation-inducible genes. The activities of these genes are coordinately reversed during differentiation. The changes in the transcriptome upon conditional KAP1 knockout in ES cells overlapped with the changes during embryoid body formation. KAP1 repressed differentiation-inducible genes and derepressed pluripotency-associated genes in ES cells. KAP1 formed complexes with polycomb repressive complexes 1 (PRC1) through an interaction that was mediated by the KAP1 coiled-coil region. KAP1 and PRC1 bound cooperatively at the promoters of differentiation-inducible genes and repressed their transcription. In contrast, KAP1 bound the transcribed and flanking sequences of pluripotency-associated genes, did not enhance PRC1 binding, and derepressed their transcription. KAP1 had opposite effects on differentiation-inducible and pluripotency-associated gene transcription both in ES cells and in differentiating embryoid bodies. The region of KAP1 that mediated the interaction with PRC1 was required for KAP1 enhancement of PRC1 binding and for KAP1 repression of transcription at differentiation-inducible promoters. This region of KAP1 was not required for KAP1 suppression of PRC1 binding or for KAP1 derepression of transcription at pluripotency-associated promoters. The opposite effects of KAP1 on the transcription of differentiation-inducible versus pluripotency-associated genes contributed to the reciprocal changes in their transcription during differentiation.
INTRODUCTION
Genes that maintain the pluripotent state are expressed and genes that induce differentiation are repressed in embryonic stem (ES) cells. Pluripotency-associated genes are thought to be activated by sequence-specific DNA-binding proteins, and differentiation-inducible genes are thought to be repressed by epigenetic regulatory complexes. The relationship between the activation of pluripotency-associated genes and the repression of differentiation-inducible genes has been unclear. Consequently, the mechanisms that coordinate the switch from pluripotency-associated gene transcription in ES cells to differentiation-inducible gene transcription during lineage commitment were largely uncharacterized.
Polycomb group complexes repress differentiation-inducible genes in ES cells (reviewed in reference 1). The mechanisms that specify polycomb group complex binding at differentiation-inducible promoters have been investigated extensively. Both DNA sequence-dependent and chromatin modification-dependent mechanisms have been proposed to influence their binding specificities. The mechanisms that suppress polycomb group protein binding at pluripotency-associated gene promoters in ES cells were unknown.
The core subunits of canonical polycomb repressive complexes 1 (PRC1) have no known sequence-specific DNA-binding activities. PRC1 complexes can interact with many DNA- and chromatin-binding proteins (2–7). Some interaction partners can modulate PRC1 binding at a subset of differentiation-inducible genes in ES cells (6, 8–12). None of these proteins are essential for PRC1 binding to chromatin, nor are most of them required to maintain ES cell self-renewal or pluripotency.
PRC1 binding at many promoters has been correlated with histone H3 K27 trimethylation (reviewed in reference 1). However, PRC1 can bind chromatin in ES cells containing mutations that eliminate detectable H3 K27 trimethylation and PRC1 binding can be regulated independently of changes in H3 K27 trimethylation (6, 13–16).
The KRAB-associated protein 1 (KAP1/TRIM28/TIF1β) transcription coregulatory protein is essential for ES cell pluripotency (17). KAP1 has been characterized mainly as a corepressor that can interact with the KRAB domains of DNA-binding proteins through its N-terminal RING and B-box 1 and 2 regions (Fig. 1E) (18, 19). The C-terminal PXVXL, PHD and BROMO regions of KAP1 can interact with many chromatin-binding proteins, including HP1 and Setdb1, most of which are also associated with transcription repression (20). The central coiled-coil region of KAP1 can interact with E2F1 (21), but the roles of this domain in transcription regulation by KAP1 were unknown. Conditional KAP1fl/fl knockout reduces the transcription of some genes, suggesting that KAP1 can directly or indirectly activate transcription. The mechanisms whereby KAP1 represses some genes and activates others were unknown.
FIG 1.

KAP1 interaction with PRC1 in cell extracts and in living cells is mediated by the coiled-coil region. (A to D) Coprecipitation of endogenous KAP1 in association with endogenous Ring1b (A), Cbx7 (B), Cbx2 (C), and E2F6.com subunits (D). OHT, tamoxifen; DOX, doxycycline. Extracts of the wild-type (WT), conditional Ring1bfl/fl knockout (KO), constitutive Cbx7−/− and Cbx2−/− knockout, and conditional shMga knockdown mouse ES cells as indicated above the lanes were incubated with the antibodies indicated above the lanes. The immunoprecipitated (IP) proteins were analyzed by immunoblotting with the antibodies indicated to the left of each blot. Five percent (A), 0.2% (B, C), and 2% (D) of the cell extracts were analyzed in parallel (Input). The efficiencies of Ring1b, Cbx7, Cbx2, and Mga depletion were established by immunoblotting. (E) KAP1 deletion derivatives used to map the region that interacts with PRC1 (immunoblots are shown in panel F and in Fig. S1C in the supplemental material). The presence of Cbx2 coprecipitation is shown to the right of each KAP1 deletion derivative. (F) Coprecipitation of KAP1 deletion derivatives with Cbx2. The KAP1 deletion derivatives indicated above the lanes were N-terminally fused to FLAG and expressed transiently with YFP-Cbx2 in HEK293T cells. The cell extracts were incubated with anti-GFP or control (IgG) antibodies. The precipitated proteins were analyzed by immunoblotting with anti-FLAG (upper blots) and anti-GFP (lower blots) antibodies. Five percent of the cell extracts were analyzed in parallel (Input). Asterisks indicate cross-reactive bands. (G) Selectivity of PRC1 interactions mediated by the coiled-coil region of KAP1. Extracts of ES cell lines that stably expressed either WT KAP1 or KAP1 lacking the coiled-coil region (KAP1ΔCC) fused to the FLAG epitope, were incubated with anti-FLAG antibodies. The precipitated proteins were analyzed by immunoblotting with antibodies directed against Ring1b, Mel18, HP1β and FLAG. 1% of the extracts were analyzed in parallel (Input). The asterisks indicates the mouse immunoglobulin heavy chain, which is detected by the anti-mouse IgG secondary antibody. The data in panels A to G are representative of results of two or more experiments. (H) BiFC analysis of interactions between KAP1 and Cbx family proteins in living HEK293T cells. KAP1 fused to the C-terminal fragment of YFP (YC-KAP1) was coexpressed with individual Cbx family proteins fused to the N-terminal fragment of YFP (YN-Cbx). The fluorescence intensities of cells that expressed the BiFC fusion proteins together with an internal normalization standard (cyan fluorescent protein [CFP]) were measured by flow cytometry. The data shown represent the mean and standard deviation of the BiFC/CFP ratio of three separately transfected cell populations. The Cbx family proteins, as well as KAP1 and the KAP1 deletion derivatives, were expressed at similar levels (see Fig. S1 in the supplemental material). The fluorescence microscopy images show the subnuclear distributions of BiFC complexes formed by KAP1 with the Cbx family proteins indicated above the adjacent bar graphs.
Both KAP1 and the Ring1b subunit of PRC1 are essential for gastrulation in mice (22, 23). The early developmental arrest of mouse embryos that lack KAP1 or Ring1b is likely to reflect the essential roles of KAP1 and PRC1 in ES cell proliferation and in the regulation of genes that control the switch between pluripotency and differentiation (8, 24–26). KAP1 and several PRC1 subunits can also repress the transcription of endogenous retroelements in mouse ES cells (26–28). The PXVXL motif of KAP1, which mediates the interaction with HP1, is not required for gastrulation or for early mouse development (29). It is therefore likely that KAP1 interactions with other proteins are important in early development.
Both KAP1 and PRC1 subunits are essential for the replenishment of many cell types in adult mice (30–37, 52). Many mechanisms have been proposed to mediate the effects of KAP1 and PRC1 on the proliferation and differentiation of multipotent progenitor cells. However, the mechanisms of KAP1 and PRC1 action had not been investigated in parallel in the same cells.
The genome-wide locations of KAP1 and PRC1 binding have been investigated in different cell types. PRC1 binds many promoters that contain CpG islands, as well as promoters with no detectable CpG enrichment (38, 39). KAP1 binds both promoters and transcribed and intergenic sequences (17, 40). Several other proteins bind to a subset of the regions that are bound by KAP1 or PRC1 (10–12, 17, 40–42). The relationship between the chromatin binding specificities of KAP1 and PRC1 and their potential interactions had not been investigated.
Here, we identify an interaction between KAP1 and PRC1 and characterize the effects of this interaction on their binding at differentiation-inducible versus pluripotency-associated genes and on the transcription of these genes in ES cells.
MATERIALS AND METHODS
Plasmid expression vectors.
The FLAG-hemagglutinin (HA)-KAP1 and YC-KAP1 expression vectors were prepared by substituting the FLAG and yellow fluorescent protein (YFP) (nucleotides 173 to 238) coding sequences for the sequence encoding green fluorescent protein (GFP) in GFP-HA-KAP1 (43). For the deletions in KAP1, see Materials and Methods in the supplemental material. The YN-Cbx family expression vectors were as described previously (44).
Cell lines.
Kap1fl/fl ES cells were provided by Didier Trono (School of Life Sciences, École Polytechnique Fédérale de Lausanne [EPFL], Lausanne, Switzerland) and were cultured as described previously (26). Ring1a−/−; Ring1bfl/fl; Rosa26::CreERT2 (8), Ring1bfl/fl; Rosa26::CreERT2 (8), Bmi1−/−Mel18−/− (45), Cbx2−/− (46), Cbx2#1, Cbx4#11, Cbx6#7, Cbx7#3, and Cbx8#1 (44) ES cell lines were described previously. Cbx7−/− ES cell lines were isolated from blastocyst embryos of Cbx7tm1b/tm1b mice generated by Cre-mediated recombination in Cbx7tm1a/tm1a mice obtained from the Welcome Trust Sanger Institute. Stable Kap1fl/fl & KAP1 and Kap1fl/fl & KAP1ΔCCES cell lines (cell lines with KAP1 or KAP1ΔCC added into a background of Kap1fl/fl) were selected by using G418 and screened by immunoblotting. Embryoid bodies were prepared by seeding ES cells into petri dishes in ES cell medium without leukemia inhibitory factor (LIF).
Analysis of protein-protein interactions, chromatin binding, and transcript levels.
BiFC analysis, immunoprecipitation, immunoblotting, chromatin immunoprecipitation, and transcript quantification were performed as described in the supplemental material.
Microarray and ChIP-seq analysis.
Transcripts were isolated from wild-type (WT) and KAP1fl/fl knockout ES cells after 24 h of tamoxifen treatment, followed by 48 h of culture without tamoxifen. The transcripts were reverse transcribed, and the labeled cDNAs were analyzed by hybridization to the Affymetrix Mouse Gene ST 1.1 array in accordance with the manufacturer's instructions.
Chromatin was cross-linked in WT, KAP1fl/fl, Ring1bfl/fl, and Ring1bfl/fl Ring1a−/− ES cells after 24 h of tamoxifen treatment, followed by 48 h of culture without tamoxifen. The sonicated chromatin was precipitated by using antibodies against KAP1 and Ring1b. The precipitated DNA was purified and amplified with the Illumina ChIP-seq Sample Prep kit, and the DNA fragments were sequenced with the Illumina Genome Analyzer.
The ChIP-seq tags were mapped to the mouse genome (mm10) by using Bowtie2, and the regions that were bound by Ring1b and KAP1 were identified with MACS14. The coverage at genes and promoters (within 1 kb of transcription start sites) was analyzed with Ngsplot. The overlaps between the genes and promoters that were bound by Ring1b and KAP1 and those whose transcription was altered upon Kap1fl/fl knockout were identified by determining the intersect between Ring1b and KAP1 binding regions and Refgenes with Bedtools. For more detailed descriptions of the materials and methods used, see the supplemental material.
Microarray data accession number.
The data from the high-throughput assays are available in the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) under superseries number GSE56629.
RESULTS
To identify proteins that could interact with PRC1, we purified complexes containing different Cbx family PRC1 subunits from mouse ES cells (6). Mass spectrometry analysis of a band with a mobility corresponding to the molecular weight of KAP1 identified 19 peptides with fragmentation products consistent with the amino acid sequence of KAP1 (see Fig. S1A in the supplemental material).
Interactions between KAP1 and PRC1 subunits in ES cell extracts and in living cells.
We examined if endogenous KAP1 and PRC1 interacted with each other by immunoprecipitating complexes containing different PRC1 subunits from ES cell extracts. KAP1 was coprecipitated by antibodies directed against Ring1b, Cbx7, and Cbx2 from WT ES cell extracts (Fig. 1A to C). KAP1 coprecipitation was eliminated or markedly reduced when extracts from Ring1bfl/fl, Cbx7−/−, and Cbx2−/− knockout cells were precipitated with the respective antibodies, confirming the specificity of KAP1 coprecipitation with each of these proteins. KAP1 was also coprecipitated with each of the Cbx family fusion proteins when they were stably expressed in mouse ES cells (see Fig. S1B in the supplemental material). Since KAP1 coprecipitated with each of the core PRC1 subunits tested, it is likely that KAP1 interacted with intact PRC1 complexes.
To identify the PRC1 subunit(s) that could mediate the interaction with KAP1, we examined KAP1 coprecipitation with subunits of complexes that have proteins in common with the canonical PRC1 complex. KAP1 coprecipitated with the Mga, E2F6, L3MBTL2, and Max subunits of the E2F6.com complex (Fig. 1D). Mga depletion by short hairpin RNA knockdown reduced KAP1 coprecipitation with Mga, E2F6, and L3MBTL2 but not with Max, indicating that Mga depletion selectively reduced KAP1 interaction with E2F6.com subunits. Since Ring1b is the only protein known to be common to E2F6.com and the canonical PRC1 complexes containing Cbx family proteins (47), it is likely that the interactions between these complexes and KAP1 required Ring1b. We have focused on KAP1 interactions with Ring1b and with Cbx family proteins in order to examine interactions both with a ubiquitous PRC1 subunit and with subunits that are present in specific PRC1 complexes.
We mapped the region of KAP1 that is required for its association with PRC1. Deletions that removed the coiled-coil region of KAP1 eliminated its coprecipitation with Cbx2, as well as with other Cbx family proteins (Fig. 1E and F; see Fig. S1C and D in the supplemental material). Deletions that did not overlap this region had small effects on Cbx2 coprecipitation that were not consistently observed in repeated experiments. To establish if the coiled-coil region of KAP1 is required for its interaction with endogenous PRC1 in ES cells, we generated mouse ES cell lines that express epitope-tagged intact KAP1 or KAP1 lacking the coiled-coil region (KAP1ΔCC). Both endogenous Ring1b and Mel18 subunits of PRC1 coprecipitated with intact KAP1 but not with KAP1ΔCC (Fig. 1G). In contrast, HP1β, which interacts with the PXVXL motif in KAP1 (20), coprecipitated with both intact KAP1 and KAP1ΔCC. The coiled-coil region of KAP1 was therefore selectively required for its interaction with endogenous PRC1.
We investigated if KAP1 and PRC1 form complexes in living cells and determined their subcellular distributions by using bimolecular fluorescence complementation (BiFC) analysis of KAP1 and Cbx family proteins. The BiFC assay is based on the formation of a fluorescent complex by two fragments of a fluorescent protein when they are brought together by an interaction between proteins fused to the fragments (48). Coexpression of KAP1 with Cbx2, Cbx4, Cbx6, or Cbx7 fused to fluorescent protein fragments produced BiFC complexes in the nuclei of HEK293T cells (Fig. 1H). Deletion of the coiled-coil region of KAP1 reduced the efficiency of BiFC complex formation by more than 20-fold, whereas deletion of the B1 region of KAP1 had no detectable effect on BiFC fluorescence. Thus, the coiled-coil region of KAP1 was required for a specific interaction between KAP1 and PRC1 both in vitro and in living cells.
Mutual enhancement of binding and parallel repression by KAP1 and PRC1 at selected promoters.
To investigate potential functional consequences of KAP1 interaction with PRC1, we compared KAP1 and PRC1 subunit binding and changes in transcription upon conditional KAP1fl/fl and Ring1bfl/fl knockout. We used both targeted analysis of selected genes that were predicted to be repressed by KAP1 and PRC1 (Fig. 2) and unbiased genome- and transcriptome-wide analyses (Fig. 3 and 4). First, we tested if KAP1 and PRC1 subunits bound to promoters that were selected on the basis of results of previous high-throughput analyses (17, 39). Since KAP1 has been characterized as a repressor of transcription, we focused on genes that were predicted to be repressed in ES cells. KAP1 and PRC1 subunits bound a majority (12/15) of the promoters tested (Fig. 2A). KAP1, but not the PRC1 subunits, also bound a region adjacent to an endogenous retroelement.
FIG 2.

Cooperative KAP1 and PRC1 binding and transcription repression at selected promoters. (A) Effects of KAP1fl/fl knockout on PRC1 binding at selected promoters. KAP1 and PRC1 subunit binding and histone modifications were measured by ChIP analysis at the promoters indicated below all of the bar graphs in KAP1fl/fl knockout cells left untreated (−OHT) or treated with tamoxifen for 24 h (+OHT), followed by 48 h of culture without tamoxifen. (B) Effects of Ring1bfl/fl knockout on KAP1 binding at selected promoters. KAP1 and PRC1 subunit binding and histone modifications were measured by ChIP analysis at the promoters indicated below all of the bar graphs in Ring1bfl/fl Ring1a−/− cells left untreated (−OHT) and after culture with tamoxifen for 3 days (+OHT). (C) Effects of KAP1fl/fl knockout on PRC1 subunit expression. The levels of KAP1 and of PRC1 subunits were measured in KAP1fl/fl ES cells either left untreated (−OHT) or after 24 h of treatment with tamoxifen (+OHT), followed by 48 h of culture without tamoxifen. Extracts of the cells indicated above the lanes were analyzed by immunoblotting with the antibodies directed against the proteins indicated to the left of the blots. (D) Effects of Ring1bfl/fl knockout on the level of KAP1 expression. The levels of Ring1b and KAP1 were measured in Ring1bfl/fl; Ring1a−/− ES cells either left untreated (−OHT) or after 3 days of culture with tamoxifen (+OHT). Extracts of the cells indicated above the lanes were analyzed by immunoblotting with the antibodies directed against the proteins indicated to the left of the blots. (E) Effects of KAP1fl/fl or PRC1 subunit knockout on the transcription of selected genes. The levels of the transcripts indicated below all of the bar graphs were measured in ES cell lines containing the mutations indicated above each bar graph and in the corresponding control ES cells. The bars show the ratios of the transcript levels in mutant cells to the respective control cells (+OHT/−OHT or mutant/control). The data in all of the panels represent the means and standard deviations of three replicate qPCR measurements and are representative of at least two experiments with independently cultured ES cells.
FIG 3.
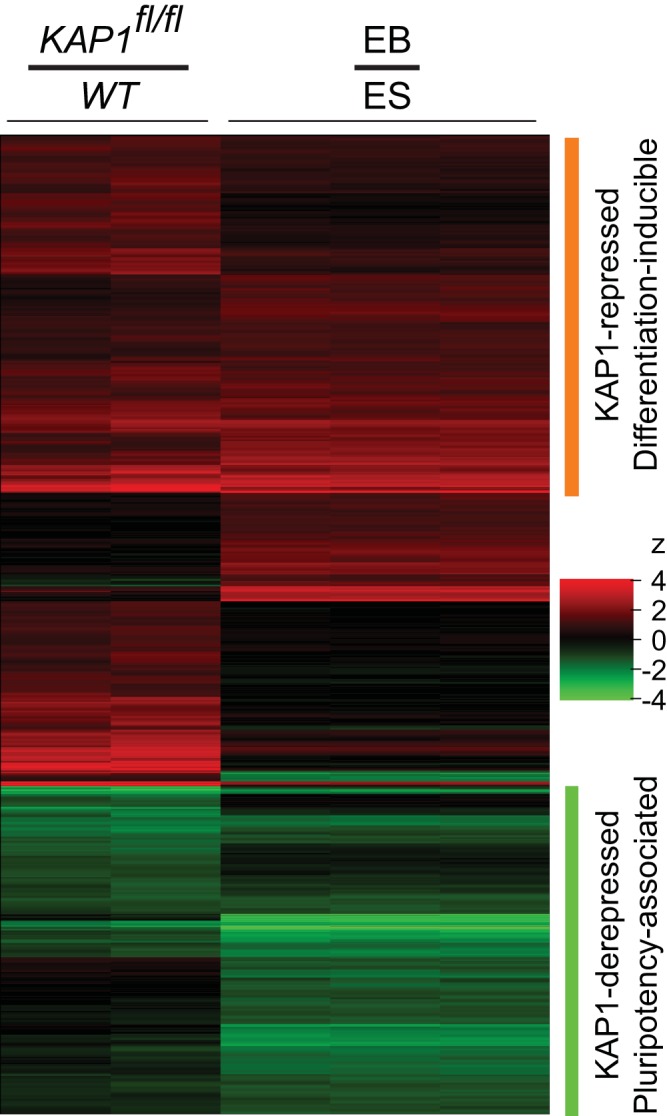
Changes in the transcriptome upon KAP1fl/fl knockout and upon embryoid body (EB) formation. Comparison of the changes in transcript levels upon KAP1fl/fl knockout and upon embryoid body formation. The heat maps show the ratios of transcript levels in KAP1fl/fl knockout cells to WT cells (KAP1fl/fl/WT) and of transcript levels in embryoid bodies to those in ES cells (EB/ES) determined previously (49). Levels of transcripts that increased or decreased at least 2-fold either upon KAP1fl/fl knockout or upon embryoid body formation are shown by the color code on the right. The orange bar indicates transcripts whose levels increased more than 2-fold upon KAP1fl/fl knockout and upon embryoid body formation. The green bar indicates transcripts whose levels decreased at least 2-fold upon KAP1fl/fl knockout and upon embryoid body formation. The columns show the ratios of the transcript levels for two KAP1fl/fl knockout and two WT cell populations and for three embryoid body and three ES cell populations. Transcript levels were quantified by microarray (Affymetrix Mouse Gene ST 1.1) analysis of transcripts isolated from WT and KAP1fl/fl knockout ES cells after 24 h of tamoxifen treatment, followed by 48 h of culture without tamoxifen.
FIG 4.

Genome-wide comparison of KAP1 and Ring1b binding at genes that were repressed versus derepressed by KAP1. (A and B) Comparison of the sets of genes (A) and promoters (B) that were bound by KAP1 and by Ring1b and of the sets of genes and promoters whose transcription increased (KAP1 repressed) or decreased (KAP1 derepressed) at least 2-fold upon KAP1fl/fl knockout. The genes (transcribed region ± 1 kb) and promoters (transcription start site ± 1 kb) that were bound by KAP1 and by Ring1b and those whose transcription was altered more than 2-fold upon KAP1fl/fl knockout were identified on the basis of their intersects with KAP1 and Ring1b binding regions. (C) Comparison of the distributions of KAP1 and Ring1b ChIP-seq tags near annotated transcription start sites (TSS). The traces indicate the average levels of ChIP-seq tags isolated from KAP1fl/fl and Ringb1fl/fl cells cultured in the absence (upper two traces) or in the presence (lower three traces) of tamoxifen as a function of the distance from the TSS. The widths of the shaded areas in panels C, E, and F reflect the standard deviations of the mean ChIP-seq tag levels determined with Ngsplot. (D) Distances between KAP1 and Ring1b ChIP-seq tag peaks at all of the promoters that bound both KAP1 and Ring1b (orange bars). The bars show the percentages of all of the promoters in which the maxima of the KAP1 and Ring1b ChIP-seq tag distributions were separated by the indicated distances. For comparison, we plotted the distances from the Ring1b ChIP-seq tag peaks to simulated KAP1 ChIP-seq tag peaks that were transposed to the opposite side of the transcription start site without changing the distance from the transcription start site (purple bars). The diagram illustrates an example of the measurement of distances between observed (orange bar) and simulated (purple bar) binding locations. (E and F) Comparison of the overall levels and distributions of KAP1 and Ring1b ChIP-seq tags at genes whose transcription increased (E) or decreased (F) more than 2-fold upon KAP1fl/fl knockout. The average levels of ChIP-seq tags were plotted as a function of the position within the genes (scaled to a constant length) and the distances from the TSS and the poly(A) cleavage sites. (G) Comparison of KAP1 and Ring1b ChIP-seq tag distributions in genomic regions encompassing KAP1-repressed (orange) or KAP1-derepressed (green) genes. The tracks display the levels of ChIP-seq tags that were isolated from KAP1fl/fl and Ringb1fl/fl cells cultured in the absence (upper two tracks) or presence (lower three tracks) of tamoxifen across large genomic regions. RPM, reads per million.
To determine if KAP1 and PRC1 affected binding by each other at the selected promoters, we tested the effects of conditional KAP1fl/fl and Ring1bfl/fl knockouts on PRC1 and KAP1 binding, respectively. Conditional KAP1fl/fl knockout reduced Ring1b, Mel18, and Cbx7 binding at all of the promoters tested that bound both KAP1 and PRC1 in WT cells (Fig. 2A). Conversely, conditional Ring1bfl/fl knockout in Ring1a−/− cells reduced KAP1 binding at all of the promoters tested that bound both KAP1 and PRC1 and that had high levels of KAP1 binding (Fig. 2B). KAP1fl/fl knockout did not reduce the levels of PRC1 subunit expression, and Ring1bfl/fl knockout did not reduce the level of KAP1 expression (Fig. 2C and D). Thus, KAP1 and PRC1 enhanced binding by each other at most of the promoters that were examined.
We examined the effects of KAP1fl/fl and PRC1 subunit knockouts on the transcription of the genes whose promoters were bound by both KAP1 and PRC1. The levels of 11 of the 12 transcripts examined increased more than 2-fold upon KAP1fl/fl knockout and more than 3-fold upon Ring1bfl/fl knockout (Fig. 2E). The levels of about half of the transcripts increased in Cbx7−/−, Cbx2−/−, and Bmi1−/−; Mel18−/− knockout cells. Both KAP1 and the ubiquitous Ring1b subunit of PRC1 were required for efficient repression of most of the selected genes, whereas other PRC1 subunits were required for the repression of a subset of the genes. These results indicate that KAP1 and PRC1 enhanced binding by each other and repressed transcription at most of the genes that were selected for analysis on the basis of the results of previous studies.
Effects of KAP1 on differentiation-inducible versus pluripotency-associated gene transcription.
To illuminate the functions of KAP1 and potential roles for its interaction with PRC1 in ES cells, we determined the effects of KAP1fl/fl knockout on the ES cell transcriptome by microarray analysis. The hybridization signals for about 800 transcripts increased and about 300 transcripts decreased more than 2-fold upon KAP1fl/fl knockout (see Table S1 in the supplemental material). We compared the changes in ES cell transcription observed upon KAP1fl/fl knockout with the changes in transcription associated with a variety of developmental transitions and signaling responses reported previously. We observed a close correspondence between the changes in ES cell transcription upon KAP1fl/fl knockout and upon embryoid body formation (49) (Fig. 3).
The levels of 57% of the transcripts whose levels increased more than 2-fold upon KAP1fl/fl knockout also increased more than 2-fold upon embryoid body formation. Likewise, the levels of 71% of the transcripts whose levels decreased at least 2-fold upon KAP1fl/fl knockout also decreased at least 2-fold upon embryoid body formation. Our data do not distinguish between derepression and activation as mechanisms of KAP1 regulation of the latter genes. We use the term derepression since Ring1b repressed the activities of many of the promoters and the level of Ring1b binding increased at some promoters whose activities decreased upon KAP1fl/fl knockout (see below). Only 3% of the transcripts whose levels increased more than 2-fold and a single transcript whose level decreased more than 2-fold upon KAP1fl/fl knockout exhibited the opposite change in transcription upon embryoid body formation. KAP1fl/fl knockout had a profound effect on the ES cell transcriptome; the average level of all transcripts that were repressed by KAP1 was lower than the average level of all transcripts that were derepressed by KAP1 in WT cells, but their relative levels were reversed upon KAP1fl/fl knockout. The extensive overlap between the changes in transcription upon KAP1fl/fl knockout and upon embryoid body formation identified KAP1 as a key regulator of both differentiation-inducible and pluripotency-associated gene transcription in ES cells.
Genome-wide analysis shows overlap between KAP1 and Ring1b binding regions.
To determine if the genes whose transcription was affected by KAP1fl/fl knockout were regulated by direct KAP1 or Ring1b binding, we determined the genome-wide locations of KAP1 and Ring1b binding in ES cells. More than 150 million ChIP-seq tags were retrieved with antibodies directed against KAP1 and Ring1b from KAP1fl/fl and Ring1bfl/fl cells cultured in the absence or presence of tamoxifen. More than 85% of the KAP1 and Ring1b ChIP-seq tags were mapped to the mouse genome (mm10) with Bowtie2 (see Table S2 in the supplemental material). Between 21% (Ring1b) and 29% (KAP1) of the ChIP-seq tags mapped to multiple locations in the genome and were distributed randomly among these locations. The inclusion or exclusion of the tags that mapped to multiple locations did not materially affect the conclusions presented here.
We identified KAP1 and Ring1b binding regions throughout the genome by mapping all of the sequences that had statistically significant differences in the levels of ChIP-seq tags isolated from KAP1fl/fl and Ring1bfl/fl cells cultured in the absence or presence of tamoxifen with MACS14. This is a conservative strategy that minimizes the risk of false positives that would artificially augment the genome-wide overlap in KAP1 and Ring1b binding regions. Some KAP1 and Ring1b binding regions may not be detected (false negatives) because of residual KAP1 and Ring1b binding in KAP1fl/fl and Ring1bfl/fl knockout cells.
Ring1b and KAP1 bound many of the same genes and promoters. 93% of the genes and 86% of the promoters that were bound by Ring1b were also bound by KAP1 (Fig. 4A and B; see Tables S3 and S4 in the supplemental material). Conversely, 48% of the genes and 43% of the promoters that were bound by KAP1 were also bound by Ring1b. The KAP1 and Ring1b binding regions overlapped each other at 97% of the promoters and at 91% of the genes that were bound by both proteins. Ring1b binding regions were concentrated at promoters; 53% were located within 1 kb of an annotated transcription start site. KAP1 binding regions were also enriched at promoters, but a large proportion (73%) of the KAP1 binding regions were scattered throughout transcribed and intergenic sequences.
About 60% of the Ring1b and 20% of the KAP1 binding regions overlapped partially with about half of all of the CpG islands. Because of the prevalence and large sizes of CpG islands near promoters, it was not clear whether this overlap reflected preferential Ring1b or KAP1 binding at CpG islands in particular or at promoters in general.
Ring1b and KAP1 bound a majority of the genes that were deregulated upon KAP1fl/fl knockout. Fifty-nine percent of the genes whose transcription increased upon KAP1fl/fl knockout were bound by both KAP1 and Ring1b, 19% were bound by KAP1 alone, and 7% were bound by Ring1b alone (Fig. 4A). Sixty-seven percent of the genes whose transcription decreased upon KAP1fl/fl knockout were bound by both KAP1 and Ring1b, 31% were bound by KAP1 alone, and none were bound by Ring1b alone. Ring1b and KAP1 also bound a majority of the promoters for these genes (Fig. 4B). The microarray analysis underestimated the magnitudes of changes in transcription upon KAP1fl/fl knockout (see Fig. 7A), which might contribute to the observation that KAP1 and Ring1b bound many genes whose hybridization signals changed less than 2-fold upon KAP1fl/fl knockout. The enrichment of Ring1b and KAP1 binding regions within the genes and promoters that were derepressed or repressed by KAP1 suggests that both groups of genes and promoters were regulated by direct KAP1 and Ring1b binding.
FIG 7.

Quantitative relationships between the levels and distributions of KAP1 and Ring1b binding and the changes in Ring1b binding and transcription upon KAP1fl/fl knockout (KO). (A) Comparison of the changes in transcript levels upon KAP1fl/fl knockout measured by RT-qPCR and by microarray analysis. The levels of 24 transcripts were measured in KAP1fl/fl knockout and in WT cells by RT-qPCR and by microarray analysis. The changes in the levels of all of the transcripts measured with both assays were plotted as a scatterplot. The microarray data represent the means and standard deviations of two biological replicates. The RT-qPCR data represent the means and standard deviations of three replicate qPCR measurements. Both axes are plotted on logarithmic scales. (B) Comparison of the changes in Ring1b binding upon KAP1fl/fl knockout measured by ChIP-Seq and ChIP-qPCR analyses. The levels of Ring1b binding at 16 promoters were measured by ChIP-seq and ChIP-qPCR analyses. The changes in the Ring1b ChIP-qPCR signals were plotted as a function of the changes in Ring1b ChIP-seq tag counts upon KAP1fl/fl knockout for all of the differentiation-inducible and pluripotency-associated promoters examined. The Ring1b ChIP-qPCR signals were normalized by the signals for histone H3. The Ring1b ChIP-seq tag counts were calculated on the basis of regions encompassing the ChIP-qPCR amplicons combined with contributions from flanking sequences that decreased linearly to a distance of 250 bp from the amplicon. Both axes were plotted on logarithmic scales. (C) Correlation between the enrichment of KAP1 bound to the promoter and the enrichment of KAP1 bound to the transcribed region and the average change in Ring1b binding upon KAP1fl/fl knockout. All of the genes that were bound by KAP1 and Ring1b were grouped on the basis of the density of KAP1 ChIP-seq tags at the promoter relative to the density in the downstream transcribed region. The average change in Ring1b ChIP-seq tag enrichment at the promoter upon KAP1fl/fl knockout was calculated for each group of 100 genes and was plotted as a function of the ratio of KAP1 ChIP-seq tag enrichment at the promoter to that in the downstream transcribed region. (D to F) The changes in transcription upon KAP1fl/fl knockout were plotted as a function of the enrichment of KAP1 (D) or of Ring1b (E) ChIP-seq tags or of the change in Ring1b ChIP-seq tag enrichment upon KAP1fl/fl knockout (F). All of the genes that were bound by KAP1 and Ring1b were divided into groups of 100 on the basis of the enrichment of KAP1 ChIP-seq tags in the gene locus (D), the enrichment of Ring1b ChIP-seq tags at the promoters (E), or the change in the enrichment of Ring1b ChIP-seq tags at the promoters upon KAP1fl/fl knockout (F). The average change in transcription upon KAP1fl/fl knockout was calculated for each group of genes on the basis of the microarray data and was plotted as a function of the average ChIP-seq tag enrichment or the change in ChIP-seq tag enrichment in each group.
Proximity of KAP1 and Ring1b binding to each other at promoters.
To investigate if the locations of KAP1 and Ring1b binding at promoters are related to each other, we compared the distributions of KAP1 and Ring1b ChIP-seq tags in the vicinity of all of the annotated transcription start sites. The overall distributions of KAP1 and Ring1b ChIP-seq tags averaged among all of the promoters were similar to each other, with peaks located 14 and 27 bp downstream from the transcription start site, respectively (Fig. 4C). The overall level of Ring1b ChIP-seq tags near transcription start sites was reduced upon KAP1fl/fl knockout.
To establish if KAP1 and Ring1b binding sites at individual promoters were in closer proximity to each other than would be predicted by chance, we measured the distances between the peaks of KAP1 and Ring1b ChIP seq tags at all of the promoters that were bound by both proteins (Fig. 4D). To determine if the observed distances were shorter than would be predicted by the fact that both KAP1 and Ring1b bound near transcription start sites, we compared the observed distances with simulated distances that were generated by transposing all of the KAP1 ChIP-seq tag peaks to the opposite side of the transcription start site. The observed distances were shorter than the simulated distances, indicating that the KAP1 and Ring1b ChIP-seq tag peaks were closer to each other than was predicted by their proximities to transcription start sites alone. The close proximity of KAP1 and Ring1b binding regions near promoters suggests that their binding at these regions was interrelated.
KAP1 binds in different locations and has distinct effects on Ring1b binding at genes that were repressed versus derepressed by KAP1.
To investigate the mechanisms whereby KAP1 had opposite effects on the transcription of differentiation-inducible and pluripotency-associated genes, we compared the relative levels and distributions of KAP1 and Ring1b ChIP-seq tags between the genes that were repressed and those that were derepressed by KAP1. Genes that were repressed by KAP1 had a higher level of Ring1b ChIP-seq tags at the promoter, KAP1 ChIP-seq tags were located mostly near the promoter, and the change in the level of Ring1b ChIP-seq tags upon KAP1fl/fl knockout was larger (Fig. 4E). In contrast, genes that were derepressed by KAP1 had a lower level of Ring1b ChIP-seq tags at the promoter, KAP1 ChIP-seq tags were distributed throughout the transcribed and flanking sequences, and the change in the level of Ring1b ChIP-seq tags upon KAP1fl/fl knockout was smaller (Fig. 4F). We quantified the enrichment of KAP1 and of Ring1b ChIP-seq tags within the promoter region, as well as in transcribed and flanking sequences for each gene. The quantitative analysis confirmed that the Ring1b ChIP-seq tag enrichment was 3-fold higher, on average, at the promoters for genes that were repressed by KAP1 than at the promoters for genes that were derepressed by KAP1. In contrast, the KAP1 ChIP-seq tag enrichment was 4-fold higher, on average, in the transcribed and flanking sequences of genes that were derepressed by KAP1 than in the transcribed and flanking sequences of genes that were repressed by KAP1. It is unlikely that these differences in KAP1 and Ring1b ChIP-seq tag enrichment were consequences of differences in the transcription of genes that were repressed versus derepressed by KAP1 since the absolute levels of these transcripts varied over wide and overlapping ranges. Thus, the genes that were repressed by KAP1 had higher levels of Ring1b binding at the promoters whereas genes that were derepressed by KAP1 had higher levels of KAP1 binding in transcribed and flanking sequences.
We also quantified the change in Ring1b ChIP seq tag enrichment upon KAP1fl/fl knockout at each gene. At genes that were repressed by KAP1, the average Ring1b ChIP-seq tag enrichment was reduced 4-fold, on average, at the promoter and throughout the transcribed region upon KAP1fl/fl knockout. At genes that were derepressed by KAP1, the Ring1b ChIP-seq tag enrichment was reduced 2.5-fold, on average, at the promoter, and there was no significant change in Ring1b ChIP-seq tag enrichment within the transcribed region upon KAP1fl/fl knockout (Fig. 4F). Thus, KAP1 had a larger effect on Ring1b binding at genes that were repressed by KAP1 and a smaller and more localized effect on Ring1b binding at genes that were derepressed by KAP1.
We examined the distributions of KAP1 and Ring1b ChIP-seq tags at individual genes that were repressed or derepressed by KAP1. Among these genes, we selected two sets that represented the distinct distributions and changes in ChIP-seq tag enrichment of genes that were repressed versus derepressed by KAP1 for further investigation. In most genes that were repressed by KAP1, the KAP1 ChIP-seq tags were concentrated near the promoter and colocalized with a high level of Ring1b ChIP-seq tags (genes on the left in Fig. 4G and in Fig S2A in the supplemental material). At these promoters, the level of Ring1b ChIP-seq tags was reduced upon KAP1fl/fl knockout. In contrast, in most genes that were derepressed by KAP1, the KAP1 ChIP-seq tags were distributed throughout the transcribed and flanking sequences and a low level of Ring1b ChIP-seq tags was found at the promoter (genes on the right in Fig. 4G and in Fig. S2A in the supplemental material). At these promoters, the level of Ring1b ChIP-seq tags was reduced to a lesser extent or was enhanced upon KAP1fl/fl knockout. These groups of genes were used to test the validity of the genome- and transcriptome-wide data and to evaluate models for the opposite effects of KAP1 on differentiation-inducible versus pluripotency-associated gene transcription.
Distinct effects of KAP1 on PRC1 binding and on transcription at individual differentiation-inducible versus pluripotency-associated promoters.
The opposite effects of KAP1fl/fl knockout on the transcription of differentiation-inducible versus pluripotency-associated genes (Fig. 3) and the distinct distributions of KAP1 binding across these genes (Fig. 4A, B, and G; see Fig. S2A in the supplemental material) suggested a hypothesis for the relationship between KAP1 and PRC1 binding and their effects on transcription (Fig. 5F). According to this hypothesis, KAP1 interaction with PRC1 facilitates their binding in close proximity to each other at differentiation-inducible gene promoters and represses their transcription. In contrast, KAP1 binding to transcribed and flanking sequences of pluripotency-associated genes does not facilitate PRC1 binding at the promoters of these genes and derepresses transcription.
FIG 5.

Opposite effects of KAP1fl/fl knockout on PRC1 binding and on transcription at differentiation-inducible versus pluripotency-associated promoters in ES cells and in embryoid bodies. (A and B) Comparison of effects of KAP1fl/fl (A) and Ring1bfl/fl (B) knockouts on KAP1 and PRC1 subunit binding and on histone modifications at selected differentiation-inducible and pluripotency-associated promoters. The levels of Ring1b, Mel18, Cbx7, KAP1, H3 K4 trimethylation, and H3 K27 trimethylation were measured at the promoters indicated below the bar graphs in KAP1fl/fl and Ring1bfl/fl; Ring1a−/−cells cultured in the absence (−OHT) or presence (+OHT) of tamoxifen. The genes to the left of the dashed lines are differentiation-inducible genes that were repressed by KAP1. The genes to the right of the dashed lines are pluripotency-associated genes that were derepressed by KAP1. (C and D) Comparison of the effects of KAP1fl/fl (C) and Ring1bfl/fl (D) knockouts on the transcription of selected differentiation-inducible and pluripotency-associated genes. The levels of the transcripts indicated below the bars were quantified in WT and KAP1fl/fl or Ring1bfl/fl; Ring1a−/− ES cells cultured in the presence of tamoxifen. The bars show the ratio of the levels of the transcripts in mutant cells to those in WT cells plotted on a logarithmic scale. (E) Comparison of the effects of embryoid body (EB) formation and of KAP1fl/fl knockout in embryoid bodies on differentiation-inducible and pluripotency-associated gene transcription. The changes in the transcription of the genes indicated below the bars were quantified and KAP1fl/fl embryoid bodies that were cultured with tamoxifen for 48 h commencing at the time of LIF withdrawal, followed by culture in the absence of tamoxifen. The changes in transcript levels upon embryoid body formation were calculated by determining the ratios of transcript levels in embryoid bodies to the transcript levels in WT ES cells cultured with tamoxifen for 48 h, followed by 48 h of culture in the absence of tamoxifen. The data shown in each panel represent the means and standard deviations of three replicate qPCR measurements and are representative of two or more experiments with independently prepared embryoid bodies. (F) Model that depicts the opposite effects of KAP1 on PRC1 binding and on transcription at differentiation-inducible (orange) or pluripotency-associated (green) genes. Cooperative binding by KAP1 (blue) and PRC1 (red) at closely juxtaposed sites mediated by the coiled-coil (CC) region of KAP1 represses differentiation-inducible gene promoters. Conversely, KAP1 binding within transcribed and flanking sequences counteracts PRC1 binding independently of the coiled-coil region and derepresses pluripotency-associated gene promoters. Genetic depletion of KAP1 or the induction of differentiation shifts the balance of PRC1 binding and transcription at differentiation-inducible versus pluripotency-associated gene promoters. The sizes of the ovals reflect the relative levels of PRC1 binding in the presence or absence of KAP1.
To validate the results of ChIP-seq analysis and to test the predictions of this hypothesis, we quantified the effects of KAP1fl/fl knockout on PRC1 binding and on transcription at selected genes that were repressed by KAP1 and activated in embryoid bodies (differentiation-inducible genes) and at genes that were derepressed by KAP1 and repressed in embryoid bodies (pluripotency-associated genes). KAP1fl/fl knockout had converse effects on Ring1b and on Mel18 binding at the promoters of the differentiation-inducible versus the pluripotency-associated genes examined (Fig. 5A, upper graphs). KAP1fl/fl knockout reduced PRC1 binding at the differentiation-inducible promoters and either increased or had no significant effect on PRC1 binding at the pluripotency-associated promoters examined. Thus, both chromatin immunoprecipitation-sequencing (ChIP-seq) and ChIP-quantitative PCR (ChIP-qPCR) analyses corroborated the distinct effects of KAP1fl/fl knockout on Ring1b binding at differentiation-inducible versus pluripotency-associated promoters (see Fig. 7B).
We also compared the effects of Ring1bfl/fl knockout on KAP1 binding at the differentiation-inducible versus pluripotency-associated promoters. Ring1bfl/fl knockout reduced KAP1 binding at the differentiation-inducible promoters but increased or had no significant effect on KAP1 binding at the pluripotency-associated promoters examined (Fig. 5B, middle graph). Thus, Ring1b and KAP1 mutually facilitated binding by each other at the differentiation-inducible promoters, but they had antagonistic or neutral effects on binding by each other at the pluripotency-associated promoters tested.
There was no detectable difference in the direct effects of KAP1fl/fl knockout on KAP1 binding or of Ring1bfl/fl knockout on PRC1 binding at the differentiation-inducible versus the pluripotency-associated promoters (Fig. 5A and B). The distinct effects of KAP1fl/fl knockout on PRC1 binding and of Ring1bfl/fl knockout on KAP1 binding at the differentiation-inducible versus pluripotency-associated promoters were therefore not caused by differences in the direct effects of KAP1fl/fl or Ring1bfl/fl knockout on KAP1 or on PRC1 binding at these promoters.
To determine the functional consequences of the opposite effects of KAP1fl/fl knockout on PRC1 binding at the differentiation-inducible and pluripotency-associated genes and to test the predictions of our hypothesis, we quantified the changes in the transcription of these genes upon KAP1fl/fl knockout. KAP1fl/fl knockout increased the transcription of the differentiation-inducible genes, whereas it reduced the transcription of the pluripotency-associated genes examined (Fig. 5C). Thus, both microarray and reverse transcription (RT)-qPCR analyses corroborated the opposite effects of KAP1fl/fl knockout on differentiation-inducible versus pluripotency-associated gene transcription (see Fig. 7A). The opposite effects of KAP1fl/fl knockout on the transcription of differentiation-inducible versus pluripotency-associated genes correlated with the converse effects of KAP1fl/fl knockout on PRC1 binding at these promoters.
We also compared the effects of Ring1bfl/fl knockout on the transcription of differentiation-inducible versus pluripotency-associated genes. Ring1bfl/fl knockout increased the transcription of both the differentiation-inducible and the pluripotency-associated genes examined (Fig. 5D). These results suggest that PRC1 repressed the transcription of both the differentiation-inducible and pluripotency-associated genes examined, and that KAP1 had opposite effects on the transcription of these genes by enhancing PRC1 binding at differentiation-inducible promoters and by suppressing PRC1 binding or otherwise activating pluripotency-associated promoters. KAP1fl/fl knockout increased the level of Ring1b ChIP-seq tags at a minority of all of the pluripotency-associated promoters in the genome. It is therefore likely that additional mechanisms contributed the decrease in pluripotency-associated gene transcription upon KAP1fl/fl knockout. Nonetheless, the opposite effects of KAP1 on the transcription of differentiation-inducible versus pluripotency-associated genes correlated with converse effects of KAP1fl/fl knockout on PRC1 binding at the genes that were tested individually. Taken together, these results were consistent with cooperative KAP1 and PRC1 binding and synergistic transcription repression at the differentiation-inducible promoters, and with anti- or noncooperative KAP1 and PRC1 binding and transcription derepression by KAP1 at the pluripotency-associated promoters (Fig. 5F).
Effects of KAP1fl/fl knockout during differentiation on differentiation-inducible and pluripotency-associated gene transcription.
To determine if KAP1 regulated transcription during differentiation, we examined the changes in transcription upon embryoid body formation and the effects of KAP1fl/fl knockout on transcription in embryoid bodies. As expected on the basis of the microarray analysis (Fig. 3), the changes in transcription during embryoid body formation by WT cells were similar to the changes in transcription that were observed upon KAP1fl/fl knockout in ES cells (compare crosshatched bars in Fig. 5E and C). The level of KAP1 transcripts decreased during embryoid body formation, but the decrease was smaller than that caused by conditional KAP1fl/fl knockout in ES cells. The close correspondence between the changes in transcription upon KAP1fl/fl knockout and upon embryoid body differentiation suggest that the changes in transcription during differentiation were mediated in part by changes in KAP1 transcriptional activity.
To determine if the changes in transcription upon KAP1fl/fl knockout were affected by differentiation, we examined the effects of KAP1fl/fl knockout on transcription in differentiating cells. When KAP1fl/fl knockout was combined with embryoid body formation there was a larger increase in the transcription of the genes whose transcription was induced and a larger decrease in the transcription of the genes whose transcription was suppressed upon embryoid body formation (Fig. 5E). Parallel effects of KAP1fl/fl knockout and of embryoid body formation were observed on the transcription of almost all of the differentiation-inducible, as well as pluripotency-associated genes tested. KAP1fl/fl knockout depleted the level of KAP1 with similar efficiencies in ES cells and during embryoid body formation (see Fig. S3A in the supplemental material). Thus, KAP1fl/fl knockout had opposite effects on the transcription of differentiation-inducible versus pluripotency-associated genes during embryoid body formation, as well as in ES cells.
The effects of KAP1fl/fl knockout on differentiation-inducible and pluripotency-associated gene transcription could, in principle, be due to changes in cell proliferation. KAP1fl/fl knockout reduced ES cell proliferation, but it had no detectable effect on cell proliferation during embryoid body formation (see Fig. S3B in the supplemental material). The effects of KAP1fl/fl knockout on transcription in embryoid bodies are therefore unlikely to be due to changes in cell proliferation. Multiple independent lines of evidence are consistent with the hypothesis that KAP1 and PRC1 regulate both differentiation-inducible and pluripotency-associated gene transcription through direct binding.
Effects of the interaction with KAP1 on PRC1 binding and on transcription at differentiation-inducible versus pluripotency-associated promoters.
We investigated the roles of the interaction between KAP1 and PRC1 at the differentiation-inducible and pluripotency-associated promoters. We depleted endogenous KAP1 by tamoxifen treatment of KAP1fl/fl cells that expressed either intact KAP1 or KAP1ΔCC, which does not interact with PRC1 (Fig. 1G and H). We compared the effects of intact KAP1 versus KAP1ΔCC expression on PRC1 binding and on transcription at the differentiation-inducible versus pluripotency-associated gene promoters before and after endogenous KAP1fl/fl knockout. Intact KAP1 and KAP1ΔCC were expressed at levels that were comparable to each other and slightly lower than the level of endogenous KAP1 (see Fig. S4A in the supplemental material).
At the differentiation-inducible promoters, intact KAP1 and KAP1ΔCC expression had little effect on the overall level of KAP1 binding before KAP1fl/fl knockout. After endogenous KAP1fl/fl knockout, both intact KAP1 and KAP1ΔCC expression partially restored KAP1 binding with similar efficiencies (Fig. 6A, left side of bottom bar graph). Intact KAP1 expression had little effect on Ring1b or Cbx7 binding at the differentiation-inducible promoters before KAP1fl/fl knockout and partially restored both Ring1b and Cbx7 binding after KAP1fl/fl knockout (Fig. 6A, left side of upper bar graphs). In contrast, KAP1ΔCC expression reduced endogenous Ring1b and Cbx7 binding at the differentiation-inducible promoters before KAP1fl/fl knockout and did not enhance Ring1b or Cbx7 binding after KAP1fl/fl knockout. The reduction in endogenous Ring1b and Cbx7 binding in cells that expressed both endogenous KAP1 and ectopic KAP1ΔCC is consistent with the competition for chromatin binding by the native and mutant proteins. Independent ES cell clones that expressed intact KAP1 versus KAP1ΔCC exhibited the same differences in PRC1 binding at the differentiation-inducible promoters upon KAP1fl/fl knockout (see Fig. S4B in the supplemental material). Deletion of the coiled-coil region of KAP1 therefore eliminated the enhancement of PRC1 binding at the differentiation-inducible promoters that was produced by WT KAP1 expression. These results suggest that direct interaction with KAP1 enhanced endogenous PRC1 binding at differentiation-inducible promoters (Fig. 5F).
FIG 6.

Effects of the coiled-coil interaction interface of KAP1 on PRC1 binding and on transcription at differentiation-inducible versus pluripotency-associated promoters. (A) Effects of intact KAP1 and KAP1ΔCC expression in KAP1fl/fl knockout cells on PRC1 binding at differentiation-inducible versus pluripotency-associated promoters. The levels of Ring1b, Cbx7, and KAP1 binding and of H3 K27 trimethylation were measured at the genes indicated below the bar graphs. The ChIP-qPCR signals were compared in KAP1fl/fl cells that expressed no ectopic KAP1 (white and black), intact KAP1 (red; clone 13), or KAP1ΔCC (blue; clone 15). These cells were cultured in the absence (−OHT) and in the presence (+OHT) of tamoxifen. (B) Effects of intact KAP1 and KAP1ΔCC expression in KAP1fl/fl knockout cells on the transcription of differentiation-inducible or pluripotency-associated genes. The changes in the transcription of the genes indicated below the bars upon KAP1fl/fl knockout (+OHT/−OHT) were compared between KAP1fl/fl cells that expressed no ectopic KAP1 (black), intact KAP1 (red; clone 13), or KAP1ΔCC (blue; clone 15). The data in each panel represent the means and standard deviations of three replicate qPCR measurements and are representative of two experiments that were performed with different ES cell clones.
At the pluripotency-associated promoters, the expression of either intact KAP1 or KAP1ΔCC increased the overall level of KAP1 binding both before and especially after endogenous KAP1fl/fl knockout (Fig. 6A, right side of bottom bar graph). Both intact KAP1, as well as KAP1ΔCC expression reduced endogenous Ring1b and Cbx7 binding at the pluripotency-associated promoters before and after KAP1fl/fl knockout. Deletion of the coiled-coil region of KAP1 therefore did not prevent the inhibition of PRC1 binding at the pluripotency-associated promoters by KAP1 expression. These observations indicate that KAP1 inhibition of PRC1 binding at the pluripotency-associated genes did not require a direct interaction between KAP1 and PRC1 (Fig. 5F).
We also compared the effects of intact KAP1 and of KAP1ΔCC expression on the transcription of the differentiation-inducible and pluripotency-associated genes. Intact KAP1 expression repressed the differentiation-inducible genes and derepressed the pluripotency-associated genes following KAP1fl/fl knockout (Fig. 6B). In contrast, KAP1ΔCC expression did not repress the differentiation-inducible genes but derepressed the pluripotency-associated genes as efficiently as intact KAP1 expression following KAP1fl/fl knockout. The same differences between the effects of KAP1 and KAP1ΔCC expression on differentiation-inducible versus pluripotency-associated gene transcription were observed at 12 additional genes, as well as in independent ES cell clones (Fig. 6B; see Fig. S4C in the supplemental material). The coiled-coil region was required for KAP1 repression of differentiation-inducible gene transcription, but it was not required for KAP1 derepression of pluripotency-associated gene transcription. The distinct roles of the coiled-coil region in KAP1 repression of differentiation-inducible genes and in KAP1 derepression of pluripotency-associated genes further demonstrate that these effects were independent of each other and were therefore not indirect consequences of each other.
Genome-wide relationships between KAP1 and Ring1b binding and the changes in transcription upon KAP1fl/fl knockout.
In order to investigate the relationships between the levels of KAP1 and Ring1b binding and of the changes in transcription upon KAP1fl/fl knockout throughout the genome, it was necessary to first determine if the changes in microarray signals and ChIP-seq tag counts were valid proxies for the changes in transcription and protein binding. We compared the changes in transcript levels upon KAP1fl/fl knockout as detected by microarray analysis with the changes in the transcription of overlapping sequences as detected by RT-qPCR at 24 genes. Opposite changes in the levels of differentiation-inducible and pluripotency-associated transcripts upon KAP1fl/fl knockout were observed for almost all of the transcripts examined with both assays, although the absolute changes in transcript levels detected by ChIP-qPCR were generally larger than the changes detected by microarray analysis (Fig. 7A). We also compared the changes in the levels of Ring1b binding upon KAP1fl/fl knockout detected by ChIP-qPCR with the changes in Ring1b ChIP-seq tag enrichment at 16 promoters. Opposite changes in Ring1b binding at differentiation-inducible versus pluripotency-associated promoters upon KAP1fl/fl knockout were observed at almost all of the promoters in both assays (Fig. 7B). The magnitudes of the changes in transcript levels and in Ring1b binding at individual genes varied when they were measured with different assays. To overcome this experimental variability, we compared the average changes in Ring1b binding and in transcription upon KAP1fl/fl knockout between groups of genes that had similar distributions or levels of KAP1 binding and between groups of genes with similar levels of Ring1b binding or changes in Ring1b binding upon KAP1fl/fl knockout (Fig. 7C to E).
We investigated the relationship between the distribution of KAP1 binding and the change in Ring1b binding upon KAP1fl/fl knockout by grouping all of the genes that were bound by KAP1 and Ring1b on the basis of the ratio of the enrichment of KAP1 ChIP-seq tags at the promoter to the enrichment of KAP1 ChIP-seq tags in the downstream transcribed sequence. We then plotted the average change in the level of Ring1b ChIP-seq tags upon KAP1fl/fl knockout for each group of genes as a function of the ratio of the density of KAP1 ChIP-seq tags at the promoter to the density of KAP1 ChIP-seq tags in the transcribed region (Fig. 7C). Genes with a high proportion of KAP1 binding at the promoter exhibited a larger decrease in Ring1b binding upon KAP1fl/fl knockout, whereas genes with a low proportion of KAP1 binding at the promoter exhibited a smaller decrease in Ring1b binding upon KAP1fl/fl knockout, on average. The relationship between the distribution of KAP1 binding and the change in Ring1b binding upon KAP1fl/fl knockout suggested that KAP1 had distinct effects on Ring1b binding at genes where it bound preferentially to different regions of the genes.
We investigated the relationships between the levels of KAP1 and Ring1b binding and the changes in transcription upon KAP1fl/fl knockout by grouping all of the genes that were bound by KAP1 and Ring1b on the basis of the enrichment of KAP1, as well as of Ring1b ChIP-seq tags in the gene locus and within the promoter, respectively. We then plotted the average change in transcription upon KAP1fl/fl knockout for each group of genes measured by microarray analysis as a function of the enrichment of KAP1, as well as of Ring1b ChIP-seq tags (Fig. 7D and E). The transcription of genes that had higher levels of KAP1 binding in transcribed and flanking sequences decreased upon KAP1fl/fl knockout, whereas the transcription of genes that had lower levels of KAP1 binding in these regions increased upon KAP1fl/fl knockout, on average (Fig. 7D). Conversely, KAP1fl/fl knockout increased the transcription of genes that had higher levels of Ring1b binding at the promoter, whereas KAP1fl/fl knockout had little effect on transcription from promoters that had lower levels of Ring1b binding, on average (Fig. 7E). Consequently, higher levels of KAP1 binding in transcribed and flanking sequences were associated with transcription derepression by KAP1, whereas higher levels of Ring1b binding at promoters were associated with transcription repression by KAP1, on average.
We investigated the relationship between the changes in Ring1b binding and the changes in transcription upon KAP1fl/fl knockout by grouping all of the genes that were bound by KAP1 and Ring1b on the basis of the change in Ring1b ChIP-seq tags at the promoter upon KAP1fl/fl knockout. We then plotted the average change in transcription upon KAP1fl/fl knockout for each group of genes measured by microarray analysis as a function of the change in Ring1b ChIP-seq tag enrichment (Fig. 7F). KAP1fl/fl knockout increased transcription from promoters where it caused a larger decrease in Ring1b binding, on average. In contrast KAP1fl/fl knockout reduced transcription from promoters where it caused a smaller decrease or an increase in Ring1b binding, on average. The opposite effects of KAP1fl/fl knockout on the transcription of differentiation-inducible versus pluripotency-associated genes correlated with distinct changes in Ring1b binding at these promoters.
Integration of positive and negative effects of KAP1 on PRC1 binding and transcription.
The effects of KAP1 on PRC1 binding and on transcription varied continuously from strong enhancement of PRC1 binding and repression of transcription to strong suppression of PRC1 binding and derepression of transcription at different genes. Ectopic expression of KAP1 and KAP1ΔCC had opposite effects on PRC1 binding at differentiation-inducible gene promoters, suggesting that KAP1 had both positive and negative effects on PRC1 binding at individual genes (Fig. 6A, left side of upper graphs). KAP1 bound both the promoter and the transcribed and flanking sequences of many genes (Fig. 4G; see Fig. S2A in the supplemental material). The overall effect of KAP1 on PRC1 binding at each promoter likely reflected a combination of positive effects of KAP1 interaction with PRC1 at the promoter, and of negative effects of KAP1 binding throughout transcribed and flanking sequences.
Relationships between KAP1 and PRC1 binding and histone modifications at differentiation-inducible and pluripotency-associated genes.
KAP1fl/fl knockout reduced H3 K27 trimethylation at the differentiation-inducible promoters and increased it at the pluripotency-associated promoters (Fig. 5A). Intact KAP1 but not KAP1ΔCC expression restored H3 K27 trimethylation at the differentiation-inducible promoters, whereas both intact KAP1 and KAP1ΔCC expression suppressed H3 K27 trimethylation at the pluripotency-associated promoters (Fig. 6A). Ring1bfl/fl knockout reduced H3 K27 trimethylation at both the differentiation-inducible and pluripotency-associated promoters (Fig. 5B). Since both KAP1fl/fl and Ring1bfl/fl knockouts reduced H3 K27 trimethylation at the differentiation-inducible promoters and since the coiled-coil region that mediated KAP1 interaction with PRC1 was required for KAP1 to enhance both PRC1 binding and H3 K27 trimethylation at these promoters, it is likely that KAP1 and Ring1b enhanced H3 K27 trimethylation through cooperative binding at differentiation-inducible promoters. Conversely, since KAP1fl/fl knockout increased and Ring1bfl/fl knockout reduced H3 K27 trimethylation at the pluripotency-associated promoters and since the coiled-coil region of KAP1 was not required for KAP1 inhibition of either PRC1 binding or H3 K27 trimethylation at these promoters, it is likely that KAP1 suppressed both PRC1 binding and H3 K27 trimethylation at pluripotency-associated promoters through a mechanism that did not require direct KAP1-PRC1 interaction.
KAP1fl/fl knockout increased H3 K4 trimethylation at the differentiation-inducible promoters and reduced it at the pluripotency-associated promoters (Fig. 5A). It is likely that the reciprocal effects of KAP1fl/fl knockout on H3 K4 and H3 K27 trimethylation at the differentiation-inducible versus pluripotency-associated promoters were consequences of the changes in transcription at these promoters resulting from the effects of KAP1 on PRC1 binding.
KAP1fl/fl knockout had no consistent effect on the low levels of H3 K9 trimethylation observed at the promoters examined (Fig. 2A). In contrast, KAP1fl/fl knockout reduced the much higher level of H3 K9 trimethylation observed near an endogenous retrovirus. Consequently, it is unlikely that the opposite effects of KAP1 and PRC1 on binding by each other and on transcription at the differentiation-inducible and pluripotency-associated promoters were mediated by changes in these histone modifications.
DISCUSSION
Many independent experimental approaches identified an interaction between KAP1 and PRC1 and demonstrated that this interaction was required for cooperative binding and for concerted transcription repression at differentiation-inducible genes. KAP1 was required both for the repression of differentiation-inducible genes and for the derepression of pluripotency-associated genes in ES cells. The opposite effects of KAP1 on the transcription of these genes were determined in part by cooperative KAP1 and PRC1 binding at the differentiation-inducible promoters and by anti- or noncooperative KAP1 and PRC1 binding at the pluripotency-associated genes.
The interaction between KAP1 and PRC1 was independent of the interactions between KAP1 and other DNA- and chromatin-binding proteins that can interact with the N- and C-terminal domains of KAP1. Whereas KAP1 interactions with KRAB domain zinc finger proteins, HP1, Setdb1, and other partners could contribute to the regulation of differentiation-inducible and pluripotency-associated gene transcription in ES cells, only the central coiled-coil region of KAP1 was essential for KAP1 interaction with PRC1. Since the coiled-coil region of KAP1 was essential for KAP1-PRC1 interaction but was not required for chromatin binding, the fact that both KAP1 and PRC1 bind to chromatin was not sufficient to produce the interactions detected in cell extracts and in living cells. The coiled-coil region of KAP1 can also interact with E2F1 (21), but E2F1 was not detected in the PRC1-KAP1 complexes isolated from ES cells. The interaction between KAP1 and PRC1 is a novel mechanism that regulates their chromatin binding specificities and transcriptional activities.
The opposite effects of KAP1 both on PRC1 binding and on transcription at many differentiation-inducible versus pluripotency-associated genes indicated that both transcription repression and derepression by KAP1 correlated with changes in PRC1 binding (Fig. 5F). Moreover, both enhanced PRC1 binding and transcription repression at the differentiation-inducible genes required the coiled-coil region of KAP1, whereas this region was not required for either KAP1 inhibition of PRC1 binding or KAP1 derepression of transcription at pluripotency-associated genes. The functions of KAP1 in the repression of differentiation-inducible genes and in the derepression of pluripotency-associated genes were both associated with changes in PRC1 binding.
The opposite effects of KAP1 on PRC1 binding and on transcription at the differentiation-inducible versus pluripotency-associated genes correlated with the distinct distributions of KAP1 binding at these genes. At differentiation-inducible genes, KAP1 binding regions overlapped Ring1b binding regions near promoters. KAP1 and PRC1 reciprocally enhanced binding by each other at these promoters, suggesting that both complexes contributed to chromatin binding. Both KAP1 and PRC1 repressed the transcription of the differentiation-inducible genes. The enhancement of PRC1 binding and the repression of transcription by KAP1 at differentiation-inducible promoters required the coiled-coil region that mediated KAP1-PRC1 interaction. The close juxtaposition of the KAP1 and Ring1b binding regions and the requirement for their direct interaction are consistent with synergistic transcription repression by cooperative KAP1 and PRC1 binding at adjacent sites on chromatin.
At pluripotency-associated genes, KAP1 binding regions were dispersed throughout the transcribed and flanking sequences and often overlapped regions that had low levels of Ring1b binding. KAP1 antagonized PRC1 binding and Ring1b did not affect KAP1 binding at many pluripotency-associated gene promoters. KAP1 derepressed the transcription of the pluripotency-associated genes, whereas Ring1b repressed their transcription. The inhibition of PRC1 binding and the derepression of pluripotency-associated gene transcription by KAP1 did not require the coiled-coil region that mediated KAP1-PRC1 interaction. The wide dispersion of KAP1 binding regions and the lack of a requirement for direct interaction with PRC1 at pluripotency-associated genes are consistent with long-range effects of KAP1 on PRC1 binding and on transcription through interactions with additional factors. The genes at which KAP1 repressed and activated transcription therefore differed in the distributions of KAP1 binding, the regions of KAP1 that were required, and the effects of KAP1 on PRC1 binding. The mechanisms that specify the distinct distributions of KAP1 binding at differentiation-inducible versus pluripotency-associated genes are unknown but are likely to include interactions with different sequence-specific DNA-binding proteins.
Previous studies of KAP1 binding have shown that KAP1 can bind to promoters in ES cells (17), to promoters and the 3′ ends of transcribed sequences in HEK293T cells (40), and to endogenous retroviral sequences in ES cells (50). Of the 3,331 KAP1 binding regions that were identified previously by microarray analysis of promoter regions in ES cells (17), more than 90% overlapped KAP1 binding regions identified in our genome-wide ChIP-seq analysis and more than 85% overlapped both KAP1 and Ring1b binding regions. About 90 and 60% of the mouse homologues of the genes whose transcription increased or decreased the most upon KAP1 knockdown in Ntera2 cells and that were bound by KAP1 in HEK293T cells (40) contained KAP1 and Ring1b binding regions identified in our genome-wide analysis. Lastly, whereas only 36 ES cell promoters were interpreted previously to bind KAP1 through sequences that were not endogenous retroviruses (50), our genome-wide analysis identified KAP1 and Ring1b binding regions in 83% of these promoters in addition to 4,427 other promoters that did not overlap retroviral sequences as defined by Repeatmasker. Likewise, a majority (78%) of the intragenic KAP1 binding regions did not overlap retroviral sequences, whereas a large proportion (58%) of the intergenic KAP1 binding regions overlapped retroviral sequences. Thus, the KAP1 binding regions in promoter and transcribed sequences that determined the influence of KAP1 on PRC1 binding and on transcription were predominantly nonretroviral in origin.
The opposite effects of KAP1 on PRC1 binding and on transcription at different genes indicated that PRC1 binding is regulated by mechanisms that are more complex than simple recruitment. Previous studies of the effects of REST on PRC1 binding and on transcription at different genes demonstrated that REST has opposite effects on PRC1 binding and on transcription at regulatory elements that are located at different distances from promoters (6). KRAB domain fusion proteins that can interact with KAP1 also had distinct effects on transcription when they bound to reporter constructs that were integrated at different genomic locations in HeLa cells (51). The opposite effects of both KAP1 and REST on PRC1 binding and on transcription at different genes indicate that they regulate PRC1 binding through multiple mechanisms that have both positive and negative effects on PRC1 binding. The level of PRC1 binding at each promoter is likely to be determined by the combined synergistic and antagonistic effects of many different interaction partners.
KAP1 bound directly both to the differentiation-inducible genes whose transcription increased upon KAP1fl/fl knockout and to the pluripotency-associated genes whose transcription decreased upon KAP1fl/fl knockout. The derepression of pluripotency-associated genes by KAP1 was not an indirect consequence of the repression of differentiation-inducible genes or vice versa since deletion of the coiled-coil region of KAP1 had distinct effects on both PRC1 binding and transcription at these groups of genes. The repression of differentiation-inducible genes and the derepression of pluripotency-associated genes by KAP1 were also not indirect consequences of differentiation since KAP1 had similar effects on their transcription in ES cells and during embryoid body formation. The requirement for KAP1 in the maintenance of ES cell pluripotency is likely to be due to its roles both in the repression of differentiation-inducible genes and in the derepression of pluripotency-associated genes.
The simultaneous repression and derepression of different genes by KAP1 can coordinate opposite changes in transcription during differentiation. KAP1 and PRC1 are required for the self-renewal or differentiation of many classes of multipotent stem cells in mice (30–37, 52). Most transitions between progenitor and differentiated cell types involve the coordinated derepression of differentiation-inducible genes and the repression of progenitor-specific genes. The derepression or repression of one group of genes without a concomitant change in the transcription of the other group of genes could cause aberrant differentiation. The reciprocal regulation of genes that promote stemness versus differentiation by KAP1 and other transcription factor complexes can control transitions between successive stages of development.
Supplementary Material
ACKNOWLEDGMENTS
We thank Didier Trono, Haruhiko Koseki, Yael Ziv, and Danny Reinberg for materials; Yanqing Liu for generating the PTRE-shMga ES cell line and preparing the anti-Mga antibodies; Thom Saunders of the University of Michigan Transgenic Animal Model Core for assistance with the isolation of Cbx7−/− ES cells; Craig Johnson of the University of Michigan DNA sequencing core for assistance with microarray analysis; and members of the Kerppola laboratory for sharing reagents and for constructive criticisms.
This work was funded by grants from the National Institute of General Medical Sciences (GM086213) and the National Institute on Drug Abuse (DA030339).
Footnotes
Published ahead of print 31 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01729-13.
REFERENCES
- 1.Kerppola TK. 2009. Polycomb group complexes—many combinations, many functions. Trends Cell Biol. 19:692–704. 10.1016/j.tcb.2009.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ogawa H, Ishiguro K, Gaubatz S, Livingston DM, Nakatani Y. 2002. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G(0) cells. Science 296:1132–1136. 10.1126/science.1069861 [DOI] [PubMed] [Google Scholar]
- 3.Gearhart MD, Corcoran CM, Wamstad JA, Bardwell VJ. 2006. Polycomb group and SCF ubiquitin ligases are found in a novel BCOR complex that is recruited to BCL6 targets. Mol. Cell. Biol. 26:6880–6889. 10.1128/MCB.00630-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sánchez C, Sánchez I, Demmers JAA, Rodriguez P, Strouboulis J, Vidal M. 2007. Proteomics analysis of Ring1B/Rnf2 interactors identifies a novel complex with the Fbxl10/Jhdm1B histone demethylase and the Bcl6 interacting corepressor. Mol. Cell. Proteomics 6:820–834. 10.1074/mcp.M600275-MCP200 [DOI] [PubMed] [Google Scholar]
- 5.Boukarabila H, Saurin AJ, Batsche E, Mossadegh N, van Lohuizen M, Otte AP, Pradel J, Muchardt C, Sieweke M, Duprez E. 2009. The PRC1 polycomb group complex interacts with PLZF/RARA to mediate leukemic transformation. Genes Dev. 23:1195–1206. 10.1101/gad.512009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ren X, Kerppola TK. 2011. REST interacts with Cbx proteins and regulates polycomb repressive complex 1 occupancy at RE1 elements. Mol. Cell. Biol. 31:2100–2110. 10.1128/MCB.05088-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vandamme J, Volkel P, Rosnoblet C, Le Faou P, Angrand PO. 2011. Interaction proteomics analysis of polycomb proteins defines distinct PRC1 complexes in mammalian cells. Mol. Cell. Proteomics 10:M110.002642. 10.1074/mcp.M110.002642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Endoh M, Endo TA, Endoh T, Fujimura YI, Ohara O, Toyoda T, Otte AP, Okano M, Brockdorff N, Vidal M, Koseki H. 2008. Polycomb group proteins Ring1A/B are functionally linked to the core transcriptional regulatory circuitry to maintain ES cell identity. Development 135:1513–1524. 10.1242/dev.014340 [DOI] [PubMed] [Google Scholar]
- 9.Dietrich N, Lerdrup M, Landt E, Agrawal-Singh S, Bak M, Tommerup N, Rappsilber J, Sodersten E, Hansen K. 2012. REST-mediated recruitment of polycomb repressor complexes in mammalian cells. PLoS Genet. 8:e1002494. 10.1371/journal.pgen.1002494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farcas AM, Blackledge NP, Sudbery I, Long HK, McGouran JF, Rose NR, Lee S, Sims D, Cerase A, Sheahan TW, Koseki H, Brockdorff N, Ponting CP, Kessler BM, Klose RJ. 2012. KDM2B links the polycomb repressive complex 1 (PRC1) to recognition of CpG islands. eLife 1:e00205. 10.7554/eLife.00205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu X, Johansen Jens V, Helin K. 2013. Fbxl10/Kdm2b recruits polycomb repressive complex 1 to CpG islands and regulates H2A ubiquitylation. Mol. Cell 49:1134–1146. 10.1016/j.molcel.2013.01.016 [DOI] [PubMed] [Google Scholar]
- 12.He J, Shen L, Wan M, Taranova O, Wu H, Zhang Y. 2013. Kdm2b maintains murine embryonic stem cell status by recruiting PRC1 complex to CpG islands of developmental genes. Nat. Cell Biol. 15:373–384. 10.1038/ncb2702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schoeftner S, Sengupta AK, Kubicek S, Mechtler K, Spahn L, Koseki H, Jenuwein T, Wutz A. 2006. Recruitment of PRC1 function at the initiation of X inactivation independent of PRC2 and silencing. EMBO J. 25:3110–3122. 10.1038/sj.emboj.7601187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vincenz C, Kerppola TK. 2008. Different polycomb group CBX family proteins associate with distinct regions of chromatin using nonhomologous protein sequences. Proc. Natl. Acad. Sci. U. S. A. 105:16572–16577. 10.1073/pnas.0805317105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Puschendorf M, Terranova R, Boutsma E, Mao XH, Isono KI, Brykczynska U, Kolb C, Otte AP, Koseki H, Orkin SH, van Lohuizen M, Peters AHFM. 2008. PRC1 and Suv39h specify parental asymmetry at constitutive heterochromatin in early mouse embryos. Nat. Genet. 40:411–420. 10.1038/ng.99 [DOI] [PubMed] [Google Scholar]
- 16.Tavares L, Dimitrova E, Oxley D, Webster J, Poot R, Demmers J, Bezstarosti K, Taylor S, Ura H, Koide H, Wutz A, Vidal M, Elderkin S, Brockdorff N. 2012. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell 148:664–678. 10.1016/j.cell.2011.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu G, Kim J, Xu Q, Leng Y, Orkin SH, Elledge SJ. 2009. A genome-wide RNAi screen identifies a new transcriptional module required for self-renewal. Genes Dev. 23:837–848. 10.1101/gad.1769609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Friedman JR, Fredericks WJ, Jensen DE, Speicher DW, Huang XP, Neilson EG, Rauscher FJ., III 1996. KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev. 10:2067–2078. 10.1101/gad.10.16.2067 [DOI] [PubMed] [Google Scholar]
- 19.Kim SS, Chen YM, O'Leary E, Witzgall R, Vidal M, Bonventre JV. 1996. A novel member of the RING finger family, KRIP-1, associates with the KRAB-A transcriptional repressor domain of zinc finger proteins. Proc. Natl. Acad. Sci. U. S. A. 93:15299–15304. 10.1073/pnas.93.26.15299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ryan RF, Schultz DC, Ayyanathan K, Singh PB, Friedman JR, Fredericks WJ, Rauscher FJ., III 1999. KAP-1 corepressor protein interacts and colocalizes with heterochromatic and euchromatic HP1 proteins: a potential role for Krüppel-associated box-zinc finger proteins in heterochromatin-mediated gene silencing. Mol. Cell. Biol. 19:4366–4378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang C, Rauscher FJ, III, Cress WD, Chen J. 2007. Regulation of E2F1 function by the nuclear corepressor KAP1. J. Biol. Chem. 282:29902–29909. 10.1074/jbc.M704757200 [DOI] [PubMed] [Google Scholar]
- 22.Cammas F, Mark M, Dolle P, Dierich A, Chambon P, Losson R. 2000. Mice lacking the transcriptional corepressor TIF1beta are defective in early postimplantation development. Development 127:2955–2963 [DOI] [PubMed] [Google Scholar]
- 23.Voncken JW, Roelen BA, Roefs M, de Vries S, Verhoeven E, Marino S, Deschamps J, van Lohuizen M. 2003. Rnf2 (Ring1b) deficiency causes gastrulation arrest and cell cycle inhibition. Proc. Natl. Acad. Sci. U. S. A. 100:2468–2473. 10.1073/pnas.0434312100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leeb M, Wutz A. 2007. Ring1B is crucial for the regulation of developmental control genes and PRC1 proteins but not X inactivation in embryonic cells. J. Cell Biol. 178:219–229. 10.1083/jcb.200612127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van der Stoop P, Boutsma EA, Hulsman D, Noback S, Heimerikx M, Kerkhoven RM, Voncken JW, Wessels LF, van Lohuizen M. 2008. Ubiquitin E3 ligase Ring1b/Rnf2 of polycomb repressive complex 1 contributes to stable maintenance of mouse embryonic stem cells. PLoS One 3:e2235. 10.1371/journal.pone.0002235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rowe HM, Jakobsson J, Mesnard D, Rougemont J, Reynard S, Aktas T, Maillard PV, Layard-Liesching H, Verp S, Marquis J, Spitz F, Constam DB, Trono D. 2010. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 463:237–240. 10.1038/nature08674 [DOI] [PubMed] [Google Scholar]
- 27.Leeb M, Pasini D, Novatchkova M, Jaritz M, Helin K, Wutz A. 2010. Polycomb complexes act redundantly to repress genomic repeats and genes. Genes Dev. 24:265–276. 10.1101/gad.544410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maksakova IA, Thompson PJ, Goyal P, Jones SJ, Singh PB, Karimi MM, Lorincz MC. 2013. Distinct roles of KAP1, HP1 and G9a/GLP in silencing of the two-cell-specific retrotransposon MERVL in mouse ES cells. Epigenetics Chromatin 6:15. 10.1186/1756-8935-6-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herzog M, Wendling O, Guillou F, Chambon P, Mark M, Losson R, Cammas F. 2011. TIF1beta association with HP1 is essential for post-gastrulation development, but not for Sertoli cell functions during spermatogenesis. Dev. Biol. 350:548–558. 10.1016/j.ydbio.2010.12.014 [DOI] [PubMed] [Google Scholar]
- 30.Molofsky AV, Pardal R, Iwashita T, Park IK, Clarke MF, Morrison SJ. 2003. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature 425:962–967. 10.1038/nature02060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park IK, Qian DL, Kiel M, Becker MW, Pihalja M, Weissman IL, Morrison SJ, Clarke MF. 2003. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 423:302–305. 10.1038/nature01587 [DOI] [PubMed] [Google Scholar]
- 32.Iwama A, Oguro H, Negishi M, Kato Y, Morita Y, Tsukui H, Ema H, Kamijo T, Katoh-Fukui Y, Koseki H, van Lohuizen M, Nakauchi H. 2004. Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity 21:843–851. 10.1016/j.immuni.2004.11.004 [DOI] [PubMed] [Google Scholar]
- 33.Pietersen AM, Evers B, Prasad AA, Tanger E, Cornelissen-Steijger P, Jonkers J, van Lohuizen M. 2008. Bmi1 regulates stem cells and proliferation and differentiation of committed cells in mammary epithelium. Curr. Biol. 18:1094–1099. 10.1016/j.cub.2008.06.070 [DOI] [PubMed] [Google Scholar]
- 34.Zhou X-F, Yu J, Chang M, Zhang M, Zhou D, Cammas F, Sun S-C. 2012. TRIM28 mediates chromatin modifications at the TCRα enhancer and regulates the development of T and natural killer T cells. Proc. Natl. Acad. Sci. U. S. A. 109:20083–20088. 10.1073/pnas.1214704109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barde I, Rauwel B, Marin-Florez RM, Corsinotti A, Laurenti E, Verp S, Offner S, Marquis J, Kapopoulou A, Vanicek J, Trono D. 2013. A KRAB/KAP1-miRNA cascade regulates erythropoiesis through stage-specific control of mitophagy. Science 340:350–353. 10.1126/science.1232398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santoni de Sio FR, Massacand J, Barde I, Offner S, Corsinotti A, Kapopoulou A, Bojkowska K, Dagklis A, Fernandez M, Ghia P, Thomas JH, Pinschewer D, Harris N, Trono D. 2012. KAP1 regulates gene networks controlling mouse B-lymphoid cell differentiation and function. Blood 119:4675–4685. 10.1182/blood-2011-12-401117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bojkowska K, Aloisio F, Cassano M, Kapopoulou A, Santoni de Sio F, Zangger N, Offner S, Cartoni C, Thomas C, Quenneville S, Johnsson K, Trono D. 2012. Liver-specific ablation of Krüppel-associated box-associated protein 1 in mice leads to male-predominant hepatosteatosis and development of liver adenoma. Hepatology 56:1279–1290. 10.1002/hep.25767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, Bell GW, Otte AP, Vidal M, Gifford DK, Young RA, Jaenisch R. 2006. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441:349–353. 10.1038/nature04733 [DOI] [PubMed] [Google Scholar]
- 39.Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS, Adli M, Kasif S, Ptaszek LM, Cowan CA, Lander ES, Koseki H, Bernstein BE. 2008. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 4:e1000242. 10.1371/journal.pgen.1000242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iyengar S, Ivanov AV, Jin VX, Rauscher FJ, III, Farnham PJ. 2011. Functional analysis of KAP1 genomic recruitment. Mol. Cell. Biol. 31:1833–1847. 10.1128/MCB.01331-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quenneville S, Verde G, Corsinotti A, Kapopoulou A, Jakobsson J, Offner S, Baglivo I, Pedone PV, Grimaldi G, Riccio A, Trono D. 2011. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol. Cell 44:361–372. 10.1016/j.molcel.2011.08.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu M, Mazor T, Huang H, Huang HT, Kathrein KL, Woo AJ, Chouinard CR, Labadorf A, Akie TE, Moran TB, Xie H, Zacharek S, Taniuchi I, Roeder RG, Kim CF, Zon LI, Fraenkel E, Cantor AB. 2012. Direct recruitment of polycomb repressive complex 1 to chromatin by core binding transcription factors. Mol. Cell 45:330–343. 10.1016/j.molcel.2011.11.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker-Jensen S, Bartek J, Shiloh Y. 2006. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol. 8:870–876. 10.1038/ncb1446 [DOI] [PubMed] [Google Scholar]
- 44.Ren X, Vincenz C, Kerppola TK. 2008. Changes in the distributions and dynamics of polycomb repressive complexes during embryonic stem cell differentiation. Mol. Cell. Biol. 28:2884–2895. 10.1128/MCB.00949-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elderkin S, Maertens GN, Endoh M, Mallery DL, Morrice N, Koseki H, Peters G, Brockdorff N, Hiom K. 2007. A phosphorylated form of mel-18 targets the Ring1B histone H2A ubiquitin ligase to chromatin. Mol. Cell 28:107–120. 10.1016/j.molcel.2007.08.009 [DOI] [PubMed] [Google Scholar]
- 46.Katoh-Fukui Y, Tsuchiya R, Shiroishi T, Nakahara Y, Hashimoto N, Noguchi K, Higashinakagawa T. 1998. Male-to-female sex reversal in M33 mutant mice. Nature 393:688–692. 10.1038/31482 [DOI] [PubMed] [Google Scholar]
- 47.Gao Z, Zhang J, Bonasio R, Strino F, Sawai A, Parisi F, Kluger Y, Reinberg D. 2012. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell 45:344–356. 10.1016/j.molcel.2012.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu CD, Chinenov Y, Kerppola TK. 2002. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 9:789–798. 10.1016/S1097-2765(02)00496-3 [DOI] [PubMed] [Google Scholar]
- 49.Doss M, Chen S, Winkler J, Hippler-Altenburg R, Odenthal M, Wickenhauser C, Balaraman S, Schulz H, Hummel O, Hubner N, Ghosh-Choudhury N, Sotiriadou I, Hescheler J, Sachinidis A. 2007. Transcriptomic and phenotypic analysis of murine embryonic stem cell derived BMP2+ lineage cells: an insight into mesodermal patterning. Genome Biol. 8:R184. 10.1186/gb-2007-8-9-r184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rowe HM, Kapopoulou A, Corsinotti A, Fasching L, Macfarlan TS, Tarabay Y, Viville S, Jakobsson J, Pfaff SL, Trono D. 2013. TRIM28 repression of retrotransposon-based enhancers is necessary to preserve transcriptional dynamics in embryonic stem cells. Genome Res. 23:452–461. 10.1101/gr.147678.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meylan S, Groner AC, Ambrosini G, Malani N, Quenneville S, Zangger N, Kapopoulou A, Kauzlaric A, Rougemont J, Ciuffi A, Bushman FD, Bucher P, Trono D. 2011. A gene-rich, transcriptionally active environment and the pre-deposition of repressive marks are predictive of susceptibility to KRAB/KAP1-mediated silencing. BMC Genomics 12:378. 10.1186/1471-2164-12-378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hosoya T, Clifford M, Losson R, Tanabe O, Engel JD. 2013. TRIM28 is essential for erythroblast differentiation in the mouse. Blood 122:3798–3807. 10.1182/blood-2013-04-496166 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.