

Abstract
Unregulated Ca2+ entry is thought to underlie muscular dystrophy. Here, we generated skeletal-muscle-specific transgenic (TG) mice expressing the Na+-Ca2+ exchanger 1 (NCX1) to model its identified augmentation during muscular dystrophy. The NCX1 transgene induced dystrophy-like disease in all hind-limb musculature, as well as exacerbated the muscle disease phenotypes in δ-sarcoglycan (Sgcd−/−), Dysf−/−, and mdx mouse models of muscular dystrophy. Antithetically, muscle-specific deletion of the Slc8a1 (NCX1) gene diminished hind-limb pathology in Sgcd−/− mice. Measured increases in baseline Na+ and Ca2+ in dystrophic muscle fibers of the hind-limb musculature predicts a net Ca2+ influx state due to reverse-mode operation of NCX1, which mediates disease. However, the opposite effect is observed in the diaphragm, where NCX1 overexpression mildly protects from dystrophic disease through a predicted enhancement in forward-mode NCX1 operation that reduces Ca2+ levels. Indeed, Atp1a2+/− (encoding Na+-K+ ATPase α2) mice, which have reduced Na+ clearance rates that would favor NCX1 reverse-mode operation, showed exacerbated disease in the hind limbs of NCX1 TG mice, similar to treatment with the Na+-K+ ATPase inhibitor digoxin. Treatment of Sgcd−/− mice with ranolazine, a broadly acting Na+ channel inhibitor that should increase NCX1 forward-mode operation, reduced muscular pathology.
INTRODUCTION
Muscular dystrophy (MD) is characterized by myofiber degeneration that results in muscle loss, functional impairment, and eventually death. MD is generally caused by genetic mutations in genes encoding proteins that are either part of the membrane-stabilizing dystrophin-glycoprotein complex (DGC) or otherwise impact some aspect of sarcolemmal integrity and membrane channel activity (1). Such alterations cause enhanced Ca2+ entry through microtears or Ca2+ channels/exchangers (2). Downstream consequences of increased Ca2+ entry include altered signaling, calpain activation leading to unregulated intracellular protein degradation, and induction of necrosis through opening of the mitochondrial permeability transition pore with mitochondrial rupture (3, 4). However, the hypothesis that Ca2+ elevations directly induce myofiber necrosis and lead to MD is controversial (2). While some groups have indeed reported global or even subsarcolemmal increases in Ca2+ in dystrophic myofibers (5–10), such measurements are often technically difficult, which may be the reason why other studies have not observed a significant increase (11–13). Recent studies in transgenic (TG) mice have supported the Ca2+ hypothesis of disease. For example, overexpression of dominant-negative transient receptor potential canonical 6 (dnTRPC6) or dnTRPV2 was sufficient to abrogate the dystrophic phenotype in mice by inhibiting a type of store-operated Ca2+ entry that characterizes these channels (14, 15). TRPC3 overexpression in skeletal muscle, which dramatically enhanced Ca2+ entry, was sufficient to induce MD in mice (14). Finally, overexpression of the sarcoplasmic reticulum (SR) Ca2+ ATPase 1 (SERCA1) in skeletal muscle, which increases the rate of Ca2+ clearance back into the SR, abrogated many pathological indexes of MD in mice (16).
In addition to Ca2+, Na+ is also presumed to enter the dystrophic muscle fiber through microtears and channels. Importantly, cytosolic Na+ levels appear to be elevated in myofibers from the mdx mouse, a model of Duchenne MD (7, 17–20). Indeed, recent evidence suggests that intracellular Na+ levels are also elevated in Duchenne MD patients (21, 22). Increased intracellular Na+ could potentially impact dystrophic pathology through cellular edema or reversal of Na+-Ca2+ exchange, thereby secondarily leading to even greater Ca2+ entry or less clearance (20, 22).
As Ca2+ levels rise with contraction, some of the Ca2+ is removed during relaxation by the Na+-Ca2+ exchanger (NCX), whereby the Na+ gradient generated by the Na+-K+ ATPase (NKA) is used to promote Ca2+ efflux. While the exchanger typically functions in forward mode to clear Ca2+ from the intracellular milieu, reverse mode is also possible, leading to Ca2+ entry in exchange for Na+ (23). The direction of exchange is determined by the Na+ gradient, the Ca2+ gradient, and the membrane potential. Forward-mode exchange (net Ca2+ removal) is favored by low intracellular Na+, high intracellular Ca2+, and a hyperpolarized membrane potential. Reverse mode is favored by high intracellular Na+, low intracellular Ca2+, and a more depolarized membrane potential. The predominant direction or impact of Na+-Ca2+ exchange in adult muscle pathology is not currently known, nor is the role of Ca2+ entry or efflux by this system understood in MD.
Previous studies with NCX isoforms in skeletal muscle suggest that NCX1 is expressed at high levels early in rat embryonic development and postnatal maturation but is gradually downregulated as NCX3 becomes more highly expressed, when it becomes the primary adult isoform (24). Genetic deletion of the gene encoding NCX3 protein caused myofiber and nerve degeneration due to defects at the neuromuscular junction (25). Studies of NCX function in human myotubes from Duchenne MD patients showed enhancement of reverse-mode Na+-Ca2+ exchange activity (26). This was also observed in myotubes from dystrophic mice; however, adult muscle fibers failed to show significant reverse-mode activity (25, 27, 28). Thus, the role of Na+-Ca2+ exchange in MD remains uncertain.
MATERIALS AND METHODS
Animals.
Skeletal-muscle-specific expression of NCX1 was driven by a modified human skeletal α-actin promoter with a slow troponin enhancer, as described previously (29). A canine NCX1 cDNA was used for overexpression because it is allosterically activated by Ca2+ (unlike mouse NCX1 cDNA), similar to the human protein (30, 31). High and low transgenic lines (FVB/N background) were chosen for subsequent analysis, each of which showed skeletal-muscle-specific expression. Sgcd−/− mice on the C57BL/6 background were a gift from Elizabeth McNally (University of Chicago). mdx mice on a C57BL/10 background and Dysf−/− mice (A/J background) were obtained from the Jackson Laboratory (Bar Harbor, ME). Atp1a2 heterozygous mice were described previously, as were the Slc8a1 loxP-targeted mice (32, 33). Finally, mice containing the Cre recombinase cDNA within the myosin light chain 1 (Mlc1 gene) locus in the C57BL/6 background were described previously (34). Experiments involving animals were approved by the Institutional Animal Care and Use Committee of the Cincinnati Children's Hospital Medical Center.
Western blotting.
Muscles were homogenized in modified RIPA buffer (150 mM NaCl, 50 mM Tris, pH 7.4, 1% Triton X-100, 0.5% Na deoxycholate, 0.1% SDS, 5 mM EDTA, 2 mM EGTA, 1 mM dithiothreitol [DTT], and 1× Roche Complete protease inhibitor). Extracts were cleared by centrifugation at 14,000 × g for 10 min, and 5 to 30 μg of protein was separated on 10% SDS-polyacrylamide gels. The gels were transferred to polyvinylidene difluoride (PVDF) membranes and blocked with 5% milk for 1 h. The blots were incubated overnight at 4°C with the following antibodies: anti-NCX1 mouse monoclonal at 1:1,000 (Swant, Marly, Switzerland; R3F1), anti-NKA mouse monoclonal at 1:1,000 (Developmental Hybridoma Database, Iowa City, IA; a5), anti-NKAα1 mouse monoclonal at 1:1,000 (Developmental Hybridoma Database, Iowa City, IA; a6f), anti-NKAα2 rabbit polyclonal at 1:1,000 (Millipore, Billerica, MA; AB9094), SERCA1 mouse monoclonal at 1:1,000 (Affinity Bioreagents, Rockford, IL; MA3911), and dihydropyridine receptor (DHPR) mouse monoclonal at 1:1,000 (Pierce, Rockford, IL; MA3912). The blots were washed and incubated with the appropriate secondary antibody conjugated to alkaline phosphatase at 1:2,500. The blots were exposed using chemiluminescence detection reagent (GE Healthcare Biosciences, Piscataway Township, NJ).
Reverse transcription (RT)-PCR.
RNA was isolated using the RNeasy Fibrous Tissue minikit from Qiagen (Valencia, CA) following the manufacturer's instructions. The RNA was reverse transcribed using the SuperScript III First Strand synthesis kit from Life Technologies (Carlsbad, CA). The cDNA was diluted 1:100, and 5 μl of solution was used per reaction. The results were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA and quantified using the 2−ΔΔCT method (35).
Immunohistochemistry and immunocytochemistry.
Whole muscles were excised and fixed in 4% paraformaldehyde for 4 h at 4°C. Muscles were then placed in phosphate-buffered saline (PBS) with 150 mM sucrose buffer overnight for cryopreservation. Muscles were bisected, embedded in optimal cutting temperature compound (O.C.T.; VWR, Arlington Heights, IL), and rapidly frozen in liquid-nitrogen-cooled isopentane. Seven-micrometer cryosections were cut on a cryomicrotome. Isolated fibers were plated on laminin-coated glass bottom coverslips, fixed for 15 min in 4% paraformaldehyde in PBS, and then washed for 5 min in PBS, permeabilized for 15 min in 0.1% Triton X-100 in PBS, and blocked for 1 h with 5% goat serum in PBS. The sections were incubated overnight at 4°C with primary antibody in PBS supplemented with 5% goat serum. The following antibodies and dilutions were used: anti-NKAα2 at 1:100 (Millipore, Billerica, MA; AB9094) and anti-NCX1 mouse monoclonal at 1:100 (Swant, Marly, Switzerland; R3F1). The sections were washed 3 times for 5 min each time in PBS and then blocked for 30 min with 5% goat serum in PBS. Secondary antibody was applied in PBS with 5% goat serum for 1 h at room temperature. Goat anti-mouse secondary antibody conjugated to tetramethyl rhodamine isocyanate (TRITC) and goat anti-rabbit secondary antibody conjugated to fluorescein isothiocyanate (FITC) were used at a concentration of 1:400 (Molecular Probes, Eugene, OR). The slides were washed 3 times with PBS for 5 min each time. DAPI (4′,6-diamidino-2-phenylindole) stain was added for 10 min in the second wash, followed by imaging on a Nikon A1 laser scanning confocal microscope.
TUNEL staining.
Terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) was accomplished according to the manufacturer's instructions as previously described (36). Tissue sections were incubated in a humidified chamber at 37°C with the TUNEL reaction mixture from an in situ cell death detection kit (Roche, Indianapolis, IN).
Photometry experiments.
The flexor digitorum brevis (FDB) muscle was dissected from the footpad in Ringer's solution (145 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, and 10 mM HEPES, pH 7.4). The FDB was then transferred to a solution containing 0.1% collagenase A for 1 h at 37°C. The dissected fibers were loaded with 5 μM Fura-2-AM (Life Technologies, Carlsbad, CA) in Ringer's solution for 30 min and transferred to a perfusion chamber. The fibers were excited at 340 nm and 380 nm, while emission was detected at 510 nm through a Nikon Ti-U inverted microscope equipped with a 40× Fluor objective leading to a photomultiplier tube (Photon Technology International, Birmingham, NJ). Forward-mode activity measurements were adapted from previous work (25, 28). Baseline readings were acquired in Ca2+- and Mg2+-free (CMF) Ringer's solution (151 mM NaCl, 5 mM KCl, 10 mM HEPES, pH 7.4) for 1 min. The solution was then exchanged for CMF Ringer's solution containing 30 μM cyclopiazonic acid (CPA) (Sigma-Aldrich, St. Louis, MO), 50 μM N-benzyl-p-toluenesulfonamide (BTS) (Tocris, Bristol, United Kingdom), and 200 μM EGTA for 100 s. The solution was then exchanged again to contain 750 μM 4-chloro-m-cresol (4-CMC) (Sigma-Aldrich, St. Louis, MO) to elicit Ca2+ efflux through the ryanodine receptor. Traces were recorded for 15 min after treatment with 4-CMC. Stimulated transient experiments (0.2 Hz) were performed in Ringer's solution containing 5 μM BTS.
Calpain activity experiments.
Calpain activity experiments were performed using the Calpain Glo protease assay (Promega, Madison, WI) according to the manufacturer's instructions. Samples were incubated for 5 min at 37°C, and luminescence was measured using the Synergy 2 plate reader (BioTek, Winooski, VT).
Ca2+-selective microelectrodes.
Double-barreled Ca2+-selective microelectrodes were prepared as described previously (37). The 1.51-mm (outside diameter [o.d.]) barrel of the pulled microelectrode was back-filled first with the neutral carrier ETH 129 (21193; Fluka-Sigma-Aldrich, St. Louis, MO) and 24 h later with a solution containing calcium at 10−7 M. Each Ca2+-selective microelectrode was individually calibrated as described previously (38), and only those with a linear relationship between 10−3 and 10−7 M calcium-containing solutions were used. After making measurements of intracellular Ca2+ concentration ([Ca2+]i), all electrodes were recalibrated.
Na+-selective microelectrodes.
Double-barreled Na+-selective microelectrodes were made as previously described (39). The microelectrodes were calibrated 24 h later in solutions containing different [Na+] and 1 mM MgCl2. The microelectrodes gave virtually Nernstian responses at free [Na+] between 100 and 10 mM. However, at between 10 and 1 mM [Na+], the electrodes had a sub-Nernstian response (40 to 45 mV), but their response was stable and of a sufficient amplitude to measure the [Na+]i. After making measurements of resting [Na+]i, all electrodes were recalibrated, and if the two calibration curves did not agree within 3 mV, data from that microelectrode were discarded.
[Ca2+]i and [Na+]i determination in mice in vivo.
NCX1 TG and wild-type (WT) mice were anesthetized (ketamine at 100 mg/kg of body weight and xylazine at 5 mg/kg), intubated, and placed on a mouse ventilator (Minivent 845; Hugo Sachs Elektronik, Germany). A small incision was made in the skin over the gastrocnemius muscle, and the superficial fibers were exposed and perfused with rodent Ringer's solution (37°C). The determination of [Ca2+]i or [Na+]i was carried out using ion-selective microelectrodes as described above.
Digoxin treatment.
Nontransgenic (NTG) mice and NCX1 TG littermates were randomized to a digoxin treatment group or a control group at 6 weeks of age. Digoxin was produced by Roxane Laboratories (Columbus, OH) at a concentration of 50 mg/ml and obtained from the Cincinnati Children's Hospital Pharmacy. For this study, digoxin was administered in water by mixing the drug formulation with water in a 1:3 ratio. The treatment was continued for 8 weeks, and levels in plasma were obtained at sacrifice and determined to be 2 ng/ml on average.
Ranolazine treatment.
Ranolazine was given at 2 mg and ketoconazole at 0.18 mg per gram of food formulation (40). Ketoconazole was included as a p450 inhibitor to decrease the metabolism of ranolazine and increase its levels in plasma. Based on food consumption, mice were dosed with ∼320 mg/kg/day of ranolazine (Gilead, Foster City, CA) and ∼28.8 mg/kg/day of ketoconazole (Teva Pharmaceuticals, Petach Tikva, Israel). Mice in the control arm of the study received ∼28.8 mg/kg/day of ketoconazole. The mouse food was formulated by Research Diets (New Brunswick, NJ).
Statistics.
All results are means ± standard errors of the mean (SEM). Statistical analysis was performed with unpaired 2-tailed t tests (for 2 groups) and 1-way analysis of variance (ANOVA) with Bonferroni correction (for groups of 3 or more). P values of <0.05 were considered significant.
RESULTS
NCX1 expression is increased in dystrophic skeletal muscle.
Isoform switching occurs in many different contractile and Ca2+-handling genes during development to augment muscle function as the organism matures (41). With injury to heart or skeletal muscle, there is a reversion to a fetal-like gene program in the expression of contractile and Ca2+-handling gene isoforms (42, 43). NCX1 is highly expressed in developing skeletal muscle (24), where it is maintained during the first few weeks of life in rats, but is thereafter strongly downregulated in early adulthood, which we also observed in the mouse (Fig. 1A). However, NCX1 protein expression is maintained during juvenile development through 6 weeks of age, and even into adulthood in dystrophic skeletal muscle from Sgcd−/− mice, which is a mouse model of MD due to loss of the membrane-spanning δ-sarcoglycan protein (Fig. 1A). Immunofluorescence staining of histological sections from skeletal muscle of 3-month-old mice showed very low levels of membrane-localized NCX1 protein in WT mice but noticeably higher levels in Sgcd−/− muscles (Fig. 1B). We also observed upregulation of NCX1 after cardiotoxin injury of adult WT skeletal muscle, where new fibers are generated similarly to MD (Fig. 1C).
FIG 1.

Generation of transgenic mice with skeletal-muscle-specific NCX1 overexpression. (A) Western blotting for endogenous NCX1 expression at different ages in quadriceps muscles of WT and Sgcd−/− mice. GAPDH is a loading control. (B) Histological immunofluorescence images (×200 magnification) of NCX1 expression (red) and DAPI (blue) from quadriceps of Sgcd−/− and WT mice at 3 months of age. (C) Western blotting for endogenous NCX1 during a time course of regeneration at 3, 7, and 14 days (d) after cardiotoxin (CTX) injection into skeletal muscle. (D) Schematic of the human skeletal α-actin (HSA) promoter upstream of the canine NCX1 cDNA used to make TG mice. (E) Western blotting in quadriceps (Quad.) and diaphragm (Diaph.) of NCX1 TG and NTG mice at 8 weeks of age for NCX1, SERCA1, and NKA (Pan). The asterisks indicate differences between the quadriceps and diaphragm in baseline SERCA1 and NKA expression. (F) Immunofluorescence image (×200 magnification) of NCX1 (red) versus membranes stained with wheat germ agglutinin (WGA, green stained)) from histological sections of quadriceps muscle from NTG and NCX1 TG mice. (G) Immunocytochemical staining (×600 magnification) of NCX1 (red) and NKAα2 (green) in isolated myofibers from NCX1 TG and NTG mice.
Generation of mice with increased skeletal-muscle-specific expression of NCX1.
To determine the effect of sustained expression of NCX1 in MD, we generated skeletal-muscle-specific transgenic mice expressing the canine NCX1 cDNA (30, 44, 45) under the control of the human skeletal α-actin promoter (Fig. 1D). NCX1 overexpression was confirmed in the diaphragm and quadriceps by Western blotting from adult transgenic mice, and proper plasma membrane localization was shown by immunofluorescence (Fig. 1E and F). NCX1 expression was increased 4.4-fold over WT in the transgenic mice versus 3.3-fold-increased endogenous expression in injured WT muscle and 2.3-fold-increased expression in muscle from Sgcd−/− mice at 6 weeks of age. Overexpressed NCX1 protein due to the transgene was shown to properly localize to the t-tubules and endplate, similar to NKAα2 (Fig. 1G).
We next measured Na+-Ca2+ exchange activity in isolated FDB muscle fibers from NCX1 TG and WT littermates, as previously described (Fig. 2A) (25, 28). Ca2+ was measured using the ratiometric Ca2+-sensitive dye Fura-2, where baseline measurements were first obtained for 30 s in CMF. This solution was then exchanged for CMF containing 30 μM CPA to inhibit SERCA function and 50 μM BTS to block contraction for 100 s. The solution was then replaced with one containing 30 μM CPA and 750 μM 4-CMC, a ryanodine receptor agonist that elicits Ca2+ release from the SR, which was similar between the groups (Fig. 2B). Because SERCA is also inhibited, the decay of the Ca2+ transient (the pulse of intracellular calcium that mediates contraction) represents Ca2+ membrane efflux from the fibers, which is significantly faster in fibers from NCX1 transgenic mice or dystrophic fibers from mdx mice that also contain the NCX1 transgene (Fig. 2C). Thus, the transgenic mice express functional NCX1 that greatly increases Na+-Ca2+ exchange activity within muscle fibers.
FIG 2.

NCX1 overexpression increases Na+-Ca2+ exchange. (A) Representative Fura-2 Ca2+ traces of forward-mode activity in WT and NCX1 TG (gray and red traces) and mdx and mdx NCX1 (green and blue traces) FDB muscle fibers at 2 to 4 months of age. The arrows indicate buffer changes. (B) SR Ca2+ store calculated from peak heights of 4-CMC Ca2+ transient in WT, NCX1 TG, mdx, and mdx NCX1 TG mice. (C) Time for 1/2 of the Fura-2 signal decay of the 4-CMC-induced Ca2+ transient during the forward-mode Na+-Ca2+ exchange experiment. *, P < 0.0001 versus WT. (D) Peak height of an electrically stimulated (paced) Ca2+ transient in Ringer's solution with 5 μM BTS using the Ca2+-sensitive dye Fura-2. *, P < 0.001 versus WT. (E) Time constant (Tau) fit to electrically stimulated transients for WT, NCX1 TG, mdx, and mdx NCX1 TG. (F) Amplitude of the repolarization of the membrane (afterhyperpolarization) of the action potential recorded in WT, NCX1 TG, mdx, and mdx NCX1 TG EDL muscle. *, P < 0.05 versus WT. The error bars indicate SEM. Numbers in the bars represent the number of fibers analyzed.
We also examined the effect of greater NCX1 activity on excitation contraction-coupling, which showed a significant increase in the amplitude of the electrically evoked Ca2+ transient in NCX1 TG mice (Fig. 2D); however, no change in the decay of the Ca2+ transient was observed (Fig. 2E). Interestingly, NCX1 overexpression had a similar effect on the transient amplitude in both the WT and mdx backgrounds. The increased store size suggested that NCX1 could be functioning to facilitate greater Ca2+ entry in the transgenic mice (see below). The observed alterations in Ca2+ handling due to NCX1 overexpression did not result from compensatory changes in SERCA1 or NKA levels in either the quadriceps or diaphragm of TG mice compared with the WT (Fig. 1E).
We also observed a significant alteration in the membrane voltage relationship of the action potential during repolarization (Fig. 2F). Specifically, action potentials from NCX1 extensor digitorum longus (EDL) showed increased repolarization of the membrane compared with WT fibers, although this relationship was not further altered when crossed into the mdx-mouse background (Fig. 2F). These results are yet another indirect line of evidence that increased Ca2+ entry occurs in myofibers from NCX1 TG mice, given that its known electrogenic properties would hyperpolarize the membrane when acting in reverse mode. Indeed, dystrophic fibers from mice lacking laminin 2 displayed membrane hyperpolarization due to presumed reverse-mode NCX activity (46).
NCX1 transgenic mice develop progressive muscle pathology in their limbs.
NCX1 overexpression induced aspects of a dystrophic phenotype in the limb muscles, a phenotype that is consistent with increased Ca2+ entry. At 2 months of age, we observed a pseudohypertrophic response in the quadriceps and gastrocnemius muscles of TG mice (Fig. 3A), which remained significantly increased in the gastrocnemius at 6 months of age (Fig. 3B). Pseudohypertrophy is due to the increased fibrosis and inflammation that typifies MD, not increased muscle fiber size or number. The increased inflammation and fibrosis due to NCX1 TG was confirmed by histological analysis at 2, 6, and 15 months of age in NCX1 TG quadriceps (Fig. 3C). Quantitation of histological pathology at 2 and 6 months of age revealed significantly greater fibrosis and central nucleation of myofibers in TG mice compared with NTG mice (Fig. 3D to H). An increase in calpain activity was also observed in NCX1 TG mice, suggesting that necrotic pathways are activated due to greater Ca2+ levels (Fig. 3I). These results show that NCX1 overexpression is sufficient to induce skeletal-muscle pathology that is reminiscent of MD.
FIG 3.
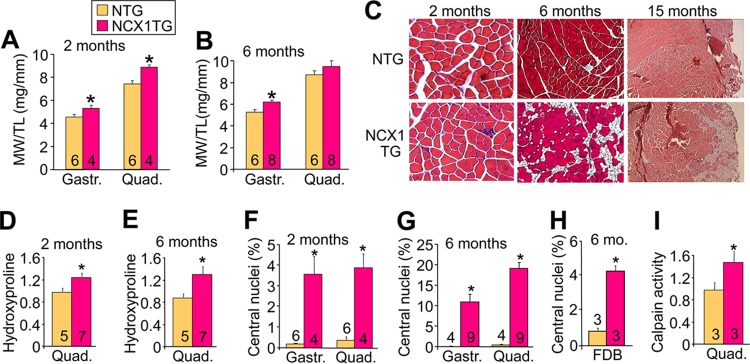
NCX1 overexpression induces progressive skeletal-muscle pathology. (A and B) Muscle weight-to-tibia length (MW/TL) ratios measured at 2 and 6 months of age in the gastrocnemius (Gastr.) and quadriceps (Quad). *, P < 0.05 versus NTG. (C) Representative hematoxylin and eosin (H&E)-stained histological sections from NCX1 TG and NTG mice at 2 (×200 magnification), 6 (Masson's trichrome; ×100 magnification), and 15 (H&E; ×40 magnification) months of age. (D and E) Results of hydroxyproline assay to measure collagen contents of quadriceps from NTG and NCX1 TG mice at 2 and 6 months of age. *, P < 0.05 versus NTG mice. (F and G) Percentages of fibers with centrally localized nuclei at 2 and 6 months of age in NCX1 TG and NTG histological sections from the indicated muscles. *, P < 0.05 versus NTG mice. (H) Percentages of fibers with centrally localized fibers in 6-month-old FDB muscles from NTG and NCX1 TG skeletal muscle. *, P < 0.05 versus NTG mice. (I) Calpain activity measured by luciferase assay at 3 months of age in the indicated muscles from NTG and NCX1 TG mice. *, P < 0.05 versus NTG mice. The error bars indicate SEM.
NCX1 overexpression exacerbates mouse models of MD.
The NCX1 protein is reexpressed in dystrophic muscle of Sgcd−/− mice well into adulthood (Fig. 1A), although it was uncertain if this was protective or deleterious in the disease process. To address this issue, we first crossed NCX1 TG mice into the Sgcd−/− background. The results showed a much more prominent MD, so that by 6 months of age muscle weights were significantly lower than in Sgcd−/− or NTG mice due to extreme necrosis of the myofibers (Fig. 4A versus Fig. 3B). NCX1 TG Sgcd−/− mice also showed a progressive fibrotic effect that was most prominent at 6 months compared with 6 weeks, likely because endogenous NCX1 is already expressed during the neonatal period (Fig. 4B). Fatty replacement was also greater in NCX1 TG Sgcd−/− mice at 6 months compared with Sgcd−/−-only mice and, along with the accumulation of fibrosis, was clearly visible in representative histological sections from the quadriceps (Fig. 4C and E). TUNEL, an assay that suggests myofiber death, was significantly increased in muscle from Sgcd−/− mice by the presence of the NCX1 transgene at 6 weeks (Fig. 4D). The NCX1 transgene also significantly hastened the demise of Sgcd−/− mice, which appears to be due to extreme loss and failure of the musculature as the mice age (Fig. 4F).
FIG 4.

NCX1 overexpression exacerbates hind-limb pathology in Sgcd−/− mice. (A) MW/TL ratios measured in Sgcd−/− and Sgcd−/− NCX1 TG mice at 6 months of age. TA, tibialis anterior; Dia., diaphragm. *, P < 0.05 versus Sgcd−/− mice. (B) Hydroxyproline content of collagen in quadriceps of Sgcd−/− and Sgcd−/− NCX1 TG mice measured at 6 weeks and 6 months of age. *, P < 0.05 versus Sgcd−/− mice. (C) Histological measurements of areas replaced by adipose tissue in quadriceps muscles of Sgcd−/− and Sgcd−/− NCX1 TG mice at 6 months of age. *, P < 0.05 versus Sgcd−/− mice. (D) Quantification of TUNEL-positive muscle fibers between WT, NCX1 TG, Sgcd−/−, and Sgcd−/− NCX1 TG mice at 6 weeks of age in histological sections from quadriceps. *, P < 0.05 versus WT mice; #, P < 0.05 versus Sgcd−/− mice. (E) Representative H&E and Masson's trichrome (Tri) images of quadriceps muscles from Sgcd−/− and Sgcd−/− NCX1 TG mice at 6 months of age. (F) Survival plot of Sgcd−/− and Sgcd−/− NCX1 TG mice. The error bars indicate SEM.
We also crossed the NCX1 TG mice to the Dysf−/− and mdx mouse models of MD. Dysf−/− mice are a model of dysferlinopathy associated with defective membrane repair (47). mdx mice lack the protein dystrophin, which biochemically models Duchenne MD, leading to an unstable membrane and dysregulation of Ca2+ (48, 49). We observed that pathological features of the limb musculature of Dysf−/− mice became dramatically more pronounced in the presence of the NCX1 transgene at 6 months of age (Fig. 5A). Dysf−/− mice with the NCX1 transgene showed significantly greater loss of quadriceps and gastrocnemius mass at 6 months of age than did WT, NCX1 TG, or Dysf−/−-only mice (Fig. 5B), as well as dramatically elevated serum creatine kinase (CK) levels, suggesting higher ongoing rates of muscle breakdown (Fig. 5C). Indeed, rates of myofiber central nucleation were significantly higher in Dysf−/− mice containing the NCX1 transgene (Fig. 5D). NCX1 TG mice crossed into the mdx background also showed greater myofiber central nucleation and fibrosis in the gastrocnemius and quadriceps than mdx or NCX1 TG-only mice at 2 months of age (Fig. 5E and F). These data show that the effect of NCX1 overexpression is conserved across three distinct dystrophic-mouse models.
FIG 5.

NCX1 overexpression exacerbates pathology in Dysf−/− and mdx mouse models. (A) Representative H&E-stained histological sections (×100 magnification) from gastrocnemius and quadriceps of Dysf−/− and Dysf−/− NCX1 TG mice at 6 months of age. (B) MW/TL ratios of WT, NCX1 TG, Dysf−/−, and Dysf−/− NCX1 TG muscles at 6 months of age. *, P < 0.05 versus WT mice; #, P < 0.05 versus Dysf−/− mice. (C) Serum creatine kinase measurements at 6 months of age in WT, NCX1 TG, Dysf−/−, and Dysf−/− NCX1 TG mice. *, P < 0.05 versus WT mice; #, P < 0.05 versus Dysf−/− mice. (D) Percentages of muscle fibers with centrally localized nuclei in WT, NCX1 TG, Dysf−/−, and Dysf−/− NCX1 TG quadriceps and gastrocnemius histological sections at 6 months of age. *, P < 0.05 versus WT mice; #, P < 0.05 versus Dysf−/− mice. (E) Percent centrally localized nuclei from histological sections in WT, NCX1 TG, mdx, and mdx NCX1 TG mice at 2 months of age. *, P < 0.05 versus WT mice; #, P < 0.05 versus mdx mice. (F) Percent fibrosis measured by Masson's trichrome staining from histological sections at 2 months of age of the indicated muscles in mdx versus mdx NCX1 TG mice. *, P < 0.05 versus mdx mice. The error bars indicate SEM. Numbers in the bars represent the number of animals analyzed.
Deletion of NCX1 reduces the dystrophic phenotype.
The Slc8a1 (NCX1) gene was deleted specifically in skeletal muscle by crossing Slc8a1-loxP-targeted (fl/fl) mice into the Sgcd−/− genetic background with the MLC-Cre line (the heart is spared). We confirmed deletion of Slc8a1 in skeletal muscle by real-time PCR, which showed a 90% reduction of mRNA expression while SERCA1 levels were unchanged (Fig. 6A). Deletion of Slc8a1 specifically in skeletal muscle of Sgcd−/− mice led to significantly less histopathology at 6 weeks of age, including decreased central nucleation and fibrosis in both the quadriceps and the gastrocnemius (Fig. 6B to D). However, at 6 months of age, the protective effect of Slc8a1 deletion was lost (Fig. 6E and F). This is probably due to the fact that endogenous NCX1 is expressed only during the neonatal period and is gradually lost by young adulthood. Even in dystrophic mice, NCX1 expression decreases with age, making its deletion less relevant at later time points. However, the prominent protection observed in 6-week-old Sgcd−/− mice with Slc8a1 deletion suggests that early expression of NCX1 in the limb musculature contributes to MD disease.
FIG 6.

Deletion of NCX1 (Slc8a1) attenuates skeletal-muscle pathology in Sgcd−/− mice at early ages. (A) RT-PCR comparing the relative amounts of NCX1 and SERCA1 mRNAs in quadriceps from Sgcd−/− Slc8a1fl/fl and Sgcd−/− Slc8a1fl/fl MLC-Cre mice. The results were normalized to GAPDH mRNA. (B) Representative muscle histopathology (×200 magnification) in Sgcd−/− Slc8a1fl/fl (control) and Sgcd−/− Slc8a1fl/fl MLC-Cre mice at 6 weeks of age. (C) Percentages of centrally localized nuclei quantified from histological sections of the indicated muscles in Sgcd−/− Slc8a1fl/fl and Sgcd−/− Slc8a1fl/fl MLC-Cre mice at 6 weeks of age. *, P < 0.05 versus Sgcd−/− Slc8a1fl/fl mice. (D) Percent fibrotic area measured by Masson's trichrome staining from histological sections of the indicated muscles at 6 weeks of age. *, P < 0.05 versus Sgcd−/− Slc8a1fl/fl mice. (E) Percentages of fibers with centrally localized nuclei from histological sections in Sgcd−/− Slc8a1fl/fl and Sgcd−/− Slc8a1fl/fl MLC-Cre quadriceps, gastrocnemius, and diaphragm at 6 months of age. (F) Percent fibrotic area in histological sections in Sgcd−/− Slc8a1fl/fl and Sgcd−/− Slc8a1fl/fl MLC-Cre quadriceps, gastrocnemius, and diaphragm at 6 months of age. The error bars indicate SEM. Numbers in the bars represent the number of mice analyzed.
Intracellular elevations in Na+ exacerbate NCX1-mediated pathology.
The Na+ gradient is a key determinant of net Ca2+ influx or efflux by NCX1, which is directly regulated by the NKA pump. Hence, we reasoned that genetic reduction of the NKAα2 gene product using targeted mice should slightly elevate Na+, leading to even greater reverse-mode Ca2+ entry from NCX1. Accordingly, we crossed NKAα2-heterozygous (Atp1a2+/−) mice into the NCX1 TG background. The gene for the α2 isoform of NKA is the predominant gene expressed in skeletal muscle (50), and it is tightly coupled to NCX activity (51). We observed that NCX1 TG Atp1a2+/− mice had increased central nucleation and increased serum CK levels compared with NCX1 TG mice, as well as more obvious histopathology at 3 months of age (Fig. 7A to C). Moreover, treatment of mice with the NKA inhibitor digoxin, which is known to elevate Na+ levels, also significantly exacerbated dystrophic pathology in hind-limb muscle from NCX1 TG mice (Fig. 7D and E). These results further support the hypothesis that the adverse effects of NCX1 expression occur through enhanced Ca2+ entry due to reverse-mode activity of the exchanger.
FIG 7.
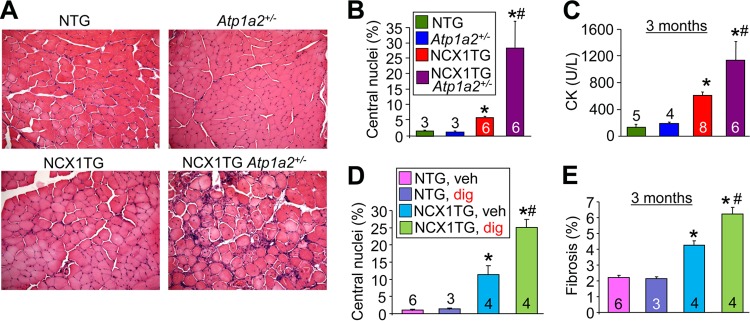
Haploinsufficiency of Atp1a2 or digoxin exacerbates MD in NCX1 TG mice. (A) Representative H&E-stained histological images of the quadriceps (×200 magnification) of NTG, Atp1a2+/−, NCX1 TG, and NCX1 TG Atp1a2+/− mice at 3 months of age. (B) Percentages of fibers with centrally localized nuclei at 3 months of age from the same muscles shown in panel A. *, P < 0.05 versus WT mice; #, P < 0.05 versus NCX1 TG mice. (C) Serum CK levels measured in the indicated groups at 3 months of age. *, P < 0.05 versus WT mice; #, P < 0.05 versus NCX1 TG mice. (D) Percent centrally localized nuclei in histological sections from quadriceps muscles of the groups shown. *, P < 0.05 versus NTG-vehicle-treated (veh) mice; #, P < 0.05 versus NCX1 TG-digoxin-treated (dig) mice. (E) Percent fibrosis measured by Masson's trichrome in histological sections of quadriceps muscles of the groups shown. *, P < 0.05 versus NTG-vehicle treated mice; #, P < 0.05 versus NCX1 TG-digoxin-treated mice. The error bars indicate SEM. Numbers in the bars represent the number of mice analyzed.
Transgenic NCX1 expression reduces pathology in the diaphragm due to MD.
The diaphragm is specialized to contract rhythmically and hence has more features in common with the heart than with the limb musculature (52–55). In the heart, NCX1 is known to operate predominantly in forward mode to remove Ca2+ after each contractile cycle, which we suspected could also be the case in the diaphragm. Indeed, the diaphragm has higher levels of NKA protein and lower SERCA1 expression than the quadriceps, suggesting that the diaphragm is more specialized for sarcolemmal Ca2+ handling (Fig. 1E). Consistent with these observations, NCX1 overexpression in the diaphragm did not lead to pathology, and it actually protected Sgcd−/− mice from dystrophic changes at 6 weeks and 6 months of age (Fig. 8A). Quantitation of central nucleation and fibrosis showed significantly lower levels due to the NCX1 transgene in the Sgcd−/− background (Fig. 8B and C), and there were no longer fibers with Ca2+ deposits due to Ca2+ overload, as assessed histologically with von Kossa staining (Fig. 8D). Rates of myofiber death assessed by TUNEL were also decreased by the NCX1 transgene mice in the Sgcd−/− background (Fig. 8E). These results provide evidence that NCX1 functions in forward mode in the diaphragm to remove Ca2+.
FIG 8.
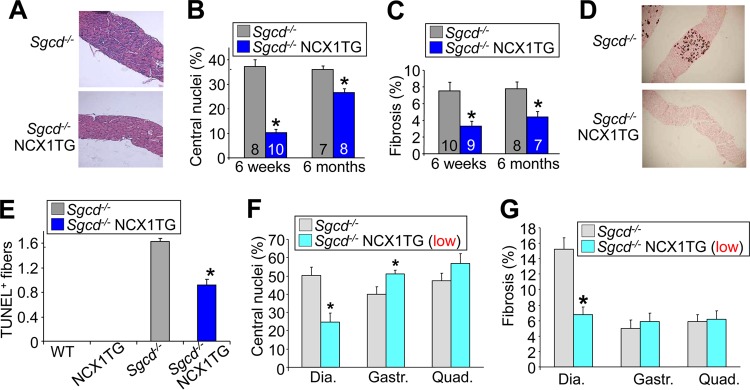
NCX1 overexpression rescues histopathology of the diaphragm of Sgcd−/− mice. (A) Representative H&E-stained histological images (×40 magnification) from the diaphragms of Sgcd−/− and Sgcd−/− NCX1 TG mice at 6 weeks of age. (B) Percentages of fibers with centrally localized nuclei quantified from the diaphragm in Sgcd−/− and Sgcd−/− NCX1 TG mice. *, P < 0.05 versus Sgcd−/− mice. Numbers in the bars represent the number of mice analyzed. (C) Fibrotic areas (percent) from trichrome-stained histological sections of diaphragms at 6 weeks and 6 months in Sgcd−/− and Sgcd−/− NCX1 TG mice. (D) Images (×40 magnification) of von Kossa-stained histological sections of Sgcd−/− and Sgcd−/− NCX1 TG diaphragm muscle at 6 weeks of age. (E) TUNEL-positive myofibers in the diaphragms of WT, NCX1 TG, Sgcd−/−, and Sgcd−/− NCX1 TG mice at 6 weeks of age. *, P < 0.05 versus Sgcd−/− mice. (F) Central nucleation measured in Sgcd−/− and Sgcd−/− NCX1 TG-low mice in histological sections from diaphragm, gastrocnemius, and quadriceps. (G) Percent fibrosis measured in Masson's trichrome-stained histological sections from the indicated muscles of Sgcd−/− and Sgcd−/− NCX1 TG-low mice. *, P < 0.05 versus Sgcd−/− mice. The error bars indicate SEM.
The diaphragm expresses substantially less NCX1 protein from the transgene than the limb musculature; hence, we were concerned that the rescue observed in this muscle was only a reflection of lower levels of NCX1 overexpression. To determine if this was indeed the case, we generated and crossed a much lower-expressing NCX1 transgenic line with Sgcd−/− mice. Here, as in the high-expressing line utilized throughout, lower levels of NCX1 overexpression still rescued the diaphragm at the 6-week time point but either had no protective effect or continued to exacerbate the hind-limb phenotype (Fig. 8F and G). Thus, even a smaller increase in NCX1 overexpression in the peripheral musculature is maladaptive toward MD and still protective in the diaphragm.
The diaphragm of the mdx mouse displays substantial pathology that is highly reminiscent of Duchenne MD (56). To extend the clinical relevance of the partial rescue that was observed in the diaphragms of Sgcd−/− mice with the NCX1 transgene, we examined the diaphragms of mdx and mdx NCX1 TG mice at 6 months of age. Examination of muscle weights, as an indication of inflammation and pseudohypertrophy disease, showed that the NCX1 transgene increased disease in the gastrocnemius and quadriceps, but it had the opposite effect in the diaphragm and was protective (Fig. 9A). Gross histological analysis also showed dramatic protection in the diaphragms of mdx mice carrying the NCX1 transgene (Fig. 9B), and quantitation of this effect revealed significantly fewer centrally localized nuclei and less fibrosis at 6 months of age than with mdx alone (Fig. 9C and D). The diaphragm showed increased expression of both NKAα1 and NKAα2 isoforms (which tends to lower Na+ to maintain forward-mode NCX1 operation) and decreased SERCA expression compared with the quadriceps (Fig. 9E). L-type Ca2+ channel levels were unchanged (DHPR), which was used as a loading control.
FIG 9.

NCX1 overexpression rescues histopathology of the diaphragms of mdx mice. (A) MW/TL ratios measured at 6 months of age in WT, NCX1 TG, mdx, and mdx NCX1TG mice for the indicted muscles. *, P < 0.05 versus WT mice; #, P < 0.05 versus mdx mice. (B) Representative H&E-stained histological images (×40 magnification) of diaphragms from mdx and mdx NCX1 TG mice at 2 months of age. (C) Percent fibrosis measured from Masson's trichrome-stained histological sections of diaphragms from mdx and mdx NCX1 TG mice. *, P < 0.05 versus mdx mice. (D) Percentages of fibers with centrally localized nuclei from diaphragm histological sections at 6 months of age in mdx and mdx NCX1 TG mice. *, P < 0.05 versus mdx mice. (E) Western blots for the indicated proteins from quadriceps versus diaphragm in WT mice at 6 weeks of age. DHPR, L-type Ca2+ channel. The error bars indicate SEM. Numbers in the bars represent the number of mice analyzed.
The Na+ channel inhibitor ranolazine decreases MD pathology.
NCX1 overexpression induced an increase in both resting Na+ and Ca2+ in FDB myofibers compared with NTG (Fig. 10A and B). While this increase in Ca2+ did not reach the level observed in mdx mice, it was nearly double the resting Ca2+ level of NTG mice. With regard to Na+ homeostasis, the NCX1 transgene elevated this ion to a level nearly equivalent to that measured in myofibers from mdx and mdx NCX1 TG mice (Fig. 10B). Myofibers from mdx NCX1 TG mice were found to have a small but significant increase in Na+ levels relative to mdx mice. Elevated Na+ levels poise a cell to utilize NCX1 as a Ca2+ entry mechanism (reverse mode), especially when the cell is depolarized.
FIG 10.

Pharmacological treatment of altered Na+ decreases dystrophic pathology. (A) Intracellular resting Ca2+ measured with a Ca2+-sensitive microelectrode in gastrocnemius muscles of NTG, NCX1 TG, mdx, and mdx NCX1 TG mice. *, P < 0.05 versus NTG mice; #, P < 0.05 versus NCX1 TG mice. The numbers in the bars represent the number of fibers analyzed (B) Intracellular resting Na+ measured in vivo with an Na+ microelectrode as for panel A. *, P < 0.05 versus NTG mice; #, P < 0.05 versus mdx mice. (C) Schematic for pharmacological trial of ranolazine in Sgcd−/− mice. w, weeks. (D) Representative H&E-stained histological images (×200) of gastrocnemius muscles of control and ranolazine-treated Sgcd−/− mice. (E) Percent fibrotic area in histological sections from quadriceps and gastrocnemius of vehicle- and ranolazine-treated Sgcd−/− mice. *, P < 0.05 versus ranolazine. The number in the bars represent the number of mice analyzed. (F) Percentages of centrally localized nuclei in histological sections from quadriceps and gastrocnemius of vehicle- and ranolazine-treated Sgcd−/− mice. *, P < 0.05 versus ranolazine. The error bars indicate SEM.
The profile of Na+ and Ca2+ alterations observed in hind-limb myofibers from NCX1 TG and dystrophic mice suggested the application of a pharmacologic agent, ranolazine. This drug is known to reduce Na+ channel leak through Nav1.4, as well as other Na+ channels (57, 58). Ranolazine also antagonizes reverse-mode Na+-Ca2+ exchange in cardiomyocytes through both direct (59) and indirect (60, 61) means. Finally, ranolazine is known to result in lower resting intracellular Na+ levels in cardiomyocytes (62, 63). Hence, 4-week-old Sgcd−/− mice were treated with ranolazine in their food at 320 mg/kg/day for 8 weeks (Fig. 10C). Histological examination of quadriceps muscles showed noticeably less pathology with significantly less fibrosis and central nucleation with ranolazine treatment, as well as fewer infiltrates and less fiber size irregularity (Fig. 10D to F). These results not only suggest a novel therapeutic option to consider in treating MD, they also experimentally suggest that Na+ elevation and reverse-mode NCX1 contribute to disease.
DISCUSSION
Initial attempts to directly measure Ca2+ levels in dystrophic myofibers or myotubes led to equivocal results, with some observations supporting a general increase in total cytosolic Ca2+ or subsarcolemmal Ca2+ (5–10) and others showing no difference (11–13). These divergent accounts may reflect technical difficulties associated with measuring Ca2+ in isolated fibers or myotubes, not to mention the uncertain physiological significance of such measurements in various ex vivo preparations or culture conditions. Our direct assessment of Na+ and Ca2+ levels using microprobes showed a significant elevation of both ions in acutely isolated myofibers from mdx mice. As a second approach, we and others have adopted a genetic strategy in mice. For example, we showed that overexpression of TRPC3 in skeletal muscle, which mimics/models a known increase in store-operated Ca2+ entry in dystrophic myofibers, directly led to greater Ca2+-Na+ influx that induced MD in mice (14). Moreover, blocking this known increase in the activity of TRP channels with either a dnTRPC or a dnTRPV construct abrogated MD in mouse models of disease, suggesting that unregulated or greater activation of the TRP class of cation channels is a primary disease determinant. These channels also permeate Na+, which would enhance Ca2+ entry through NCX1 by favoring reverse-mode activity (7, 17–20). Finally, we also previously showed that overexpression of SERCA1 in skeletal muscle, which produces higher rates of Ca2+ clearance back into the SR, was protective against MD in the mouse (16).
Elevations in Ca2+ lead to myofiber necrosis and disease by at least 2 mechanisms. The first is activation of the Ca2+-sensitive protease calpain, and the second is mitochondrial swelling and rupture. For the former, Spencer and Mellgren showed that transgene-mediated expression of the calpain inhibitor calpastatin reduced disease in the mdx background (64). More recently, we showed that mice lacking cyclophilin D (the Ppif gene), which underlies Ca2+-dependent swelling of mitochondria and subsequent cellular necrosis, were protected from Sgcd−/−- or laminin α2 deficiency-based MD (4).
The results of the current study provide even more evidence to support the proposed importance of unregulated Ca2+ influx in underlying MD and myofiber necrosis. Specifically, we observed that NCX1 directly influences MD by altering the Ca2+ and Na+ balance in a myofiber. NCX1 is normally expressed in immature fibers during development or after regeneration, when the developmental program is recapitulated, likely because these immature myofibers are more dependent on membrane Ca2+ exchange than the dedicated internal Ca2+ cycling through the SR that characterizes fully mature adult myofibers (65). At early time points, we observed that enhanced NCX1 expression interacts with other MD disease determinants to worsen pathology. Specifically, dystrophic fibers already show dysregulation in TRP channels and greater intracellular Na+ levels, which more readily favors reverse-mode Ca2+ entry through NCX1 that would further exacerbate disease. However, in the diaphragm, ion-handling dynamics are different and are more likely to favor forward-mode NCX1 activity leading to sustained Ca2+ efflux, thus having a protective effect against disease. This is consistent with greater NKA levels in the diaphragm versus the quadriceps that would tend to keep Na+ levels lower, even with enhanced TRP channel activity.
With respect to treatments for human MD, our results strengthen the overall paradigm that Ca2+ removal strategies have therapeutic potential. For example, overexpression of SERCA2 in the heart using an adeno-associated virus (AAV) vector has been through clinical trials and has shown early success in treating heart failure by recharging the SR with Ca2+ and removing diastolic Ca2+ more quickly (66). Thus, a similar strategy, if applied to skeletal muscle, might represent a “universal” approach to treating MD of many different etiologies by reducing myofiber intracellular Ca2+ and reducing ongoing necrosis rates. With respect to NCX1, our results suggest that a pharmacologic inhibitor specific for reverse-mode Ca2+ entry would be a bona fide therapeutic strategy. Ranolazine might be an appropriate therapeutic agent to consider, as it is known to reduce intracellular Na+ levels, possibly by reducing the leak activity of Nav1.4, as well as possibly by inhibiting reverse-mode NCX1 activity (57–59, 62, 63). Thus, even though this inhibitor is admittedly multifactorial, 8 weeks of ranolazine treatment in Sgcd−/− mice resulted in significantly less muscle pathology. In addition to these medicinal applications, our study is the first to suggest functional implications of the way in which Na+ overload can be pathological in dystrophic fibers by leading to enhanced Ca2+ entry through NCX1.
ACKNOWLEDGMENTS
This work was supported by grants from the NIH (J.D.M., P.D.A., K.D.P., J.R.L., and J.M.). J.D.M was also supported by the Howard Hughes Medical Institute. A.R.B. was supported by a Medical Scientist Training Program training grant at the University of Cincinnati.
We also thank Jeffrey Widrick, Department of Physical Medicine and Rehabilitation, Harvard Medical School Spaulding Rehabilitation Hospital, Boston, MA, for excellent technical assistance.
Footnotes
Published ahead of print 24 March 2014
REFERENCES
- 1.Durbeej M, Campbell KP. 2002. Muscular dystrophies involving the dystrophin-glycoprotein complex: an overview of current mouse models. Curr. Opin. Genet. Dev. 12:349–361. 10.1016/S0959-437X(02)00309-X [DOI] [PubMed] [Google Scholar]
- 2.Allen DG, Whitehead NP. 2011. Duchenne muscular dystrophy—what causes the increased membrane permeability in skeletal muscle? Int. J. Biochem. Cell Biol. 43:290–294. 10.1016/j.biocel.2010.11.005 [DOI] [PubMed] [Google Scholar]
- 3.Allen DG, Whitehead NP, Yeung EW. 2005. Mechanisms of stretch-induced muscle damage in normal and dystrophic muscle: role of ionic changes. J. Physiol. 567:723–735. 10.1113/jphysiol.2005.091694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Millay DP, Sargent MA, Osinska H, Baines CP, Barton ER, Vuagniaux G, Sweeney HL, Robbins J, Molkentin JD. 2008. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 14:442–447. 10.1038/nm1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Han R, Grounds MD, Bakker AJ. 2006. Measurement of sub-membrane [Ca2+] in adult myofibers and cytosolic [Ca2+] in myotubes from normal and mdx mice using the Ca2+ indicator FFP-18. Cell Calcium 40:299–307. 10.1016/j.ceca.2006.04.016 [DOI] [PubMed] [Google Scholar]
- 6.Hopf FW, Turner PR, Denetclaw WF, Jr, Reddy P, Steinhardt RA. 1996. A critical evaluation of resting intracellular free calcium regulation in dystrophic mdx muscle. Am. J. Physiol. 271:C1325–C1339 [DOI] [PubMed] [Google Scholar]
- 7.Turner PR, Fong PY, Denetclaw WF, Steinhardt RA. 1991. Increased calcium influx in dystrophic muscle. J. Cell Biol. 115:1701–1712. 10.1083/jcb.115.6.1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Backer F, Vandebrouck C, Gailly P, Gillis JM. 2002. Long-term study of Ca(2+) homeostasis and of survival in collagenase-isolated muscle fibres from normal and mdx mice. J. Physiol. 542:855–865. 10.1113/jphysiol.2002.020487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mallouk N, Jacquemond V, Allard B. 2000. Elevated subsarcolemmal Ca2+ in mdx mouse skeletal muscle fibers detected with Ca2+-activated K+ channels. Proc. Natl. Acad. Sci. U. S. A. 97:4950–4955. 10.1073/pnas.97.9.4950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Altamirano F, Lopez JR, Henriquez C, Molinski T, Allen PD, Jaimovich E. 2012. Increased resting intracellular calcium modulates NF-kappaB-dependent inducible nitric-oxide synthase gene expression in dystrophic mdx skeletal myotubes. J. Biol. Chem. 287:20876–20887. 10.1074/jbc.M112.344929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pressmar J, Brinkmeier H, Seewald MJ, Naumann T, Rudel R. 1994. Intracellular Ca2+ concentrations are not elevated in resting cultured muscle from Duchenne (DMD) patients and in MDX mouse muscle fibres. Pflugers Arch. 426:499–505. 10.1007/BF00378527 [DOI] [PubMed] [Google Scholar]
- 12.Head SI. 1993. Membrane potential, resting calcium and calcium transients in isolated muscle fibres from normal and dystrophic mice. J. Physiol. 469:11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gailly P, Boland B, Himpens B, Casteels R, Gillis JM. 1993. Critical evaluation of cytosolic calcium determination in resting muscle fibres from normal and dystrophic (mdx) mice. Cell Calcium 14:473–483. 10.1016/0143-4160(93)90006-R [DOI] [PubMed] [Google Scholar]
- 14.Millay DP, Goonasekera SA, Sargent MA, Maillet M, Aronow BJ, Molkentin JD. 2009. Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc. Natl. Acad. Sci. U. S. A. 106:19023–19028. 10.1073/pnas.0906591106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iwata Y, Katanosaka Y, Arai Y, Shigekawa M, Wakabayashi S. 2009. Dominant-negative inhibition of Ca2+ influx via TRPV2 ameliorates muscular dystrophy in animal models. Hum. Mol. Genet. 18:824–834. 10.1093/hmg/ddn408 [DOI] [PubMed] [Google Scholar]
- 16.Goonasekera SA, Lam CK, Millay DP, Sargent MA, Hajjar RJ, Kranias EG, Molkentin JD. 2011. Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. J. Clin. Invest. 121:1044–1052. 10.1172/JCI43844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirn C, Shapovalov G, Petermann O, Roulet E, Ruegg UT. 2008. Nav1.4 deregulation in dystrophic skeletal muscle leads to Na+ overload and enhanced cell death. J. Gen. Physiol. 132:199–208. 10.1085/jgp.200810024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miles MT, Cottey E, Cottey A, Stefanski C, Carlson CG. 2011. Reduced resting potentials in dystrophic (mdx) muscle fibers are secondary to NF-kappaB-dependent negative modulation of ouabain sensitive Na+-K+ pump activity. J. Neurol. Sci. 303:53–60. 10.1016/j.jns.2011.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mathes C, Bezanilla F, Weiss RE. 1991. Sodium current and membrane potential in EDL muscle fibers from normal and dystrophic (mdx) mice. Am. J. Physiol. Cell Physiol. C718–C725 [DOI] [PubMed] [Google Scholar]
- 20.Dunn JF, Bannister N, Kemp GJ, Publicover SJ. 1993. Sodium is elevated in mdx muscles: ionic interactions in dystrophic cells. J. Neurol. Sci. 114:76–80. 10.1016/0022-510X(93)90052-Z [DOI] [PubMed] [Google Scholar]
- 21.Weber MA, Nagel AM, Jurkat-Rott K, Lehmann-Horn F. 2011. Sodium (23Na) MRI detects elevated muscular sodium concentration in Duchenne muscular dystrophy. Neurology 77:2017–2024. 10.1212/WNL.0b013e31823b9c78 [DOI] [PubMed] [Google Scholar]
- 22.Weber MA, Nagel AM, Wolf MB, Jurkat-Rott K, Kauczor HU, Semmler W, Lehmann-Horn F. 2012. Permanent muscular sodium overload and persistent muscle edema in Duchenne muscular dystrophy: a possible contributor of progressive muscle degeneration. J. Neurol. 259:2385–2392. 10.1007/s00415-012-6512-8 [DOI] [PubMed] [Google Scholar]
- 23.Blaustein MP, Lederer WJ. 1999. Sodium/calcium exchange: its physiological implications. Physiol. Rev. 79:763–854 [DOI] [PubMed] [Google Scholar]
- 24.Fraysse B, Rouaud T, Millour M, Fontaine-Perus J, Gardahaut MF, Levitsky DO. 2001. Expression of the Na(+)/Ca(2+) exchanger in skeletal muscle. Am. J. Physiol. Cell Physiol. 280:C146–C154 [DOI] [PubMed] [Google Scholar]
- 25.Sokolow S, Manto M, Gailly P, Molgo J, Vandebrouck C, Vanderwinden JM, Herchuelz A, Schurmans S. 2004. Impaired neuromuscular transmission and skeletal muscle fiber necrosis in mice lacking Na/Ca exchanger 3. J. Clin. Invest. 113:265–273. 10.1172/JCI200418688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deval E, Levitsky DO, Marchand E, Cantereau A, Raymond G, Cognard C. 2002. Na(+)/Ca(2+) exchange in human myotubes: intracellular calcium rises in response to external sodium depletion are enhanced in DMD. Neuromuscul. Disord. 12:665–673. 10.1016/S0960-8966(02)00022-6 [DOI] [PubMed] [Google Scholar]
- 27.Iwata Y, Katanosaka Y, Hisamitsu T, Wakabayashi S. 2007. Enhanced Na+/H+ exchange activity contributes to the pathogenesis of muscular dystrophy via involvement of P2 receptors. Am. J. Pathol. 171:1576–1587. 10.2353/ajpath.2007.070452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balnave CD, Allen DG. 1998. Evidence for Na+/Ca2+ exchange in intact single skeletal muscle fibers from the mouse. Am. J. Physiol. 274:C940–C946 [DOI] [PubMed] [Google Scholar]
- 29.Brennan KJ, Hardeman EC. 1993. Quantitative analysis of the human alpha-skeletal actin gene in transgenic mice. J. Biol. Chem. 268:719–725 [PubMed] [Google Scholar]
- 30.Nicoll DA, Longoni S, Philipson KD. 1990. Molecular cloning and functional expression of the cardiac sarcolemmal Na(+)-Ca2+ exchanger. Science 250:562–565. 10.1126/science.1700476 [DOI] [PubMed] [Google Scholar]
- 31.Weber CR, Ginsburg KS, Philipson KD, Shannon TR, Bers DM. 2001. Allosteric regulation of Na/Ca exchange current by cytosolic Ca in intact cardiac myocytes. J. Gen. Physiol. 117:119–131. 10.1085/jgp.117.2.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.James PF, Grupp IL, Grupp G, Woo AL, Askew GR, Croyle ML, Walsh RA, Lingrel JB. 1999. Identification of a specific role for the Na,K-ATPase alpha 2 isoform as a regulator of calcium in the heart. Mol. Cell 3:555–563. 10.1016/S1097-2765(00)80349-4 [DOI] [PubMed] [Google Scholar]
- 33.Henderson SA, Goldhaber JI, So JM, Han T, Motter C, Ngo A, Chantawansri C, Ritter MR, Friedlander M, Nicoll DA, Frank JS, Jordan MC, Roos KP, Ross RS, Philipson KD. 2004. Functional adult myocardium in the absence of Na+-Ca2+ exchange: cardiac-specific knockout of NCX1. Circulat. Res. 95:604–611. 10.1161/01.RES.0000142316.08250.68 [DOI] [PubMed] [Google Scholar]
- 34.Bothe GW, Haspel JA, Smith CL, Wiener HH, Burden SJ. 2000. Selective expression of Cre recombinase in skeletal muscle fibers. Genesis 26:165–166. [DOI] [PubMed] [Google Scholar]
- 35.Nolan T, Hands RE, Bustin SA. 2006. Quantification of mRNA using real-time RT-PCR. Nat. protoc. 1:1559–1582. 10.1038/nprot.2006.236 [DOI] [PubMed] [Google Scholar]
- 36.Davis J, Kwong JQ, Kitsis RN, Molkentin JD. 2013. Apoptosis repressor with a CARD domain (ARC) restrains Bax-mediated pathogenesis in dystrophic skeletal muscle. PLoS One 8:e82053. 10.1371/journal.pone.0082053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eltit JM, Yang T, Li H, Molinski TF, Pessah IN, Allen PD, Lopez JR. 2010. RyR1-mediated Ca2+ leak and Ca2+ entry determine resting intracellular Ca2+ in skeletal myotubes. J. Biol. Chem. 285:13781–13787. 10.1074/jbc.M110.107300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lopez JR, Alamo L, Caputo C, Dipolo R, Vergara J. 1983. Determination of ionic calcium in frog skeletal-muscle fibers. Biophys. J. 43:1–4. 10.1016/S0006-3495(83)84316-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eltit JM, Ding X, Pessah IN, Allen PD, Lopez JR. 2013. Nonspecific sarcolemmal cation channels are critical for the pathogenesis of malignant hyperthermia. FASEB J. 27:991–1000. 10.1096/fj.12-218354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lovelock JD, Monasky MM, Jeong EM, Lardin HA, Liu H, Patel BG, Taglieri DM, Gu L, Kumar P, Pokhrel N, Zeng D, Belardinelli L, Sorescu D, Solaro RJ, Dudley SC., Jr 2012. Ranolazine improves cardiac diastolic dysfunction through modulation of myofilament calcium sensitivity. Circulat. Res. 110:841–850. 10.1161/CIRCRESAHA.111.258251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, Swanson MS, Thornton CA. 2006. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 15:2087–2097. 10.1093/hmg/ddl132 [DOI] [PubMed] [Google Scholar]
- 42.Rajabi M, Kassiotis C, Razeghi P, Taegtmeyer H. 2007. Return to the fetal gene program protects the stressed heart: a strong hypothesis. Heart Fail. Rev. 12:331–343. 10.1007/s10741-007-9034-1 [DOI] [PubMed] [Google Scholar]
- 43.Chen YW, Zhao P, Borup R, Hoffman EP. 2000. Expression profiling in the muscular dystrophies: identification of novel aspects of molecular pathophysiology. J. Cell Biol. 151:1321–1336. 10.1083/jcb.151.6.1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adachi-Akahane S, Lu L, Li Z, Frank JS, Philipson KD, Morad M. 1997. Calcium signaling in transgenic mice overexpressing cardiac Na(+)-Ca2+ exchanger. J. Gen. physiol. 109:717–729. 10.1085/jgp.109.6.717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pott C, Goldhaber JI, Philipson KD. 2004. Genetic manipulation of cardiac Na+/Ca2+ exchange expression. Biochem. Biophys. Res. Commun. 322:1336–1340. 10.1016/j.bbrc.2004.08.038 [DOI] [PubMed] [Google Scholar]
- 46.Noireaud J, Leoty C. 1989. Na+-Ca-2+ exchange in limb muscles of dystrophic (C57 Bl/6j Dy2j/Dy2j) mice. Q. J. Exp. Physiol. 74:75–77 [DOI] [PubMed] [Google Scholar]
- 47.Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, McNeil PL, Campbell KP. 2003. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 423:168–172. 10.1038/nature01573 [DOI] [PubMed] [Google Scholar]
- 48.Hoffman EP, Brown RH, Kunkel LM. 1987. Dystrophin: the protein product of the Duchenne muscular-dystrophy locus. Cell 51:919–928. 10.1016/0092-8674(87)90579-4 [DOI] [PubMed] [Google Scholar]
- 49.Sicinski P, Geng Y, Rydercook AS, Barnard EA, Darlison MG, Barnard PJ. 1989. The molecular basis of muscular dystrophy in the Mdx mouse: a point mutation. Science 244:1578–1580. 10.1126/science.2662404 [DOI] [PubMed] [Google Scholar]
- 50.Orlowski J, Lingrel JB. 1988. Tissue-specific and developmental regulation of rat Na,K-ATPase catalytic alpha isoform and beta subunit mRNAs. J. Biol. Chem. 263:10436–10442 [PubMed] [Google Scholar]
- 51.Radzyukevich TL, Moseley AE, Shelly DA, Redden GA, Behbehani MM, Lingrel JB, Paul RJ, Heiny JA. 2004. The Na(+)-K(+)-ATPase alpha2-subunit isoform modulates contractility in the perinatal mouse diaphragm. Am. J. Physiol. Cell Physiol. 287:C1300–C1310. 10.1152/ajpcell.00231.2004 [DOI] [PubMed] [Google Scholar]
- 52.Gandevia SC, McKenzie DK, Neering IR. 1983. Endurance properties of respiratory and limb muscles. Respir. Physiol. 53:47–61. 10.1016/0034-5687(83)90015-4 [DOI] [PubMed] [Google Scholar]
- 53.Barr DJ, Green HJ, Lounsbury DS, Rush JW, Ouyang J. 2005. Na+-K+-ATPase properties in rat heart and skeletal muscle 3 mo after coronary artery ligation. J. Appl. Physiol. 99:656–664. 10.1152/japplphysiol.00343.2004 [DOI] [PubMed] [Google Scholar]
- 54.Pereon Y, Dettbarn C, Lu Y, Westlund KN, Zhang JT, Palade P. 1998. Dihydropyridine receptor isoform expression in adult rat skeletal muscle. Pflugers Arch. 436:309–314. 10.1007/s004240050637 [DOI] [PubMed] [Google Scholar]
- 55.Jorgensen LH, Blain A, Greally E, Laval SH, Blamire AM, Davison BJ, Brinkmeier H, MacGowan GA, Schroder HD, Bushby K, Straub V, Lochmuller H. 2011. Long-term blocking of calcium channels in mdx mice results in differential effects on heart and skeletal muscle. Am. J. Pathol. 178:273–283. 10.1016/j.ajpath.2010.11.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stedman HH, Sweeney HL, Shrager JB, Maguire HC, Panettieri RA, Petrof B, Narusawa M, Leferovich JM, Sladky JT, Kelly AM. 1991. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 352:536–539. 10.1038/352536a0 [DOI] [PubMed] [Google Scholar]
- 57.Wang GK, Calderon J, Wang SY. 2008. State- and use-dependent block of muscle Nav1.4 and neuronal Nav1.7 voltage-gated Na+ channel isoforms by ranolazine. Mol. Pharmacol. 73:940–948. 10.1124/mol.107.041541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.El-Bizri N, Kahlig KM, Shryock JC, George AL, Belardinelli L, Rajamani S. 2011. Ranolazine block of human Na(v)1.4 sodium channels and paramyotonia congenita mutants. Channels 5:161–172. 10.4161/chan.5.2.14851 [DOI] [PubMed] [Google Scholar]
- 59.Soliman D, Wang LG, Hamming KSC, Yang W, Fatehi M, Carter CC, Clanachan AS, Light PE. 2012. Late sodium current inhibition alone with ranolazine is sufficient to reduce ischemia- and cardiac glycoside-induced calcium overload and contractile dysfunction mediated by reverse-mode sodium/calcium exchange. J. Pharmacol. Exp. Ther. 343:325–332. 10.1124/jpet.112.196949 [DOI] [PubMed] [Google Scholar]
- 60.Wasserstrom JA, Sharma R, O'Toole MJ, Zheng JB, Kelly JE, Shryock J, Belardinelli L, Aistrup GL. 2009. Ranolazine antagonizes the effects of increased late sodium current on intracellular calcium cycling in rat isolated intact heart. J. Pharmacol. Exp. Ther. 331:382–391. 10.1124/jpet.109.156471 [DOI] [PubMed] [Google Scholar]
- 61.Fraser H, Belardinelli L, Wang LG, Light PE, McVeigh JJ, Clanachan AS. 2006. Ranolazine decreases diastolic calcium accumulation caused by ATX-II or ischemia in rat hearts. J. Mol. Cell. Cardiol. 41:1031–1038. 10.1016/j.yjmcc.2006.08.012 [DOI] [PubMed] [Google Scholar]
- 62.Zhang XQ, Yamada S, Barry WH. 2008. Ranolazine inhibits an oxidative stress-induced increase in myocyte sodium and calcium loading during simulated-demand ischemia. J. Cardiovasc. Pharmacol. 51:443–449. 10.1097/FJC.0b013e318168e711 [DOI] [PubMed] [Google Scholar]
- 63.Sossalla S, Wagner S, Rasenack ECL, Ruff H, Weber SL, Schoendube FA, Tirilomis T, Tenderich G, Hasenfuss G, Belardinelli L, Maier LS. 2008. Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts: role of late sodium current and intracellular ion accumulation. J. Mol. Cell. Cardiol. 45:32–43. 10.1016/j.yjmcc.2008.03.006 [DOI] [PubMed] [Google Scholar]
- 64.Spencer MJ, Mellgren RL. 2002. Overexpression of a calpastatin transgene in mdx muscle reduces dystrophic pathology. Hum. Mol. Genet. 11:2645–2655. 10.1093/hmg/11.21.2645 [DOI] [PubMed] [Google Scholar]
- 65.Takekura H, Sun X, Franzini-Armstrong C. 1994. Development of the excitation-contraction coupling apparatus in skeletal muscle: peripheral and internal calcium release units are formed sequentially. J. Muscle Res. Cell Motil. 15:102–118. 10.1007/BF00130422 [DOI] [PubMed] [Google Scholar]
- 66.Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, Borow K, Dittrich H, Zsebo KM, Hajjar RJ, Calcium Up-Regulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID) Trial Investigators 2009. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J. Card. Fail. 15:171–181. 10.1016/j.cardfail.2009.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]