

Abstract
Peroxisome proliferator-activated receptor gamma (PPARγ) coactivator 1α (PGC-1α) and PGC-1β have been shown to be intimately involved in the transcriptional regulation of cellular energy metabolism as well as other biological processes, but both coactivator proteins are expressed in many other tissues and organs in which their function is, in essence, unexplored. Here, we found that both PGC-1 proteins are abundantly expressed in maturing erythroid cells. PGC-1α and PGC-1β compound null mutant (Pgc-1c) animals express less β-like globin mRNAs throughout development; consequently, neonatal Pgc-1c mice exhibit growth retardation and profound anemia. Flow cytometry shows that the number of mature erythrocytes is markedly reduced in neonatal Pgc-1c pups, indicating that erythropoiesis is severely compromised. Furthermore, hematoxylin and eosin staining revealed necrotic cell death and cell loss in Pgc-1c livers and spleen. Chromatin immunoprecipitation studies revealed that both PGC-1α and -1β, as well as two nuclear receptors, TR2 and TR4, coordinately bind to the various globin gene promoters. In addition, PGC-1α and -1β can interact with TR4 to potentiate transcriptional activation. These data provide new insights into our understanding of globin gene regulation and raise the interesting possibility that the PGC-1 coactivators can interact with TR4 to elicit differential stage-specific effects on globin gene transcription.
INTRODUCTION
The transcriptional coactivator PPARGC1A (PGC-1α) was originally identified based on its functional interaction with the nuclear receptor peroxisome proliferator-activated receptor gamma (PPARγ) in brown fat (1, 2). Subsequently, a second closely related family member, PPARGC1B (PGC-1β), was identified that shares a similar structure with PGC-1α (3). Both PGC-1α and PGC-1β can activate a cascade of genes involved in mitochondrial biogenesis and respiratory function in adipocytes, cardiac myocytes, and myogenic cells (3–6). In addition, the PGC-1 coactivators control hepatic gluconeogenesis and lipoprotein metabolism, skeletal muscle fiber determination, circadian clock function, and angiogenesis, as well as macrophage polarization (6–16).
PGC-1α and PGC-1β exert their effects on the transcription of target genes through their interactions with a variety of nuclear receptors (e.g., PPARγ, PPARα, and ERRα) and the recruitment of chromatin-remodeling complexes (1, 17–19). Recently, we reported that PGC-1α can potentiate transcriptional activation of the orphan nuclear receptor TR4 (NR2C2) in a cell-based transfection assay (20). TR4 and its evolutionarily related homolog, TR2 (NR2C1), have been shown to play key roles in regulating the embryonic and fetal globin genes in erythroid cells and may prove to be useful for identifying therapeutic targets for sickle cell disease and β-thalassemia (21–25). Recently, we discovered that the expression of some erythroid genes was lower after short hairpin RNA (shRNA)-mediated TR4 mRNA knockdown, which indicated that TR4 also functions as a transcriptional activator (L. Shi, Y. X. Lin, M. C. Sierant, F. Zhu, S. Cui, Y. Guan, S. Maureen, O. Tanabe, K. C. Lim, and J. D. Engel, submitted for publication). Moreover, in humanized sickle cell model mice, TR2 and TR4 overexpression significantly induced fetal HbF synthesis, thereby mitigating sickle cell disease phenotypes (25). However, the molecular mechanisms that convert TR2 and/or TR4 from transcriptional repressor complexes to transcriptional activation and vice versa are unknown. The observation that transcription can be potentiated by PGC-1α through TR4 suggested that the PGC-1 coactivators function as transcriptional coactivators in erythroid cells (20).
Here, we investigated the expression of the β- and α-like globin genes in mice bearing individual or combined deficiencies in germ line loss-of-function mutations in the Pgc-1α or Pgc-1β gene. The results show that compound mutant Pgc-1α−/−:Pgc-1β−/− (referred to here as Pgc-1c) mice exhibit significantly reduced embryonic Hbb-y (εy), Hbb-bh1 (βh1), and Hba-x (ζ), as well as adult Hbb-b1 (βmaj) and Hba-a1 (α) globin gene expression in the embryonic day 10.5 (e10.5) yolk sac. The expression of these same globin genes is also compromised in the fetal livers of e14.5 embryos and in the spleens of pups at birth (p0). In addition, neonatal Pgc-1c mice exhibit anemia, and their peripheral blood smears and flow-cytometric profiles reveal erythroblastosis, thrombocytopenia, and leukopenia, demonstrating multilineage hematopoietic defects in Pgc-1 mutant animals. Hematoxylin and eosin (H&E) staining revealed necrotic cell death and cell loss in Pgc-1c livers and spleens, both of which accumulated lipid-filled adipocytes.
Here, we show that coactivator PGC-1α is only able to stably interact with orphan nuclear receptor TR2, while both PGC-1α and PGC-1β can form stable complexes with, and potentiate transcriptional activation by, TR4. Chromatin immunoprecipitation studies further revealed that PGC-1α and -1β, together with TR2/TR4, bind to the promoters of the embryonic εy- and βh1-globin genes in e11.5 erythrocytes but are bound only at the εy promoter by e14.5.
These data demonstrate that PGC-1α and -1β together play an essential role in erythropoiesis and globin gene regulation. The data are consistent with the hypothesis that PGC-1α and PGC-1β exert direct effects on transcriptional activation of the globin genes, perhaps acting through the orphan nuclear receptor TR2 and/or TR4, and that the mutation of these coactivator genes significantly reduces globin gene expression, thereby leading to anemia.
MATERIALS AND METHODS
Mice.
Compound heterozygotes (Pgc-1α+/−:Pgc-1β+/−) bred from Pgc-1α−/− and Pgc-1β−/− singly null mutant mice (26) (27) were used to generate compound homozygous mutants (Pgc-1c) in addition to all other anticipated genotypes. All procedures described were approved by the Animal Care and Use Committee of the University of Michigan.
Quantitative real-time PCR.
cDNA was synthesized from total RNA using Superscript cDNA synthesis kits (Invitrogen) (28), and quantitative real-time reverse transcription-PCR (RT-PCR) was performed on an ABI Prism 7000 instrument (Applied Biosystems) as previously described (29). The abundance of target gene mRNAs in each population was determined based on cycle threshold (CT) values of the quantitative RT-PCR and the experimentally determined amplification efficiency of each primer set (see Table S1 in the supplemental material) and normalized to the abundance of 18S rRNA or glycophorin A (GPA) as internal controls. Gpa encodes an erythroid-specific protein that is expressed in both primitive and definitive erythroid cells.
Hematologic and histological analysis.
Neonatal p0 pups were sacrificed, peripheral blood was collected, and counts were quantified as previously reported (25). Peripheral blood smears were stained with Wright-Giemsa (Sigma). Tissue preparations from the organs were fixed in 70% alcoholic formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E) (25).
Flow cytometry.
Bone marrow, spleen, and liver cells from p0 pups were stained with anti-mouse CD71, Ter119, CD8, or Mac1 (from BioLegend), CD19 or CD4 (from BD Biosciences), or B220 or Gr1 (from eBioscience) antibody on ice for 15 min in the dark. Cells were washed twice and resuspended in phosphate-buffered saline (PBS) plus 0.5% bovine serum albumin (BSA). Analytical flow cytometry was performed using a FACSCanto II instrument (BD Biosciences) (30).
ChIP assay.
Chromatin immunoprecipitation (ChIP) assays were performed as previously described (24). Circulating blood cells from e11.5 embryos and fetal liver cells from e14.5 embryos and neonatal p0 pups were harvested for ChIP assays. Rabbit sera recognizing PGC-1α (sc-13067; Santa Cruz Biotech), PGC-1β (sc-67285; Santa Cruz Biotech), and TR2 and TR4 (22) were used for immunoprecipitation. Primers used for real-time PCR in ChIP assays are listed in Table S1 in the supplemental material.
Immunoprecipitation and Western blotting.
PGC-1α, PGC-1β, TR2, and TR4 antibodies were coupled to protein G-Dynabeads (Invitrogen) and then incubated with nuclear extracts prepared from mouse erythroleukemia (MEL) cells. Immunoprecipitated proteins were eluted from the beads and then subjected to SDS-PAGE and immunoblotting. The proteins were transferred to a nitrocellulose membrane and probed with specific primary antibodies, followed by fluorescence-conjugated secondary antibodies (Li-Cor). The proteins were visualized on an Odyssey infrared imaging system (Li-Cor).
Luciferase assays.
HEK293T cells were transfected using Lipofectamine 2000 (Invitrogen) with an equal amount of DNA reporter (DR-pGL3-luc) (20) and expression plasmid (TR2, TR4, PGC-1α, or PGC-1β) (3, 21). A Renilla luciferase vector (Promega) was cotransfected as an internal control. Dual-luciferase assays were performed according to the manufacturer's instructions.
RESULTS
PGC-1α and PGC-1β are both expressed in erythroid cells during development.
To understand the roles that PGC-1α and PGC-1β might play in erythroid gene regulation, we examined their expression in differentiating erythroid cells during mouse development (Fig. 1). Total RNA and protein lysates were prepared from sorted wild-type (WT) Ter119+ (erythroid) cells on e10.5 and e14.5, at birth (p0), and 6 weeks after birth from both Ter119+ (w6) and Ter119− (w6−) cells. Quantitative RT-PCR and Western blotting were performed to document mRNA and protein expression, respectively, of PGC-1α and PGC-1β during erythroid differentiation.
FIG 1.
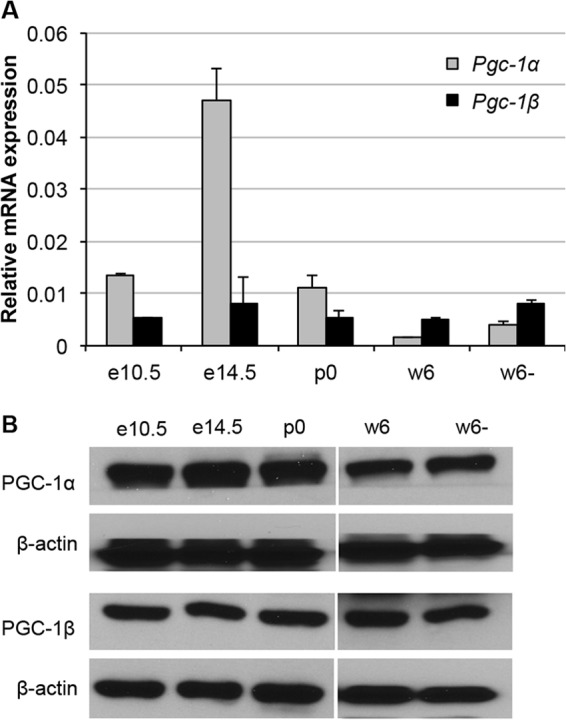
Expression of PGC-1α and PGC-1β during erythroid cell development. (A) Total RNA was extracted from flow-sorted wild-type Ter119+ cells recovered from e10.5 yolk sacs (primitive erythrocytes), e14.5 fetal livers (definitive erythroid cells), p0 fetal livers, and the circulating blood of w6 postnatal animals. Ter119− cells of w6 animals were used as a nucleated nonerythroid blood cell control. The diagram depicts the mRNA abundance of PGC-1α and PGC-1β as determined by reverse transcription and real-time quantitative PCR, normalized to 18S rRNA. (B) Western blotting was performed using the cell lysates from the samples described for panel A to show the expression of PGC-1α and PGC-1β at the protein level during erythroid cell differentiation.
PGC-1α expression was dynamically differentially regulated during erythroid cell development. PGC-1α was most abundantly expressed during embryonic stages, peaking at around e14.5 shortly after the primitive-to-definitive erythroid switch occurs, going from yolk sac to fetal liver erythropoiesis (31). After birth, PGC-1α expression in definitive erythroid cells (w6) and myeloid cells (w6−) diminished. In contrast, PGC-1β expression varies by less than 2-fold throughout erythroid development (Fig. 1A). Accumulation of the PGC-1α and PGC-1β proteins was generally consistent with their mRNA expression (Fig. 1B). The specificity of the anti-PGC-1α and anti-PGC-1β antibodies was validated (see Fig. S1 in the supplemental material).
PGC-1 loss affects expression of the globin genes at all developmental stages.
To investigate the effects of Pgc-1α or Pgc-1β loss of function on globin gene expression, quantitative RT-PCR was performed to measure the abundance of accumulated globin mRNAs in the yolk sacs of e10.5 embryos, the site of primitive erythropoiesis at this stage. Compared to WT mice, a significant reduction in the embryonic εy-, βh1-, ζ-, and adult α-globin genes was observed in compound mutant Pgc-1c mice (no βmaj-globin mRNA was detected at e10.5) (Fig. 2A). Loss of function in either Pgc-1α−/− or Pgc-1β−/− animals individually affects the expression of these same genes, but the consequences are more severe in the compound mutants (Fig. 2B, left) at e10.5, suggesting that Pgc-1α and Pgc-1β have complementary, compensatory activities at this stage. In addition, the expression of the εy- and βh1-globin genes was also statistically different in Pgc-1c compound mutants compared to single Pgc-1α or Pgc-1β mutants. The data indicate that Pgc-1α and Pgc-1β interact genetically in primitive erythroid cells at e10.5 and that both genes are required for full mouse β-like and α-like globin gene expression in primitive erythroid cells.
FIG 2.

Developmental stage-specific Pgc-1α and Pgc-1β loss-of-function effects on murine α- and β-globin gene expression. (A) The relative abundances of β-globin and α-globin transcripts (compared to glycophorin A mRNA abundance) was determined in e10.5 yolk sacs, e14.5 fetal livers, and p0 spleens of WT and compound mutant Pgc-1c mice. (B) The individual globin mRNA levels, normalized to the abundance of GPA, was set at 100% for each WT transcript. The values for expression of the same genes in various Pgc-1 mutant backgrounds are shown. Four embryos of each genotype were used to determine the mean globin/GPA mRNA ratio. Statistically significant differences in expression of the mutant embryos compared to the WT are indicated (*, P < 0.05; **, P < 0.01). Error bars represent standard deviations (SD).
At e14.5, the site of erythropoiesis shifts to the fetal liver, where definitive erythropoiesis ensues. PGC-1α and PGC-1β loss similarly results in diminished β-like and α-like globin transcript accumulation in the fetal livers of individual null mutant and in compound mutant Pgc-1c embryos (Fig. 2A). Curiously, and in contrast to what was observed in primitive erythropoiesis, there appeared to be no significant difference in globin gene expression between the individual Pgc-1 mutants and the compound mutant mice in definitive erythroid cells (Fig. 2B).
Loss of function in the Pgc-1α−/− or Pgc-1β−/− mice also significantly affected the expression of the adult βmaj- and α-globin genes in definitive p0 spleen erythroid cells (Fig. 2), but as in the fetal liver, there was no evidence for genetic interaction between the two coactivators at this stage of definitive erythropoiesis. These data indicate that PGC-1α and PGC-1β play stage-specific roles in mouse β-like and α-type globin gene expression, and that their activities differ during primitive and definitive erythropoiesis.
PGC-1 loss affects neonatal hematopoiesis.
To investigate the effect of PGC-1α and PGC-1β deficiencies on hematologic properties, complete blood counts were performed using peripheral blood from neonatal p0 pups, since the compound mutants exhibited growth retardation (see Fig. S2 in the supplemental material) and survived for only 1 or 2 days after birth. Compared to WT mice, single null mutant (Pgc-1α−/− or Pgc-1β−/−) newborns did not display significant differences in cellular phenotypes, while compound mutant Pgc-1c mice exhibited moderate anemia accompanied by a slightly lowered hematocrit, lower than average total hemoglobin, and fewer red blood cells (Table 1). White blood cells and platelet counts in the Pgc-1c mice were both significantly reduced, suggesting that the hematopoietic defects engendered on Pgc-1c dual loss of function were not restricted to erythropoiesis and anemia (Table 1).
TABLE 1.
Blood count results
| Blood count parametera | Value by cell typeb |
|||
|---|---|---|---|---|
| WT | Pgc-1α−/− | Pgc-1β−/− | Pgc-1c | |
| RBC (×106 cells/μl) | 4.8 ± 0.2 | 4.67 ± 0.2 | 4.9 ± 0.2 | 3.4 ± 0.2* |
| HGB (g/dl) | 11.5 ± 1 | 12 ± 0.6 | 11 ± 0.6 | 7.2 ± 0.5* |
| HCT (%) | 50.5 ± 2.9 | 50.33 ± 1.2 | 48 ± 2.5 | 40 ± 1.8* |
| MCV (fl) | 106.625 ± 1 | 107.98 ± 2.4 | 98.9 ± 2.2 | 119.7 ± 6 |
| MCH (pg) | 24.95 ± 0.8 | 25.63 ± 0.4 | 23.1 ± 1.2 | 21.6 ± 0.7* |
| MCHC (g/dl) | 23.4 ± 0.6 | 23.78 ± 0.6 | 23.3 ± 0.7 | 18.2 ± 0.7* |
| Retic (%) | 29.2 ± 0.4 | 21.5 ± 0.7 | 22.8 ± 0.9 | 12.9 ± 2.3* |
| RDW (%) | 18.9 ± 1.1 | 20.2 ± 1.8 | 17.3 ± 0.8 | 23.1 ± 3.1 |
| PLT (×103 cells/μl) | 500 ± 73 | 370 ± 88 | 520 ± 114 | 320 ± 63* |
| WBC (×103 cells/μl) | 4.5 ± 0.9 | 4.2 ± 0.9 | 6.2 ± 1.3 | 2.88 ± 0.5* |
| Neut (×103 cells/μl) | 1.27 ± 0.4 | 1.02 ± 0.1 | 0.6 ± 0.3 | 0.65 ± 0.1 |
| Lymph (×103 cells/μl) | 1.36 ± 0.3 | 0.87 ± 0.2 | 0.9 ± 0.5 | 0.79 ± 0.1 |
RBC, red blood cells; HGB, hemoglobin; HCT, hematocrit; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; Retic, reticulocyte count; RDW, red cell distribution width; PLT, platelet count; WBC, white blood cells; Lymph, lymphocyte count.
*, P < 0.05.
Reticulocyte counts in WT, singly null mutant (Pgc-1α−/− or Pgc-1β−/−), and compound mutant Pgc-1c pups were next examined by flow cytometry using thiazole orange for reticulocyte staining of whole peripheral blood (25). Reticulocyte counts were reduced in individual Pgc-1α−/− or Pgc-1β−/− pups and even more so in the compound mutant Pgc-1c pups compared to WT controls (Table 1; also see Fig. S3 in the supplemental material).
We next examined differentiation and developmental markers for the erythroid and other hematopoietic lineage cells in the bone marrow, spleen, and livers of WT and compound mutant Pgc-1c p0 pups (Fig. 3). Within the hematopoietic lineages, various stages of differentiation as well as differentiated cell types can be readily assessed by interrogation of a large variety of stage- and lineage-specific cell surface markers which can then be resolved by flow cytometry. The differentiation profile of WT and compound mutant erythroid cells was examined by staining for the transferrin receptor CD71, which is induced during the first two divisions after generation of erythroid CFU (32), and Ter119, which is stably incorporated into erythroid cell membranes during later stages of differentiation (33). Erythroid progenitor cells undergo several rounds of division to produce progeny that first express CD71, followed by expression of Ter119 (34); finally, CD71 is lost from the surface of mature erythroid cells.
FIG 3.

Differentiation profiles of hematopoietic lineage cells in the bone marrow (BM), spleen, and liver of WT and mutant p0 pups. Cells were stained with anti-mouse CD71 and Ter119 (A) or Gr1 and Mac1 (B) to assess the erythroid differentiation profiles in WT versus Pgc-1 singly or compound mutant mice. The numbers in each quadrant represent the fractional percentages (means ± SD from at least 3 pups of each genotype) of cells in that quadrant. Statistically significant differences in numbers for comparisons of the mutant embryos to the WT are indicated (*, P < 0.05).
When we examined the Pgc-1 mutants for possible erythroid maturation defects using the CD71 and Ter119 surface markers (Fig. 3A), we found that the number of mature red cells (found in the CD71− Ter119+ quadrant) was dramatically reduced in compound mutant pups in both the bone marrow and spleen, while the double-negative (CD71− Ter119−) population (containing early erythroid progenitors) was elevated in both tissues. Furthermore, the number of proerythroblasts (CD71+ Ter119−) was elevated in the compound mutants, compared to levels for WT pups, in the bone marrow, spleen, and liver (Fig. 3A). Interestingly, there was no significant difference between the individual Pgc-1 mutants and the WT mice in any of the three tissues. Thus, the data clearly demonstrate that compound Pgc-1 loss of function leads to a quite severe erythroid maturational deficiency not seen in either individual Pgc-1 mutant.
We next asked whether loss of Pgc-1 exclusively affected erythropoiesis or if other hematopoietic lineages also are altered. Myeloid cell differentiation was examined by flow cytometry, in this case staining cells for expression of granulocyte differentiation antigen 1 (Gr1) (35) and macrophage antigen 1 (Mac1), expressed on the surface of macrophages, granulocytes, and dendritic cells (DC) (36). Double-positive (Mac1+ Gr1+) cells represent mature granulocytic cells as well as immature myeloid cells (37). The number of Mac1+ Gr1+ cells was, like that for mature erythroid cells, dramatically reduced in the spleens and livers of Pgc-1c pups versus levels in WT or single-mutant p0 pups, but they were only slightly reduced in their bone marrows (Fig. 3B). Interestingly, there was an enormous increase in the number of Mac1+ Gr1− cells in the bone marrows of Pgc-1c p0 pups (22.6% versus 2.8%) (Fig. 3B), reminiscent of observations in animals that exhibit impaired immune reactivity and increased inflammation and infection (38).
The differentiation of lymphoid cells was also examined by staining for B220 and CD19 (to identify different stages of B cell differentiation) or CD4 and CD8 antigens (for T cell lineage markers). In both populations, there were no detectable differences in the development of either the B (see Fig. S4A in the supplemental material) or T (see Fig. S4B) cell lineages in single or compound Pgc-1 mutants versus WT p0 pups. These data are also consistent with the complete blood cell counts shown in Table 1, where only modest changes were detected in the lymphocytes of Pgc-1c versus WT p0 pups.
Histological and pathological analysis of PGC-1 mutant newborns.
To examine the histological characteristics of PGC-1 mutant neonatal whole blood, peripheral blood was examined on glass slides after fixation with Wright-Giemsa stains. Erythroblasts were detected only in the peripheral blood of Pgc-1c newborns; this erythroblastosis is not detectable in either WT or singly mutant (data not shown here) neonates (Fig. 4A). Erythroblasts (normoblasts) appeared to be immature, nucleated primitive red cells that are not normally found in the peripheral blood. Their appearance in the periphery usually (but not always) indicates that leukoerythroblastic anemia, hemolysis, hypoxia, and bone marrow infiltration or extramedullary hematopoiesis has been activated (39).
FIG 4.
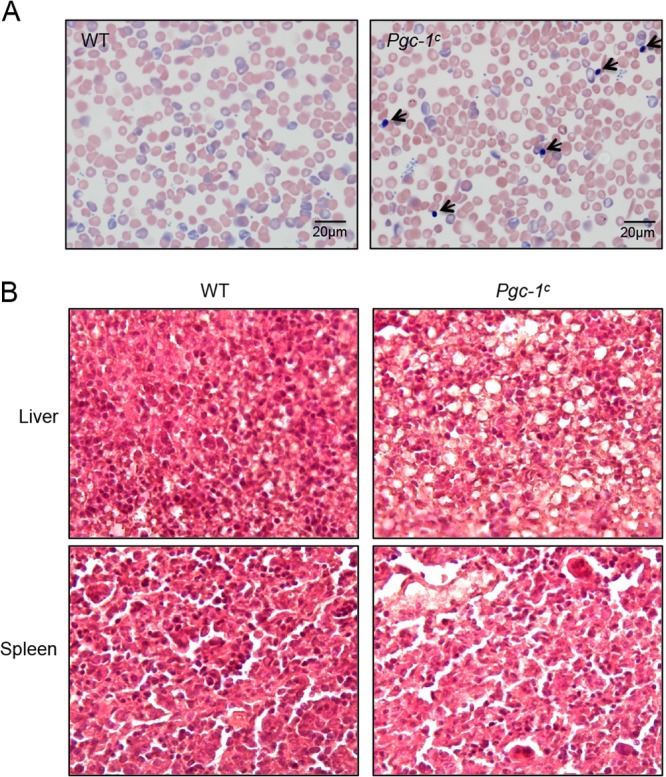
Histological and pathological analysis of PGC-1 mutant mice at birth. (A) Wright-Giemsa staining of peripheral blood smears of wild-type (WT) or PGC-1 compound mutant (Pgc-1c) newborns at p0. Arrows indicate erythroblasts (primitive nucleated erythrocytes). (B) Hematoxylin and eosin (H&E)-stained liver or spleen sections from WT and Pgc-1c mice revealed necrotic cell death and cell loss in Pgc-1c livers and spleens. Additionally, Pgc-1c livers and spleens accumulated abnormal numbers of adipocytes, displaying abundant accumulation of large lipid droplets.
We next performed pathological analysis of the livers and spleens recovered from WT and Pgc-1c p0 pups. Compared to those from the WT, H&E-stained liver sections of p0 Pgc-1c mice revealed diminished cellularity and significant necrotic cell death. Simultaneously, the number and size of adipocytes in Pgc-1c livers was increased and exhibited abundant accumulation of large lipid droplets (Fig. 4B), a feature commonly associated with impaired thermogenic function (40). H&E-stained spleen sections from the Pgc-1c mice also exhibited cell loss and fat accumulation compared to WT spleens (Fig. 4B).
PGC-1α, PGC-1β, and TR2/TR4 are associated selectively with globin gene promoters.
To address the underlying mechanism(s) that might regulate globin gene expression mediated through PGC-1α or PGC-1β, ChIP assays were performed on cells isolated from WT e11.5 murine embryonic peripheral blood (primitive red blood cells) or from e14.5 fetal livers and p0 spleens (definitive red blood cells). Equivalent cells isolated from the compound mutants served as negative controls (see Fig. S5 in the supplemental material). The data revealed a complex pattern of association of the two cofactors throughout erythroid development.
At e11.5, εy- and βh1-globin transcripts are abundantly expressed at around the time when βmaj-globin transcription initiates. Both PGC-1α and PGC-1β are bound at the e11.5 εy- and βh1-globin gene promoters, while only PGC-1β associates with the e11.5 adult βmaj-globin promoter (Fig. 5, left); neither of the cofactors is found associated with an irrelevant control sequence located 5.9 kbp 5′ to the βmaj-globin gene promoter (βmaj5.9) (Fig. 5). By e14.5, εy-globin is still being expressed, while βh1-globin is no longer transcribed (41). Consistent with this expression pattern, PGC-1α and PGC-1β remain bound to the εy-globin gene promoter but are no longer bound at the βh1 promoter (Fig. 5, center), consistent with the hypothesis that the two cofactors are dynamically promoting gene activation.
FIG 5.
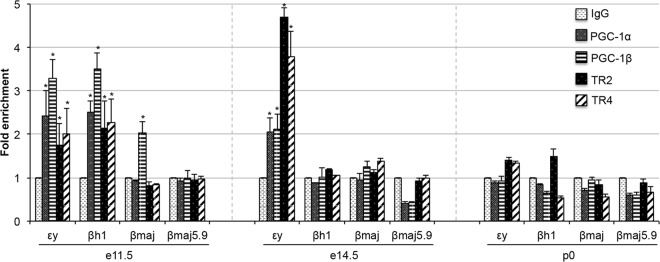
Differential PGC-1α, PGC-1β, TR2, and TR4 binding in the murine β-globin locus. The binding of PGC-1α, PGC-1β, TR2, and TR4 to select murine β-like globin gene promoters (as well as to an irrelevant control sequence located 5.9 kbp 5′ to the βmaj promoter) in e11.5 circulating blood cells (left), e14.5 fetal liver cells (center), or p0 fetal liver cells (right) was analyzed in ChIP experiments. Statistically significant enrichment of the orphan receptors and/or the coactivators at the promoters is indicated compared to the control IgG values (*, P < 0.05). Error bars represent standard errors.
By the time of birth, εy-globin transcription has completely ceased (41). In agreement with this observation, neither PGC-1α nor PGC-1β is still bound to any embryonic or adult globin gene promoters in p0 (definitive) erythroid cells (Fig. 5, right). These differential patterns of PGC-1α and PGC-1β association with the individual globin promoters suggest that PGC-1α and PGC-1β can drive often overlapping, but occasionally distinct, globin transcriptional regulatory programs.
In close correlation of the stage-specific association of PGC-1α and PGC-1β to the various globin gene promoters, we observed that both TR2 and TR4 bind to the e11.5 εy- and βh1-globin gene promoters but not to other sequences in the globin locus (Fig. 5, left). By e14.5, TR2 and TR4, like the PGC-1 coactivators, are found in association only with the εy-globin promoter but no longer with the βh1 promoter (Fig. 5, center). At the time of birth, there appears to be either no or marginal binding of either of these orphan nuclear receptors to any of the mouse β-globin promoters (Fig. 5, right). In summary, the data provide correlative evidence supporting the hypothesis that the PGC-1 coactivators are bound by the nuclear receptors TR2 and/or TR4, and that these complexes play a role in positively regulating the expression of the embryonic globin genes during erythroid cell development.
PGC-1α and PGC-1β both form stable complexes with orphan nuclear receptor TR4.
To determine whether the coactivators PGC-1α and PGC-1β can biochemically interact with either or both nuclear receptors TR2 and TR4, we performed coimmunoprecipitation assays using nuclear extracts prepared from replicating (untransfected) MEL (adult murine erythroleukemia) cells, where TR2 and TR4 were previously shown to be expressed (23). PGC-1α, PGC-1β, TR2, and TR4 were individually immunoprecipitated from MEL cell nuclear extracts and then interrogated on immunoblots to detect the expression of PGC-1 coactivators in those cells and their possible interactions with TR2 and/or TR4.
In immune complexes recovered using either the anti-PGC-1α or anti-PGC-1β antibody, TR4 was clearly coprecipitated, while TR2 was only detected in coprecipitate complexes with PGC-1α (Fig. 6). In the reciprocal experiment, when anti-TR2 or anti-TR4 antibody was used for the initial immune precipitation, both PGC-1α and PGC-1β were readily detected in complex with TR4, while only PGC-1α could be detected as a coprecipitate with TR2 (Fig. 6). These data confirm that a bona fide biochemical interaction takes place between coactivator PGC-1α or PGC-1β with nuclear receptor TR4, although the data do not provide further insight into the nature of this interaction or whether it is or is not direct. The data also demonstrate that TR2 is capable of interacting only with PGC-1α.
FIG 6.
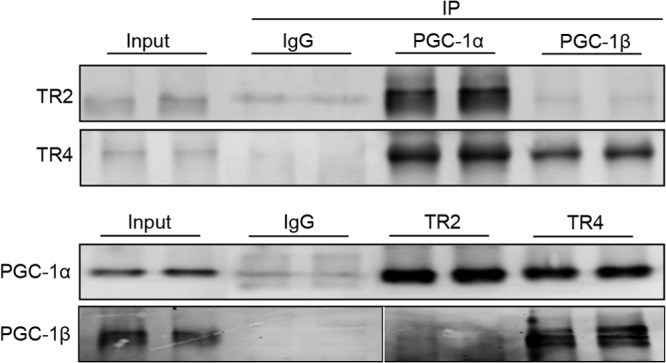
Naturally occurring interactions between TR2, TR4, PGC-1α, and PGC-1β in erythroid cells. Nuclear extracts prepared from untransfected MEL cells were incubated with protein G beads coupled to antibodies that recognize each of the proteins shown at the top or with control (nonspecific) IgG. A total of 150 μg of nuclear extract protein was electrophoresed on each lane. Each immunoprecipitate (IP) was subjected to SDS-PAGE in duplicate lanes, followed by immunoblotting and detection with the antibodies recognizing the protein indicated at the left.
PGC-1α and -1β interact with TR4 to potentiate transcriptional activation.
In order to define the interactions between PGC-1α and PGC-1β with TR2 and/or TR4 in vivo using cell-based cotransfection assays, we performed transient cotransfection of full-length TR2 or TR4 expression constructs to direct the expression of a luciferase reporter gene that was regulated by a promoter bearing five direct-repeat (DR1) sequences (AGGTCANAGGTCA [the repeated sequence is underlined]) corresponding to the optimal TR2/TR4 binding site (23). After cotransfection of either PGC-1α or PGC-1β with either TR2 or TR4 (or both) into HEK 293T cells (Fig. 7A), forcibly expressed PGC-1α or PGC-1β along with TR4 significantly enhanced reporter gene activity compared to the transfection of the individual factors, while forcibly expressed PGC-1α or PGC-1β plus TR2 quite modestly induced reporter activity (Fig. 7B). Compared to PGC-1α plus TR4 alone, which elicited maximum reporter stimulation, PGC-1α plus TR2 and TR4 modestly repressed reporter gene activity (by less than 2-fold), possibly because of competition for binding between TR2/TR4 heterodimers versus TR4 homodimers (TR2 homodimers bind only weakly to the DR1 sequence [22]). PGC-1β plus TR2/TR4 displayed reporter gene activity similar to that of PGC-1β plus TR4 alone, possibly reflecting the fact that TR2 does not interact with PGC-1β (Fig. 7B). These results suggested that both PGC-1α and PGC-1β are capable of significantly (e.g., >8-fold) potentiating transcription by TR4 in cell-based reporter assays (Fig. 7B, compare transfection of TR4 alone to TR4 plus PGC-1α). We also performed similar reporter gene assays using constructs directed by the ε-globin gene promoter (bearing two DR1 elements [22]) or the thymidine kinase (tk) promoter (no DR sequence). Activation patterns similar to those revealed using a synthetic 5× DR1 reporter were observed in assays employing the ε-globin gene promoter (Fig. 7C). In contrast, there was no activation of a tk promoter-directed construct (Fig. 7D). In summary, the data provide evidence that PGC-1α and PGC-1β exert direct transcriptional effects on the globin genes, possibly through their interaction with the orphan nuclear receptors TR2 and TR4.
FIG 7.
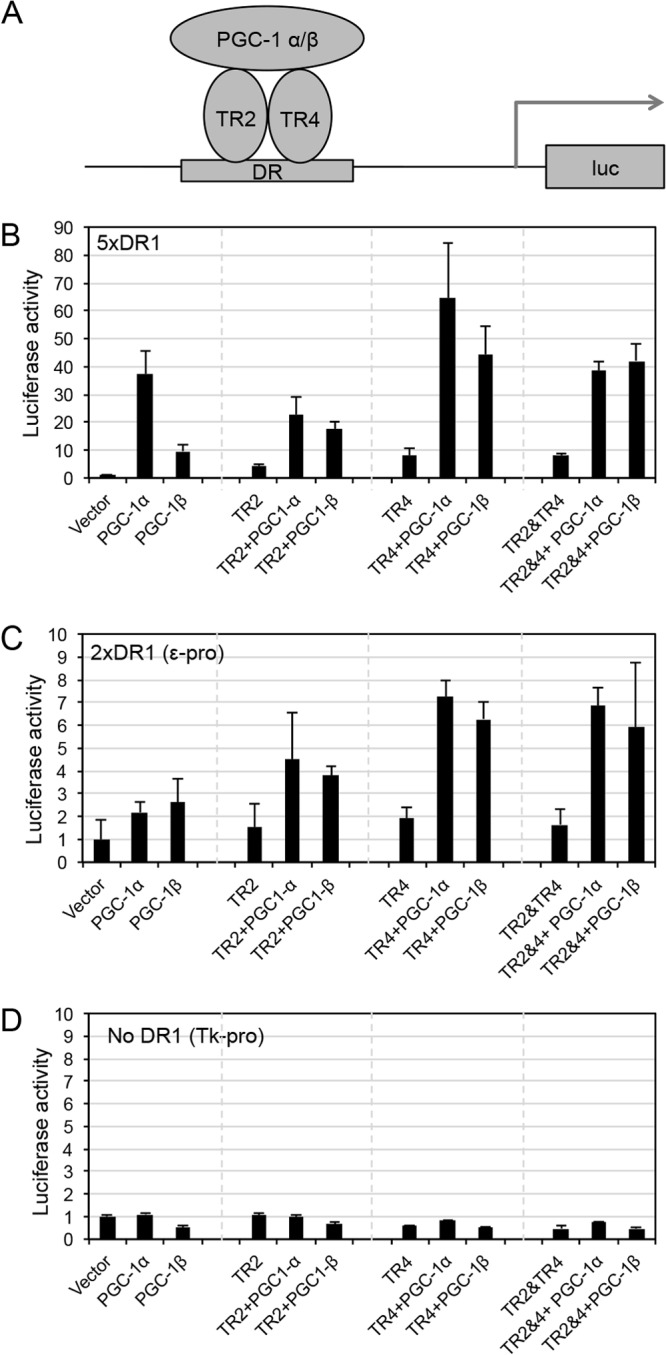
PGC-1 coactivators functionally complex with nuclear receptors TR2 and/or TR4. (A) Diagram depicting the interaction of PGC-1 and TR4 binding on a DR element. HEK293T cells were cotransfected with a luciferase reporter gene directed in cis by 5 copies of a synthetic DR1 sequence (B), the ε-globin gene promoter (containing 2 DR1 elements) (C), or the thymidine kinase (tk) gene promoter (bearing no DR1 sequences) (D) and all possible combinations of PGC-1α, PGC-1β, and TR2 or TR4 expression plasmids. The values for firefly luciferase activity were normalized to cotransfected Renilla luciferase activity. The standard errors were calculated from six independent determinations.
DISCUSSION
Differential β-globin gene expression during erythropoiesis is a complex process employing tissue-, lineage-, and developmental stage-specific transcription factor utilization during mammalian embryogenesis, with both coactivators and corepressors functioning as requisite effectors to elicit gene-specific transcriptional responses. The human β-globin locus has been extensively studied as a paradigm for genetic regulation of lineage- and stage-specific gene expression (42), as well as for its clinical relevance to β-globin disorders. Elucidation of the molecular basis for γ-globin gene silencing during definitive erythropoiesis in particular has been the focus of intense investigation, since coinheritance of genetic conditions that confer elevated γ-globin synthesis alleviates many symptoms and at least some of the pathology associated with inherited β-globin disorders (43, 44).
Over the past decade, a growing number of transcription factors (e.g., Ikaros [45], GATA1 [46], BCL11A [47], KLF1 [48, 49], FOP [50], and Myb [51]), as well as nuclear receptors COUP-TFII (52) and TR2/TR4 (22, 53, 54), have been implicated in adult-stage γ-globin gene transcriptional repression. Forced erythroid-specific transgenic expression of TR2 and TR4 led to induction of the fetal γ-globin genes in definitive erythroid cells of yeast artificial chromosome (YAC) transgenic mice (22), suggesting that TR2 and TR4 can elicit transcriptional activating responses under some experimental conditions. Consistent with that observation, forced TR2/TR4 expression in a humanized sickle cell mouse leads to elevated γ-globin expression, HbF synthesis, and alleviation of hematological and pathological indications of sickle cell disease without apparent adverse effects (25).
In this study, we detailed a new stage-specific role for the PGC-1 coactivators in erythropoiesis and globin gene regulation, with evidence that these effects are mediated through interaction with nuclear receptor TR4. In Pgc-1 germ line mutant animals, complete PGC-1 depletion reduces expression of the globin genes at all stages. PGC-1α and PGC-1β appear to act in a compensatory manner during primitive erythropoiesis, since the compound Pgc-1c mutants exhibit a more severe phenotype than either of the individual loss-of-function mutations. In contrast, in definitive erythroid cells there is no significant difference in globin expression in the singly versus compound mutant mice, suggesting that there is both gene and developmental stage specificity in PGC-1 coactivator function (Fig. 2). Furthermore, the Pgc-1 compound mutant mice had significantly lower erythroid cell hemoglobin content and the animals exhibited anemia, thrombocytopenia, and leukopenia, together suggesting multilineage myeloid hematopoietic deficiencies (Table 1) without affecting lymphopoiesis. Interestingly, the reticulocyte number in PGC-1 compound mutant mice was also reduced (see Fig. S4 in the supplemental material), suggesting that the effects of Pgc-1 compound loss of function in mature red cell production were not compensated for by increased erythropoiesis in order to produce more new red blood cells.
Flow-cytometric studies confirmed that while both erythropoiesis and myelopoiesis were affected by Pgc-1 loss, lymphopoiesis appeared to be normal (Fig. 3; also see Fig. S3 in the supplemental material). The number of mature erythroid cells (CD71− Ter119+) was significantly reduced, while the number of double-negative cells, the compartment containing the most immature erythroid progenitors, increased in Pgc-1c mutants. The number of immature intermediate erythroid cells (CD71+ Ter119−) also increased in the Pgc-1c mutant mice (Fig. 3A). The data indicated that Pgc-1 loss blocks the terminal differentiation of erythroid cells, and that the number of Mac1+ Gr1+ cells (a mixture of immature and mature granulocytes) was reduced in the hematopoietic organs of Pgc-1c mutant animals. In addition, the number of Mac1 single-positive cells (Mac1+ Gr1−) increased in the bone marrow of Pgc-1c mutant mice, a condition that is often associated with inflammation and impaired immune response (Fig. 3B), suggesting that PGC-1 activation is anti-inflammatory by reducing macrophage synthesis of proinflammatory cytokines (55).
The peripheral blood smears displayed erythroblasts (nucleated primitive red cells) only in the PGC-1 compound mutant animals (Fig. 4A). This same phenotype is observed in patients with sickle cell disease. Erythroblasts in the peripheral blood of the PGC-1 compound mutants indicates that the bone marrow has been infiltrated or that extramedullary hematopoiesis has been activated, conditions usually associated with ineffective erythropoiesis (39). In addition, significant necrotic cell death, cell loss, and abnormal adipocyte accumulation was observed in liver and spleen sections of Pgc-1c mutant pups (Fig. 4B). The abundant accumulation of lipid droplets in the livers of Pgc-1c pups is probably related to their roles in the control of energy metabolism (40).
Finally, these experiments show that the binding of PGC-1α and PGC-1β and the orphan nuclear receptors TR2 and TR4 to the various murine globin gene promoters can be quite different at different developmental stages, and that their DNA association correlated well with the expression of the εy- and βh1-globin genes. For example, the binding of the nuclear receptor orphans and the two coactivators to the εy- and βh1-globin gene promoters differed widely between e11.5 (primitive) and e14.5 (definitive) erythroid cells (Fig. 5), indicating that activation of those promoters by TR2 and TR4 with the PGC-1 coactivators was both stage and gene specific. The sole exception to this strong correlation is that PGC-1β, but not PGC-1α, was found bound to the βmaj promoter in primitive erythroid cells (at a time when βmaj-globin is not expressed), suggesting that PGC-1β plays a role in initiating the expression of the βmaj-globin gene, possibly in collaboration with a different partner (Fig. 5), or even that PGC-1β plays an active role in adult βmaj-globin gene repression in primitive erythrocytes (a role that, if true, would be unique to PGC-1 cofactors). Since PGC-1α interacts only with TR2 while both coactivators can form complexes that interact with TR4 (Fig. 6), it seems possible that these differential associations alter the affinities of the different transcription complexes for additional cofactors (56, 57). Cotransfection assays showed that compared to the expression of the individual orphan nuclear receptors, addition of PGC-1α or PGC-1β to force-expressed TR4 activated a DR1-regulated reporter gene, while addition of either coactivator to forcibly expressed TR2 induced reporter expression to a lesser extent (Fig. 7). These differences could be attributable either to competitive binding between TR2/TR4 heterodimers and the TR2 or TR4 homodimer and their different affinities for DR elements (23) or to the specificity in their association with different coactivators and corepressors.
The data described here show that (i) the PGC-1α and -1β coactivator proteins differentially associate with the orphan nuclear receptors TR2 and TR4; (ii) their ability to stimulate reporter gene transcription differs during erythroid development according to developmental stage; (iii) their combined loss-of-function effects result in anemia in vivo; (iv) their loss-of-function effects in hematopoiesis are restricted to the myeloerythroid lineages; and (v) they function as coactivators in cell-based cotransfection assays using either synthetic or natural DR-regulated promoters. Taken together with previous observations in the literature showing that TR2 and TR4 promote transcriptional activation (58–62), the present studies provide an explanation for how these orphan receptors can function as both activators (as shown here) and as repressors (21–24) during development. This hypothesis suggests that TR2 and/or TR4 serve as the scaffold for DNA binding to specific recognition sites in DNA, and that the array of coactivators (e.g., PGC-1s) or corepressors (e.g., DNMT1, LSD1, NuRD, and histone deacetylase 1 [HDAC1] to HDAC3) that differentially bind to these nuclear receptors at different stages during development or in different tissues are the true determinants of their (positive or negative) regulatory activity. If, as suggested in recent studies, TR4 is a typically ligand-regulated nuclear receptor (20), this could provide a readily distinguishable substrate to differentiate between liganded and unliganded, as well as activator or repressor, complexes.
These findings fundamentally contribute to our understanding of the molecular mechanisms that regulate the transcriptional activity of the nuclear receptors TR2 and TR4, factors that are intimately involved in the repression of the fetal γ-globin genes in adult erythroid cells (63). Nuclear receptor-mediated transcriptional regulation is controlled by ligand-induced recruitment or release of coactivators and corepressors. The balance between these differential associations determines the transcriptional output of target genes and the phenotypes associated with coactivator/corepressor binding. The work described here shows that PGC-1α and PGC-1β can participate in globin gene regulation through the nuclear receptors TR2 and TR4, suggesting that PGC-1 coactivators represent new therapeutic targets (in addition to the presumptive corepressors that we identified previously [24]) for the treatment of sickle cell disease and β-thalassemias.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to many colleagues for helpful comments on the manuscript. We thank David Ginsburg and members of the Ginsburg laboratory for instructions in the use of their automated hematology analyzer.
We are grateful for support from the Jay and Betty Van Andel Foundation and Amway (China) Limited (H.E.X.), from the American Heart Association for a Scientist Development Grant (S.C.), and from NIH grants DK86956 and HL24415 (J.D.E. and O.T.), DK071662, DK066202, and HL089301 (H.E.X.), and DK077086 (J.D.L.).
Footnotes
Published ahead of print 24 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00247-14.
REFERENCES
- 1.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. 1998. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92:829–839. 10.1016/S0092-8674(00)81410-5 [DOI] [PubMed] [Google Scholar]
- 2.Knutti D, Kaul A, Kralli A. 2000. A tissue-specific coactivator of steroid receptors, identified in a functional genetic screen. Mol. Cell. Biol. 20:2411–2422. 10.1128/MCB.20.7.2411-2422.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin J, Puigserver P, Donovan J, Tarr P, Spiegelman BM. 2002. Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta), a novel PGC-1-related transcription coactivator associated with host cell factor. J. Biol. Chem. 277:1645–1648. 10.1074/jbc.C100631200 [DOI] [PubMed] [Google Scholar]
- 4.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. 2000. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Investig. 106:847–856. 10.1172/JCI10268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.St-Pierre J, Lin J, Krauss S, Tarr PT, Yang R, Newgard CB, Spiegelman BM. 2003. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1alpha and 1beta (PGC-1alpha and PGC-1beta) in muscle cells. J. Biol. Chem. 278:26597–26603. 10.1074/jbc.M301850200 [DOI] [PubMed] [Google Scholar]
- 6.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. 1999. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98:115–124. 10.1016/S0092-8674(00)80611-X [DOI] [PubMed] [Google Scholar]
- 7.Lin JD. 2009. Minireview: the PGC-1 coactivator networks: chromatin-remodeling and mitochondrial energy metabolism. Mol. Endocrinol. 23:2–10. 10.1210/me.2008-0344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, Newgard CB, Spiegelman BM. 2001. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413:131–138. 10.1038/35093050 [DOI] [PubMed] [Google Scholar]
- 9.Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, Spiegelman B, Montminy M. 2001. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 413:179–183. 10.1038/35093131 [DOI] [PubMed] [Google Scholar]
- 10.Lin J, Handschin C, Spiegelman BM. 2005. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 1:361–370. 10.1016/j.cmet.2005.05.004 [DOI] [PubMed] [Google Scholar]
- 11.Spiegelman BM, Heinrich R. 2004. Biological control through regulated transcriptional coactivators. Cell 119:157–167. 10.1016/j.cell.2004.09.037 [DOI] [PubMed] [Google Scholar]
- 12.Liu C, Li S, Liu T, Borjigin J, Lin JD. 2007. Transcriptional coactivator PGC-1alpha integrates the mammalian clock and energy metabolism. Nature 447:477–481. 10.1038/nature05767 [DOI] [PubMed] [Google Scholar]
- 13.Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, Baek KH, Rosenzweig A, Spiegelman BM. 2008. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature 451:1008–1012. 10.1038/nature06613 [DOI] [PubMed] [Google Scholar]
- 14.Rowe GC, Jang C, Patten IS, Arany Z. 2011. PGC-1beta regulates angiogenesis in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 301:E155–E163. 10.1152/ajpendo.00681.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rhee J, Ge H, Yang W, Fan M, Handschin C, Cooper M, Lin J, Li C, Spiegelman BM. 2006. Partnership of PGC-1alpha and HNF4alpha in the regulation of lipoprotein metabolism. J. Biol. Chem. 281:14683–14690. 10.1074/jbc.M512636200 [DOI] [PubMed] [Google Scholar]
- 16.Lai L, Leone TC, Zechner C, Schaeffer PJ, Kelly SM, Flanagan DP, Medeiros DM, Kovacs A, Kelly DP. 2008. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev. 22:1948–1961. 10.1101/gad.1661708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huss JM, Kopp RP, Kelly DP. 2002. Peroxisome proliferator-activated receptor coactivator-1alpha (PGC-1alpha) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-alpha and -gamma. Identification of novel leucine-rich interaction motif within PGC-1alpha. J. Biol. Chem. 277:40265–40274. 10.1074/jbc.M206324200 [DOI] [PubMed] [Google Scholar]
- 18.Vega RB, Huss JM, Kelly DP. 2000. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell. Biol. 20:1868–1876. 10.1128/MCB.20.5.1868-1876.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. 2007. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 450:736–740. 10.1038/nature06322 [DOI] [PubMed] [Google Scholar]
- 20.Zhou XE, Suino-Powell KM, Xu Y, Chan CW, Tanabe O, Kruse SW, Reynolds R, Engel JD, Xu HE. 2011. The orphan nuclear receptor TR4 is a vitamin A-activated nuclear receptor. J. Biol. Chem. 286:2877–2885. 10.1074/jbc.M110.168740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanabe O, Shen Y, Liu Q, Campbell AD, Kuroha T, Yamamoto M, Engel JD. 2007. The TR2 and TR4 orphan nuclear receptors repress Gata1 transcription. Genes Dev. 21:2832–2844. 10.1101/gad.1593307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanabe O, McPhee D, Kobayashi S, Shen Y, Brandt W, Jiang X, Campbell AD, Chen YT, Chang C, Yamamoto M, Tanimoto K, Engel JD. 2007. Embryonic and fetal beta-globin gene repression by the orphan nuclear receptors, TR2 and TR4. EMBO J. 26:2295–2306. 10.1038/sj.emboj.7601676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanabe O, Katsuoka F, Campbell AD, Song W, Yamamoto M, Tanimoto K, Engel JD. 2002. An embryonic/fetal beta-type globin gene repressor contains a nuclear receptor TR2/TR4 heterodimer. EMBO J. 21:3434–3442. 10.1093/emboj/cdf340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cui S, Kolodziej KE, Obara N, Amaral-Psarris A, Demmers J, Shi L, Engel JD, Grosveld F, Strouboulis J, Tanabe O. 2011. Nuclear receptors TR2 and TR4 recruit multiple epigenetic transcriptional corepressors that associate specifically with the embryonic beta-type globin promoters in differentiated adult erythroid cells. Mol. Cell. Biol. 31:3298–3311. 10.1128/MCB.05310-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Campbell AD, Cui S, Shi L, Urbonya R, Mathias A, Bradley K, Bonsu KO, Douglas RR, Halford B, Schmidt L, Harro D, Giacherio D, Tanimoto K, Tanabe O, Engel JD. 2011. Forced TR2/TR4 expression in sickle cell disease mice confers enhanced fetal hemoglobin synthesis and alleviated disease phenotypes. Proc. Natl. Acad. Sci. U. S. A. 108:18808–18813. 10.1073/pnas.1104964108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM, Cui L, Manieri M, Donovan MX, Wu Z, Cooper MP, Fan MC, Rohas LM, Zavacki AM, Cinti S, Shulman GI, Lowell BB, Krainc D, Spiegelman BM. 2004. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 119:121–135. 10.1016/j.cell.2004.09.013 [DOI] [PubMed] [Google Scholar]
- 27.Vianna CR, Huntgeburth M, Coppari R, Choi CS, Lin J, Krauss S, Barbatelli G, Tzameli I, Kim YB, Cinti S, Shulman GI, Spiegelman BM, Lowell BB. 2006. Hypomorphic mutation of PGC-1beta causes mitochondrial dysfunction and liver insulin resistance. Cell Metab. 4:453–464. 10.1016/j.cmet.2006.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cui SY, Xu WH. 2006. Molecular characterization and functional distribution of N-ethylmaleimide-sensitive factor in Helicoverpa armigera. Peptides 27:1226–1234. 10.1016/j.peptides.2005.11.011 [DOI] [PubMed] [Google Scholar]
- 29.Kato K, Cui S, Kuick R, Mineishi S, Hexner E, Ferrara JL, Emerson SG, Zhang Y. 2010. Identification of stem cell transcriptional programs normally expressed in embryonic and neural stem cells in alloreactive CD8+ T cells mediating graft-versus-host disease. Biol. Blood Marrow Transplant. 16:751–771. 10.1016/j.bbmt.2010.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Sandy AR, Wang J, Radojcic V, Shan GT, Tran IT, Friedman A, Kato K, He S, Cui S, Hexner E, Frank DM, Emerson SG, Pear WS, Maillard I. 2011. Notch signaling is a critical regulator of allogeneic CD4+ T-cell responses mediating graft-versus-host disease. Blood 117:299–308. 10.1182/blood-2010-03-271940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palis J, Segel GB. 1998. Developmental biology of erythropoiesis. Blood Rev. 12:106–114. 10.1016/S0268-960X(98)90022-4 [DOI] [PubMed] [Google Scholar]
- 32.Lesley J, Hyman R, Schulte R, Trotter J. 1984. Expression of transferrin receptor on murine hematopoietic progenitors. Cell. Immunol. 83:14–25. 10.1016/0008-8749(84)90220-X [DOI] [PubMed] [Google Scholar]
- 33.Kina T, Ikuta K, Takayama E, Wada K, Majumdar AS, Weissman IL, Katsura Y. 2000. The monoclonal antibody TER-119 recognizes a molecule associated with glycophorin A and specifically marks the late stages of murine erythroid lineage. Br. J. Haematol. 109:280–287. 10.1046/j.1365-2141.2000.02037.x [DOI] [PubMed] [Google Scholar]
- 34.Shuga J, Zhang J, Samson LD, Lodish HF, Griffith LG. 2007. In vitro erythropoiesis from bone marrow-derived progenitors provides a physiological assay for toxic and mutagenic compounds. Proc. Natl. Acad. Sci. U. S. A. 104:8737–8742. 10.1073/pnas.0701829104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fleming TJ, Fleming ML, Malek TR. 1993. Selective expression of Ly-6G on myeloid lineage cells in mouse bone marrow. RB6–8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. J. Immunol. 151:2399–2408 [PubMed] [Google Scholar]
- 36.Leenen PJ, de Bruijn MF, Voerman JS, Campbell PA, van Ewijk W. 1994. Markers of mouse macrophage development detected by monoclonal antibodies. J. Immunol. Methods 174:5–19. 10.1016/0022-1759(94)90005-1 [DOI] [PubMed] [Google Scholar]
- 37.Serafini P, De Santo C, Marigo I, Cingarlini S, Dolcetti L, Gallina G, Zanovello P, Bronte V. 2004. Derangement of immune responses by myeloid suppressor cells. Cancer Immunol. Immunother. 53:64–72. 10.1007/s00262-003-0443-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gordon S, Taylor PR. 2005. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 5:953–964. 10.1038/nri1733 [DOI] [PubMed] [Google Scholar]
- 39.Constantino BT, Cogionis B. 2000. Nucleated RBCs–significance in the peripheral blood film. Lab. Med. 31:223–229. 10.1309/D70F-HCC1-XX1T-4ETE [DOI] [Google Scholar]
- 40.Lin JD, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM, Cui LB, Manieri M, Donovan MX, Wu ZD, Cooper MP, Fan MC, Rohas LM, Zavacki AM, Cinti S, Shulman GI, Lowell BB, Krainc D, Spiegelman BM. 2004. Defects in adaptive energy metabolism with CNS-Linked hyperactivity in PGC-1 alpha null mice. Cell 119:121–135. 10.1016/j.cell.2004.09.013 [DOI] [PubMed] [Google Scholar]
- 41.McConnell SC, Huo Y, Liu S, Ryan TM. 2011. Human globin knock-in mice complete fetal-to-adult hemoglobin switching in postnatal development. Mol. Cell. Biol. 31:876–883. 10.1128/MCB.00725-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kiefer CM, Hou C, Little JA, Dean A. 2008. Epigenetics of beta-globin gene regulation. Mutat. Res. 647:68–76. 10.1016/j.mrfmmm.2008.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marcus SJ, Kinney TR, Schultz WH, O'Branski EE, Ware RE. 1997. Quantitative analysis of erythrocytes containing fetal hemoglobin (F cells) in children with sickle cell disease. Am. J. Hematol. 54:40–46. [DOI] [PubMed] [Google Scholar]
- 44.Papadakis MN, Patrinos GP, Tsaftaridis P, Loutradi-Anagnostou A. 2002. A comparative study of Greek nondeletional hereditary persistence of fetal hemoglobin and beta-thalassemia compound heterozygotes. J. Mol. Med. (Berlin) 80:243–247. 10.1007/s00109-001-0312-4 [DOI] [PubMed] [Google Scholar]
- 45.Lopez RA, Schoetz S, DeAngelis K, O'Neill D, Bank A. 2002. Multiple hematopoietic defects and delayed globin switching in Ikaros null mice. Proc. Natl. Acad. Sci. U. S. A. 99:602–607. 10.1073/pnas.022412699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harju-Baker S, Costa FC, Fedosyuk H, Neades R, Peterson KR. 2008. Silencing of Agamma-globin gene expression during adult definitive erythropoiesis mediated by GATA-1-FOG-1-Mi2 complex binding at the −566 GATA site. Mol. Cell. Biol. 28:3101–3113. 10.1128/MCB.01858-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sankaran VG, Menne TF, Xu J, Akie TE, Lettre G, Van Handel B, Mikkola HK, Hirschhorn JN, Cantor AB, Orkin SH. 2008. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 322:1839–1842. 10.1126/science.1165409 [DOI] [PubMed] [Google Scholar]
- 48.Borg J, Papadopoulos P, Georgitsi M, Gutierrez L, Grech G, Fanis P, Phylactides M, Verkerk AJ, van der Spek PJ, Scerri CA, Cassar W, Galdies R, van Ijcken W, Ozgur Z, Gillemans N, Hou J, Bugeja M, Grosveld FG, von Lindern M, Felice AE, Patrinos GP, Philipsen S. 2010. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat. Genet. 42:801–805. 10.1038/ng.630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou D, Liu K, Sun CW, Pawlik KM, Townes TM. 2010. KLF1 regulates BCL11A expression and gamma- to beta-globin gene switching. Nat. Genet. 42:742–744. 10.1038/ng.637 [DOI] [PubMed] [Google Scholar]
- 50.van Dijk TB, Gillemans N, Pourfarzad F, van Lom K, von Lindern M, Grosveld F, Philipsen S. 2010. Fetal globin expression is regulated by Friend of Prmt1. Blood 116:4349–4352. 10.1182/blood-2010-03-274399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sankaran VG, Menne TF, Scepanovic D, Vergilio JA, Ji P, Kim J, Thiru P, Orkin SH, Lander ES, Lodish HF. 2011. MicroRNA-15a and -16-1 act via MYB to elevate fetal hemoglobin expression in human trisomy 13. Proc. Natl. Acad. Sci. U. S. A. 108:1519–1524. 10.1073/pnas.1018384108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aerbajinai W, Zhu J, Kumkhaek C, Chin K, Rodgers GP. 2009. SCF induces gamma-globin gene expression by regulating downstream transcription factor COUP-TFII. Blood 114:187–194. 10.1182/blood-2008-07-170712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Omori A, Tanabe O, Engel JD, Fukamizu A, Tanimoto K. 2005. Adult stage gamma-globin silencing is mediated by a promoter direct repeat element. Mol. Cell. Biol. 25:3443–3451. 10.1128/MCB.25.9.3443-3451.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tanimoto K, Liu Q, Grosveld F, Bungert J, Engel JD. 2000. Context-dependent EKLF responsiveness defines the developmental specificity of the human epsilon-globin gene in erythroid cells of YAC transgenic mice. Genes Dev. 14:2778–2794. 10.1101/gad.822500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, Chawla A. 2006. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 4:13–24. 10.1016/j.cmet.2006.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nemoto S, Fergusson MM, Finkel T. 2005. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1alpha. J. Biol. Chem. 280:16456–16460. 10.1074/jbc.M501485200 [DOI] [PubMed] [Google Scholar]
- 57.Puigserver P, Spiegelman BM. 2003. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr. Rev. 24:78–90. 10.1210/er.2002-0012 [DOI] [PubMed] [Google Scholar]
- 58.Mu XM, Young WJ, Liu YX, Uemura H, Chang CS. 1998. Induction of an intronic enhancer of the human ciliary neurotrophic factor receptor (CNTFR alpha) gene by the TR3 orphan receptor. Endocrine 9:27–32. 10.1385/ENDO:9:1:27 [DOI] [PubMed] [Google Scholar]
- 59.Wei LN, Hu XL, Chinpaisal C. 2000. Constitutive activation of retinoic acid receptor beta 2 promoter by orphan nuclear receptor TR2. J. Biol. Chem. 275:11907–11914. 10.1074/jbc.275.16.11907 [DOI] [PubMed] [Google Scholar]
- 60.Young WJ, Smith SM, Chang CS. 1997. Induction of the intronic enhancer of the human ciliary neurotrophic factor receptor (CNTFR alpha) gene by the TR4 orphan receptor–a member of steroid receptor superfamily. J. Biol. Chem. 272:3109–3116. 10.1074/jbc.272.5.3109 [DOI] [PubMed] [Google Scholar]
- 61.Khan SA, Park SW, Huq MDM, Wei LN. 2006. Ligand-independent orphan receptor TR2 activation by phosphorylation at the DNA-binding domain. Proteomics 6:123–130. 10.1002/pmic.200500068 [DOI] [PubMed] [Google Scholar]
- 62.Zhang Y, Dufau ML. 2000. Nuclear orphan receptors regulate transcription of the gene for the human luteinizing hormone receptor. J. Biol. Chem. 275:2763–2770. 10.1074/jbc.275.4.2763 [DOI] [PubMed] [Google Scholar]
- 63.Shi L, Cui S, Engel JD, Tanabe O. 2013. Lysine-specific demethylase 1 is a therapeutic target for fetal hemoglobin induction. Nat. Med. 19:291–294. 10.1038/nm.3101 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.