

Abstract
Structural maintenance of chromosomes (SMC) complexes and DNA topoisomerases are major determinants of chromosome structure and dynamics. The cohesin complex embraces sister chromatids throughout interphase, but during mitosis most cohesin is stripped from chromosome arms by early prophase, while the remaining cohesin at kinetochores is cleaved at anaphase. This two-step removal of cohesin is required for sister chromatids to separate. The cohesin-related Smc5/6 complex has been studied mostly as a determinant of DNA repair via homologous recombination. However, chromosome segregation fails in Smc5/6 null mutants or cells treated with small interfering RNAs. This also occurs in Smc5/6 hypomorphs in the fission yeast Schizosaccharomyces pombe following genotoxic and replication stress, or topoisomerase II dysfunction, and these mitotic defects are due to the postanaphase retention of cohesin on chromosome arms. Here we show that mitotic and repair roles for Smc5/6 are genetically separable in S. pombe. Further, we identified the histone variant H2A.Z as a critical factor to modulate cohesin dynamics, and cells lacking H2A.Z suppress the mitotic defects conferred by Smc5/6 dysfunction. Together, H2A.Z and the SMC complexes ensure genome integrity through accurate chromosome segregation.
INTRODUCTION
Chromosomal integrity is essential for normal growth and development. In eukaryotes, there are three essential complexes of proteins that are central to chromosome dynamics. These are cohesin, condensin, and the Smc5/6 complex, known collectively as the structural maintenance of chromosomes (SMC) complex. Each complex shares a related architecture, and central to them are a heterodimer of SMC proteins: Smc1 and -3 in cohesin, Smc2 and -4 in condensin, and Smc5 and -6 in Smc5/6. These SMC proteins are large coiled-coil molecules with globular N and C termini containing Walker A and B ATP-binding motifs. They fold and interact at a flexible hinge, with ATP acting to hold the globular domains together. A kleisin protein bridges each heterodimer to form a putative ring-shaped structure, and each complex has specific additional non-SMC proteins that serve as regulators and effectors of function (1–3).
Chromosome condensation and sister chromatid cohesion are the key roles for condensin and cohesin, respectively (1). However, defects in DNA repair have also been described for yeast strains harboring hypomorphic mutant alleles of condensin (4) and cohesin (5–7) subunits. In the case of cohesin, a role in DNA repair could stem from the fact that DNA repair by homologous recombination (HR) requires the sister chromatid to be in close proximity to the damaged chromatid. However, more recently cohesin in mammalian cells has been shown to also act as a transcriptional insulator in collaboration with CTCF (8, 9), and so the DNA repair function of cohesin may be more complex than a simple scaffolding mechanism. As its name suggests, deciphering Smc5/6 function has proved more elusive, though most studies have focused on a role for this complex in HR-mediated DNA repair (2). However, the core Smc5/6 genes are essential for cell viability, while most HR genes are not, and thus the HR defects of Smc5/6 hypomorphs, like those of cohesin and condensin, may reflect a more general role in chromosome architecture.
The association of each complex with chromosomes is highly dynamic. Condensin is mostly associated with chromosomes during mitosis, whereas cohesin and Smc5/6 are predominantly on chromosomes during interphase. Cohesin dynamics are most thoroughly understood, particularly in the budding yeast Saccharomyces cerevisiae (10). Cohesin is loaded onto individual chromatids immediately following chromosome segregation and becomes cohesive around sister chromatids coincident with DNA replication. Scc2/4 proteins control the loading of cohesin, and Eco1-catalyzed Smc3 acetylation promotes cohesiveness by antagonizing an anticohesion activity conferred by Pds5 and Wapl (11). Cohesin removal is essential for sister chromatid separation and occurs in a two-step process in most organisms. The majority of cohesin is removed from chromosome arms by prophase. The precise mechanism by which this occurs is not entirely clear but is reliant on cohesin phosphorylation by Polo and Aurora kinases (12). Cohesin at the kinetochore remains in place until the metaphase-anaphase transition, whereupon the kleisin subunit (Scc1/Rad21) is cleaved by the protease separase, allowing sister chromatid separation that is immediately followed by cohesin reloading. In S. cerevisiae, the prophase removal of cohesin does not occur; rather, most Scc1 is cleaved (13). As such, cohesin loading is also delayed in this organism because a new pool of Scc1 must be synthesized.
Smc5/6 dynamics are very similar to those of cohesin in time and space, though little is known about the mechanisms by which this occurs. Notably, there is no cleavage of the kleisin subunit Nse4 in Schizosaccharomyces pombe (14), and while Smc5/6 loading does not occur in scc2 mutants in S. cerevisiae (15), it is not clear if this is a direct effect on loading or an indirect effect via cohesin. Apart from the SMC subunits, what distinguishes Smc5/6 from cohesin complexes are additional types of non-SMC subunits. These include a SUMO ligase (Nse2) that in S. pombe is important to survive replication stress (16, 17). Further, Smc5/6 contains a vRING domain protein (Nse1) that in S. pombe is required for Smc5/6 to associate with sites of DNA damage (17) and has been suggested to have ubiquitin ligase activity (18).
The founding member of Smc5/6 was the smc6 gene in S. pombe (19), which was among the original collection of radiation-sensitive mutants that was initially named rad18 (20). The original allele, smc6-X (formerly rad18-X), is defective in HR repair, and most additional Smc5/6 genes have been studied using damage-sensitive hypomorphs. It is notable that the same collection of radiation-sensitive mutants included the rad21-P45 allele of the S. pombe cohesin kleisin gene, prompting in early studies of this gene a focus on DNA repair (5, 21).
Studies in S. pombe have shown that the terminal phenotype of Smc5/6 null mutants is a lethal inability to segregate chromosomes (22, 23). The same phenotype is observed in hypomorphic mutants following DNA damage, replication stress, and topoisomerase II dysfunction; most Smc5/6 hypomorphs are synthetically lethal with the top2 mutant top2-191. The cause of this mitotic failure is the postanaphase retention of cohesin on mitotic chromosome arms and is not associated with detectable lesions in the DNA (24, 25), suggesting any HR defect in Smc5/6 hypomorphs may be an indirect effect of altered chromosome structure. Recently, cohesin retention defects have also been described in S. cerevisiae smc5 and nse4 mutants during aberrant meiotic divisions (26), suggesting that cohesin retention is a conserved consequence of Smc5/6 dysfunction.
Deletion of the vRING domain of Nse1 causes severe damage sensitivity, mitotic defects, and lethality at 36°C. Cysteine-to-alanine mutations disrupting the vRING domain of Nse1 also cause repair defects, but more conservative cysteine-to-serine mutations do not (17, 27). Rather, the cysteine-to-serine mutations prevent the accumulation of Smc5/6 complexes (wild type or mutant) at chromosomal lesions. In the case of the Smc5/6 mutants, this suppresses their DNA repair defects and damage sensitivity, with lesions channeled into the postreplication repair (PRR) pathway. However, in cycling cells, these vRING domain mutants do not affect the localization of the Smc5/6 complex on chromatin (17). Thus, Smc5/6 recruitment to lesions is Nse1 dependent, and it is the recruitment of mutant complexes that confers the DNA damage sensitivity. Thus, the focus on HR regulation by Smc5/6 has been influenced by the phenotypes of a relatively small collection of alleles selected as damage-sensitive mutants, whereas the functional interactions between cohesin and Smc5/6 necessary for timely sister chromatid separation appear to be of greater physiological significance.
Here, we further investigated the effects of the cysteine-to-serine mutation in Nse1's vRING domain. We show that despite the absence of DNA repair defects, nse1-C216S is still synthetically lethal with top2-191, and again this is due to a failed sister chromatid arm separation that is rescued partially by separase overexpression. Thus, the mitotic and HR functions of Smc5/6 are separable. We have dissected these phenotypes through additional genetic screens coupled with genome resequencing, and we have uncovered a critical role for the histone variant H2A.Z and its regulators in mediating the mitotic defects of Smc5/6 mutants. Deleting the gene for H2A.Z (pht1) suppresses the mitotic defects of nse1 and smc6 mutants and reduces the level of cohesin on chromosome arms.
MATERIALS AND METHODS
General S. pombe methods.
All strains used were derivatives of 972 h− and 975 h+. Standard media and methods were used to propagate and create strains (28). Spot assays for drug sensitivities were plated onto rich (YES) medium. All other assays were performed on cells grown in minimal medium (EMM2). General methods for microscopy, transformation, Western blotting, and drug and radiation sensitivities were as described previously (17, 29–31). DNA was stained with 1 μg/ml 4′,6-diamino-2-phenylindole (DAPI). Microscopy was performed on a Nikon E800 microscope with a 100× 1.40-numerical-aperture Plan-Apo objective lens, together with differential interference contrast (DIC) optics. Images were captured on a Q-Click camera by using the Q-capture suite plus software and prepared for publication using Adobe Photoshop. Green fluorescent protein (GFP)-lacI foci were visualized as described previously (24) following 22 h of growth at 25°C in 0.5 μM thiamine, after which latrunculin B was added to 10 μM for 5.5 h at 25°C, or the temperature was shifted to 30°C for 3 h. Samples were fixed in 90% cold methanol before imaging. Cell extracts were made by trichloroacetic acid (TCA) precipitation as described previously (32) and quantified after dilution and boiling in 1 M Tris (pH 8.0), 1% SDS.
ChIP assays.
Chromatin immunoprecipitation (ChIP) assays were performed as described elsewhere, using previously characterized primers and Sybr green quantitative PCR (qPCR) (17, 24). Arm loci in order (left to right) were as follows: 1L, pac2; 1R, fun14; 2L, act1; 2R, zfs1; 3L, SPCC553.10; 3R, ade6. STE1 is the immediate subtelomeric region of chromosome 1 (33). In all experiments, the input was fixed to 1.5% of the IP volume, and hence the enrichment values are 80-fold higher than the percentages of DNA recovery.
Antibodies.
S129-phosphorylated histone H2A (γH2A) was detected by microscopy and Western blotting with an S. pombe phosphorylation-specific antibody (ab17353; Abcam) at a 1:1,000 dilution and detected with a 1:400 dilution of Alexa Fluor 488-coupled anti-rabbit (for microscopy) and IgG antibodies. For Western blotting, acetylation on histone H3 K56 was detected with a 1:1,000 dilution of rabbit monoclonal antibody (04-1135; Millipore), total histone H3 was detected with rabbit polyclonal antibody (ab1791; Abcam), Pht1 was detected with 1:1,000 rabbit polyclonal antibody (39640; Active Motif), acetylated Pht1 was detected with 1:1,000 rabbit polyclonal antibody (39462; Active Motif), β-tubulin was detected with 1:3,000 mouse monoclonal antibody (T5168; Sigma). Immune complexes were detected with 1:2,000 horseradish peroxidase-coupled donkey anti-mouse and anti-rabbit IgG antibodies and Clarity enhanced chemiluminescence reagent (Bio-Rad).
Gene expression analysis.
Total RNA was isolated by using TRIzol reagent (Invitrogen) following cell disruption in a mini-beadbeater (BioSpec). cDNA was synthesized using a qScript cDNA Super mix kit (Quanta Biosciences) using 100 ng of total RNA, and then 10% of each reaction mixture was used to quantitate mRNA levels relative to cdc2 via Sybr green qPCR. The primers used are shown in Table 1.
TABLE 1.
Primers used for mRNA quantification by qRT-PCRa
| Locus | Forward | Reverse |
|---|---|---|
| cdc2 | AAGTATGGCCTGGAGTCACG | CAATAGCGTCCTCTTCACCA |
| rad21 | GTACGTCGTCAACGTGCAAT | TCAAGCATAACGGTGAGGTG |
| smc1 | GAAGAGCCGTCTGAACCAGT | CTCAGAACCGTCATTGCGTA |
| mis4 | GTCCATCATGTTCTCGCGTAT | TCACGAGGCGAATAAGTGTCT |
| nse4 | CCGAATGTGCTAACTCAACCT | AGGCTGGTGTGCTTGAAGTAA |
| cut1 | CCGACGTCGATTCTAGTTCAG | TTCTGTGCTTCACGACGTATA |
| top2 | TAAGTGCACTCGTGGCAATC | TCCAACCACGTCCATTGTTA |
These primers were used for the experiment illustrated in Fig. 5A.
Genomic resequencing.
High-quality genomic DNA was extracted from saturated cultures of both parental strains and spontaneous suppressors as described previously (28). Genomic DNA was sheared to 200-bp fragments by using a Covaris E210 acoustic sonicator. NEBNext DNA sample preparation kits were used to generate Illumina sequencing libraries (E6040S; New England BioLabs), following the manufacturer's protocols for end repair, adenine tailing, and ligation. All purifications were carried out using Agencourt Ampure XP SPRI magnetic beads (Beckman Coulter). Custom-synthesized adapters and primers were used to incorporate Illumina specific adapters and 6-nucleotide (nt) molecular bar codes to allow multiplexing. The Illumina HiSeq 2000 was used to perform 100-nt sequencing of genomic DNA libraries. All samples were sequenced in multiplex with 6 to 10 samples per lane. Adapters and primers used for genomic resequencing are shown in Table 2.
TABLE 2.
Primers and adapters used for genome resequencing
| Adapter or primer | Sequencea |
|---|---|
| Multiplexing adapters | 5′-P-GATCGGAAGAGCACACGTCT |
| 5′-ACACTCTTTCCCTACACGACGCTCTTCCGATC*T | |
| Primers | |
| Illumina universal forward primer | 5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATC*T |
| Reverse primerb | 5′-CAAGCAGAAGACGGCATACGAGATXXXXXXGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC*T |
The asterisks in the sequences indicate phosphorothioate.
The Xs in the sequence indicate where specific bar code sequences are located.
Genome-wide comparison of parental versus suppressor strains was carried out using the MATCH-G software package (mutational analysis tool set comparing whole genomes; version 2.0, which can be downloaded from https://science.nichd.nih.gov/confluence/display/smcb/MATCH-G+Program) (34). The tool set is dependent on mapping and single nucleotide polymorphism (SNP) programs and is sensitive to output file formats. The S. pombe genome was downloaded in EMBL format from PomBase (35). Read mapping was performed with Bowtie, version 0.12.9 (36), and SNP calling was performed with SAMTools version 0.1.16 using the pileup command (37); recent SAMtools releases have modified the SNP calling output, which complicates analysis of haploid genomes. MATCH-G was used to call SNPs, using the default parameters.
RESULTS
Separation of the mitotic and HR roles of Smc5/6.
The lack of DNA repair defects in nse1-C216S cells was somewhat surprising, as Nse1 is the most evolutionarily conserved subunit of the Smc5/6 complex in terms of amino acid identity (17). The conservation of the vRING domain suggests an important function that must be independent of DNA repair. We thought the most likely other function would be found in mitosis, specifically, the removal of cohesin complexes from chromosome arms, a function that Smc5/6 elicits in the absence of DNA damage in cooperation with Top2. Cohesin retention occurs when Smc5/6 hypomorphs, such as smc6-74, are combined with a temperature-sensitive allele of top2 (top2-191) at the top2-191 semipermissive temperature of 30°C (24). Such defects in cohesin removal result in lethal attempts at chromosome segregation that can be resolved by overexpression of the cohesin protease separase.
We therefore constructed nse1-C216S top2-191 double mutants, and indeed these were also synthetically lethal at 30°C (Fig. 1A). Microscopic examination of cells after a 4-hour incubation at 30°C confirmed that nse1-C216S top2-191 double mutants failed in chromosome segregation, with both the “cut” and “stretched” phenotypes characteristic of smc6-74 top2-191 cells (24) (Fig. 1B), which, respectively, result from no or partial chromosome segregation prior to cytokinesis. Such cells were negative for S129 phosphorylated H2A (γH2A), a sensitive marker of DNA damage, though they were capable of phosphorylating H2A after extrinsic DNA damage (Fig. 1C and D). Additional evidence for wild-type levels of DNA damage in nse1-C216S cells includes a lack of enhanced damage sensitivity when combined with a large collection of DNA repair mutants, as well as no DNA damage checkpoint activation without extrinsic damage (17).
FIG 1.

The C216S mutation in Nse1's vRING causes synthetically lethal mitotic defects in combination with top2-191 without conferring a DNA repair defect. (A) The indicated strains were grown for either 5 days (25°C) or 4 days (30 and 36°C). (B) The indicated strains were grown at 25°C and shifted to 30°C for 4 h. Cells were fixed and DNA stained with DAPI. Arrows indicate products of failed mitoses; all other cells in this field are undergoing failed mitotic divisions, which accounts for ∼40% of total cells. Mitotic defects comprise a complete lack of chromosome segregation, known as the “cut” phenotype (i) and incompletely resolved chromosomes (ii). Bar, 10 μm. (C) Cells were grown at 25°C, shifted to 30°C for 2.5 h, and then mock irradiated or irradiated with 100 J/m2 and returned to 30°C for an additional 30 min. Cells were then fixed and stained with DAPI and antibodies to histone H2A phosphorylated on S129 (γH2A). Bar, 10 μm. (D) Cells were treated as described for panel C, and then extracts were processed for Western blotting for γH2A. (E) A micrograph showing DAPI plus GFP images of nse1-C216S top2-191 cells containing a lacO array adjacent to centromere 1 (lys1) and expressing lacI-GFP grown at 25°C and shifted to 30°C for 4 h. Arrows indicate uninuclear cells with 2 separated GFP foci (2), uninuclear with a single GFP focus (U), or a binucleated cell with a single focus in each nucleus (B). The graph quantifies uninuclear cells with 2 separated GFP foci at lys1 and at two arm loci, ade6 and ade8, located 0.28 and 3.04 Mb from centromeres 3 and 2, respectively, after growth at 30°C for 3 h. Data are means ± standard deviations (SD) for 3 samples of 300 to 500 cells. (F) top2-191 nse1-C216S cells carrying pREP81 (vector) or pREP81-Cut1 (pCut1) were grown at 25°C and then shifted to 30°C for 4 h. Cells were scored for mitotic defects (left; data are means ± SD for three counts of 100 cells) and relative viability (right; normalized to 25°C cultures; data are means ± SD [n = 3]) following colony formation over 5 days at 25°C.
We next used GFP-lacI and lacO integrated adjacent to centromere 1 (at lys1), or at two arm loci (ade6 and ade8), to assay kinetochore and chromosome arm separation. In interphase cells, the signals from sister chromatids are too close to be seen as separate foci by conventional microscopy. In keeping with wild-type growth kinetics, normal mitoses were observed in top2-191 mutant cells at both 25°C and 30°C (Fig. 1E) (24). Here, both the kinetochore (first under the power of the contracting spindle) and arm markers (second, led by the kinetochores) segregate at anaphase A, giving a small percentage (∼2%) of cells that transiently contain two GFP foci at any locus. By the time the spindle elongates in anaphase B, the nuclear masses move apart with a single GFP focus at each locus per nucleus (24) (Fig. 1E). The nse1-C216S top2-191 double mutants behaved in the same manner at 25°C. However, after 4 h at 30°C, ∼40% of these cells showed single nuclei but two widely separated GFP-lacI signals at lys1 (Fig. 1E), which was coincident with the appearance of aberrant mitotic figures. However, similar assays using the arm loci showed only ∼2% of cells with two GFP foci. Hence, while kinetochores separate in ∼40% of the cells, the arms separate in only 2% of cells. Thus, the aberrant mitoses in nse1-C216S top2-191 cells at 30°C are due to a defect in sister chromatid arm separation.
Each of the phenotypes described above are consistent with the postanaphase cohesin retention seen in smc6-74 top2-191 cells (24). Cohesin is reloaded onto chromosomes immediately upon segregation, though not all cohesin is chromosome associated. Thus, the only way to accurately assay cohesin removal is to prevent its postanaphase reloading and measure chromosome association by ChIP. Preventing reloading can be achieved by thermal inactivation of the cohesin loader Mis4 (Scc2 homolog) via use of a temperature-sensitive allele (mis4-242) (24). However, mis4-242 nse1-C216S cells showed a severe growth defect that strongly selected for suppressors, precluding a meaningful analysis of cohesin dynamics by this assay, though this interaction alone suggests a defect in the cohesin cycle. Nevertheless, the cohesin retention defects of damaged smc6-74 cells, and of smc6-74 top2-191 mutants at 30°C, are suppressed by overexpression of the cohesin protease separase (known as Cut1 in S. pombe). This reverts the hydroxyurea (HU) sensitivity of smc6-74 and the synthetic lethality of smc6-74 top2-191 double mutants (24), and it reduces the mitotic defects of UV-irradiated smc6-74 cells by ∼40% (25). Cut1 overexpression did not rescue the synthetic lethality of nse1-C216S top2-191 double mutants in terms of colony formation over 4 days (data not shown), but it did reduce the mitotic defects and lethality seen after 4 h at 30°C, each by over 50% (Fig. 1F).
As there are no other known substrates for Cut1 in S. pombe (24), this result and the lack of chromosome arm segregation (Fig. 1B and E) are consistent with cohesin retention in nse1-C216S top2-191 double mutants. Like our previous studies (24), this occurred without enhanced DNA damage, assayed by H2A phosphorylation and the absence of a checkpoint-induced cell cycle delay (Fig. 1C and D). However, unlike our other synthetic lethalities between repair-defective and damage-sensitive Smc5/6 mutants and top2-191 mutants (22–24), the mitotic defects of nse1-C216S top2-191 double mutants occur without a measurable DNA repair defect conferred by this allele of nse1. Thus, nse1-C216S is a separation-of-function mutant that confirms the physiological importance of this mitotic function for Smc5/6 to ensure chromosome segregation.
Loss of H2A.Z suppresses the mitotic and repair defects of Smc5/6 mutants.
To further understand the interaction between nse1-C216S and top2-191, we sought to identify mutations in genes that would suppress their synthetic lethality. As with many synthetic lethal combinations, nse1-C216S top2-191 mutants give rise to spontaneous suppressors at 30°C with a frequency of ∼10−7. We collected several of these suppressors that arose independently from each other and then sequenced their genomes together with that of the parental nse1-C216S top2-191 double mutant to identify the suppressor mutations. The beauty of this approach is that it enables suppressor identification without the need for an additional recessive phenotype.
This analysis identified three independent isolates with only one nucleotide substitution compared to the parent. These substitutions were in the pht1 gene, which encodes the histone variant H2A.Z (38). Each of these independently derived mutations were in the same codon (T110G), suggesting either an allele-specific phenotype or that this codon is susceptible to spontaneous mutagenesis. The T110G transversion mutation converted the invariant valine 36 to a glycine within the histone fold (39), and we refer to this allele as pht1-36 (Fig. 2A). Western blot analysis showed that pht1-36 resulted in nondetectable levels of H2A.Z protein (Fig. 2B), and consistent with this, a complete deletion of pht1 (pht1Δ) was also a strong suppressor of the synthetic lethality of nse1-C216S top2-191 double mutants (Fig. 2A). Thus, the suppression conferred by pht1-36 is not allele specific, and hence T110 within pht1 may be unusually prone to spontaneous mutations. As expected, the suppression was associated with an absence of the mitotic defects seen at 30°C (Table 3). Although the deletion of pht1 causes an increase in whole chromosome loss (assayed via chromosome 16, a derivative of the native chromosome 3) (38), it is notable that the percentage of nse1-C216S top2-191 cells with mitotic defects after only 4 h at 30°C was ∼20-fold higher than that seen for cycling pht1Δ cells (Table 3).
FIG 2.

Loss of H2A.Z suppresses the mitotic and repair defects of Smc5/6 mutants. (A) Alignment of the N-terminal sequences of S. pombe (Sp) and human (Hs) H2A.Z. Red lysines indicate residues subject to acetylation, and the valine-glycine substitution of pht1-36 is indicated above the alignment. Below are the positions of the αN-helix, and the beginning of the α1 helix of the histone fold (68). The plates under the alignment show growth at 25°C (5 days) and 30°C and 36°C (4 days). The vital dye Phloxin B stains dead cells red, and the residual colony formation of nse1-C216S top2-191 cells at 30°C shows spontaneous suppressors. Note that each pht1 allele suppressed the synthetic lethality of nse1-C216S top2-191 cells at 30°C. (B) Western blot of TCA-precipitated whole-cell extracts with anti-Pht1 and anti-β-tubulin antibodies. (C) Plates showing growth at 25°C (5 days) and 30°C and 36°C (4 days). The vital dye Phloxin B stains dead cells red, and the residual colony formation of smc6-74 top2-191 cells at 30°C represents spontaneous suppressors; all pht1 alleles suppressed the synthetic lethality of smc6-74 top2-191. (D and E) pht1Δ suppresses both the MMS (chronic exposure over 4 days at 30°C, spots are 10-fold serial dilutions) (D) and UV-C (colony formation) (E) sensitivities of smc6-74 in a ubc13-dependent manner.
TABLE 3.
Aberrant mitosis after 4 h at 30°C
| Genotype | % aberrant mitosisa |
|---|---|
| Wild type | 0.3 ± 0.6 |
| smc6-74 | 0.7 ± 0.6 |
| top2-191 | 0.7 ± 1.2 |
| pht1Δ | 1.7 ± 0.6 |
| nse1-C216S | 0.3 ± 0.6 |
| smc6-74 pht1Δ | 0.3 ± 0.6 |
| top2-191 pht1Δ | 5.7 ± 0.6 |
| nse1-C216S pht1Δ | 0.3 ± 0.6 |
| smc6-74 top2-191 | 52 ± 3.6 |
| smc6-74 top2-191 pht1Δ | 3.7 ± 0.6 |
| nse1-C216S top2-191 | 38.0 ± 3.6 |
| nse1-C216S top2-191 pht1Δ | 3.3 ± 1.5 |
| pht1-4KR | 1.3 ± 1.5 |
| pht1-4KR smc6-74 | 2.7 ± 0.6 |
| pht1-4KR top2-191 | 1.0 ± 1.0 |
| pht1-4KR top2-191 smc6-74 | 1.7 ± 1.5 |
| pht1-4KR nse1-C216S | 0.3 ± 0.6 |
| pht1-4KR top2-191 nse1-C216S | 2.3 ± 0.6 |
| pht1-36 | 1.0 ± 1.0 |
| pht1-36 smc6-74 | 2.7 ± 2.1 |
| pht1-36 top2-191 | 2.3 ± 1.5 |
| pht1-36 top2-191 smc6-74 | 2.0 ± 1.7 |
| pht1-36 nse1-C216S | 0.3 ± 0.6 |
| pht1-36 top2-191 nse1-C216S | 2.3 ± 2.1 |
| swr1Δ | 1.3 ± 0.6 |
| swr1Δ top2 | 38.7 ± 3.5 |
| swr1Δ smc6-74 | 1.7 ± 1.5 |
| swr1Δ top2 smc6-74 | 16.7 ± 2.1 |
| arp8Δ | 0.3 ± 0.6 |
| arp8Δ top2-191 | 16.0 ± 3.6 |
| arp8Δ smc6-74 | 1.0 ± 1.0 |
| arp8Δ top2-191 smc6-74 | 17.3 ± 4.0 |
| ies2Δ | 0.3 ± 0.6 |
| ies2Δ smc6-74 | 1.7 ± 1.5 |
| ies2Δ top2-191 | 6.7 ± 1.5 |
| ies2Δ top2-191 smc6-74 | 29.0 ± 2.6 |
| hst4Δ | 1.0 ± 1.0 |
| hst4Δ smc6-74 | 0.7 ± 0.6 |
| hst4Δ top2-191 | 24.3 ± 4.0 |
| hst4Δ top2-191 smc6-74 | 21.3 ± 1.5 |
Data are means ± standard deviations from triplicate counts of 100 cells.
As with all H2A.Z homologs, the acetylation of lysines in the N terminus of Pht1 is important for function (40), and indeed, nse1-C216S top2-191 was also suppressed by an allele where all of these lysine residues were converted to nonacetylatable arginines (pht1–4KR) (Fig. 2A; Table 3). Therefore, acetylated H2A.Z is required to confer the synthetic lethal mitotic defects of nse1-C216S top2-191 cells.
We next asked if the suppression of nse1-C216S top2-191 by pht1 mutants was specific to Nse1 dysfunction. To this end, we crossed pht1-36, pht1Δ, and pht1-4KR mutants with the smc6-74 top2-191 double mutant, and each allele suppressed this synthetic lethal combination and the associated mitotic defects (Fig. 2C; Table 3), without an effect on top2-191 itself (Table 3; see also Fig. S1 in the supplemental material). Further, pht1Δ also suppressed both the DNA damage sensitivity and associated mitotic defects of smc6-74 (Fig. 2D and E; Table 3) but not those conferred by deleting the rad51 HR gene (data not shown). Similar suppression was conferred by pht1-4KR (see Fig. S2 in the supplemental material). Therefore, the effects of H2A.Z depletion on Smc5/6 dysfunction are not specific to either the top2-191 mutation or the damage-independent effects of nse1-C216S. These data identify H2A.Z as a key component of the mitotic regulation conferred by Smc5/6 and Top2.
DNA repair in the presence of Smc5/6 dysfunction is dependent on the error-free branch of the PRR pathway that is controlled by the E2 ubiquitin-conjugating enzyme Ubc13. This is evidenced by the greatly enhanced sensitivity of ubc13Δ smc6-74 double mutants. Furthermore, Ubc13-dependent PRR is required in cells with the nse1-C216S mutation to suppress the damage sensitivity of smc6-74 (17). We therefore asked whether this pathway was required for the suppression of smc6-74 by pht1Δ, and indeed we found that ubc13Δ reversed the suppression of the smc6-74 mutant's methyl methanesulfonate (MMS) and UV sensitivities conferred by pht1Δ (Fig. 2D and E). Thus, error-free PRR appears to be a general backup DNA repair mechanism in the presence of Smc5/6 dysfunction. Interestingly, ubc13 was also required for MMS and UV-C resistance in pht1Δ cells, suggesting a defect in HR-mediated repair that is resolved by error-free PRR in the absence of H2A.Z.
ATP-dependent chromatin remodelers modify Smc5/6 dysfunction.
We next asked whether regulators of H2A.Z dynamics modify the phenotypes associated with Smc5/6 and Top2 dysfunction. Here, we chose to use smc6-74, as it displays both Top2-induced and (unlike nse1-C216S) DNA damage-induced mitotic defects; from extensive analysis of cohesin dynamics, we previously showed that both are attributed to postanaphase cohesin retention on chromosome arms (24).
We first considered the SWR-C chromatin-remodeling complex, which deposits H2A.Z into nucleosomes (41–43). Swr1 is the catalytic SWI/SNF family helicase of SWR-C, and in S. pombe, swr1Δ reduces total H2A.Z in chromatin and abolishes the presence of acetylated H2A.Z (40). Consistent with the effects of pht1Δ, the HU and MMS sensitivities of cells smc6-74 were efficiently suppressed by swr1Δ (Fig. 3A), as were the mitotic defects conferred by transient HU treatment of smc6-74 cells (Table 4). As with other smc6-74 suppressors, this effect was dependent on Ubc13-dependent error-free PRR (data not shown). However, swr1Δ did not suppress the synthetic lethality of the smc6-74 or nse1-C216S mutants with top2-191 (Table 3 and data not shown). Indeed, swr1Δ was actually itself synthetically lethal with top2-191 (Fig. 3B), which is associated with severe mitotic defects that are reduced by smc6-74 (Table 3). This interaction between top2-191 and swr1Δ parallels the fact that separase overexpression is a suppressor of the mitotic defects of smc6-74 and yet is also synthetically lethal with top2-191 at 30°C (24).
FIG 3.

Chromatin remodelers functionally interact with Smc5/6 and Top2. (A) swr1Δ suppresses the HU and MMS sensitivities of smc6-74. Spots are 10-fold serial dilutions grown at 30°C for 4 days. (B) swr1Δ is synthetically lethal with top2-191. Plates were incubated at 25°C (5 days) or 30°C (4 days). (C) Western blot analysis of total and acetylated Pht1 (Pht1Ac). Tubulin was used as a loading control. The dotted line indicates the boundary of omitted lanes between the wild-type and swr1Δ samples. (D) arp8Δ suppresses the HU and MMS sensitivities of smc6-74. Spots are 10-fold serial dilutions, and plates were incubated at 30°C for 4 days. (E) arp8Δ is synthetically lethal with top2-191 at 30°C. Plates were grown at 25°C for 5 days or for 4 days at 30°C and 36°C. (F) DAPI-stained DIC images of wild-type and arp8Δ cells grown at 25°C and 36°C. Bar, 10 μm.
TABLE 4.
Aberrant mitosis 4 h after release from 11 mM HU (4-h block)
| Genotype | % aberrant mitosisa |
|---|---|
| Wild type | 0.0 ± 0.0 |
| smc6-74 | 29.3 ± 4.0 |
| pht1Δ | 2.0 ± 1.0 |
| pht1Δ smc6-74 | 3.7 ± 1.5 |
| pht1-36 | 1.3 ± 0.6 |
| pht1-36 smc6-74 | 2.7 ± 1.5 |
| pht1-4KR | 1.3 ± 1.2 |
| pht1-4KR smc6-74 | 3.0 ± 1.0 |
| swr1Δ | 0.7 ± 1.2 |
| swr1Δ smc6-74 | 6.7 ± 1.2 |
| arp8Δ | 0.3 ± 0.6 |
| arp8Δ smc6-74 | 3.7 ± 1.2 |
| hst4Δ | 3.7 ± 0.6 |
| rtt109Δ | 2.7 ± 1.5 |
| smc6-74 hst4Δ | 6.3 ± 2.1 |
| smc6-74 rtt109Δ | 60 ± 4.6 |
| smc6-74 rtt109Δ pht1Δ | 5.3 ± 0.6 |
| hst4Δ pht1Δ | 0.3 ± 0.6 |
| hst4Δ pht1Δ smc6-74 | 10.7 ± 1.2 |
| hht1Δ hhf1Δ hht3Δ hhf3Δ background: | |
| Wild type | 4.3 ± 0.6 |
| smc6-74 | 15.3 ± 1.5 |
| H3K56R | 8.7 ± 1.2 |
| H3K56Q | 1.0 ± 1.0 |
| smc6-74 H3K56R | 41.7 ± 2.5 |
| smc6-74 H3K56Q | 5.3 ± 1.5 |
Data are means ± standard deviations from triplicate counts of 100 cells.
swr1Δ cells have been reported to possess unacetylated H2A.Z (40). We assayed total and acetylated H2A.Z levels and confirmed this observation. However, we also found that the pht1-4KR mutant, which abolishes acetylation, also significantly reduced the levels of total H2A.Z, even compared to swr1Δ cells (Fig. 3C). Thus, swr1Δ cells are not equivalent to pht1Δ cells, and the presence of unacetylated H2A.Z may contribute to the lethality of swr1Δ with top2-191. Indeed, pht1Δ is not synthetically lethal with top2-191 (Table 3; also, see Fig. S1 in the supplemental material).
INO80 regulates the genome-wide localization of H2A.Z and has been best characterized in S. cerevisiae. In ino80Δ cells, H2A.Z is largely unacetylated and, importantly, present in mislocalized nucleosomes (44). These cells also have defects in DSB repair (45, 46). As ino80 is an essential gene in S. pombe (47), we assayed for interactions between smc6-74 and the nonessential INO80 complex member encoded by arp8. arp8Δ does not confer DNA damage sensitivity at the range of doses used here in rich medium. However, arp8Δ does confer sensitivity to 7.5 mM HU in minimal medium (47). Nevertheless, arp8Δ suppressed the MMS and HU sensitivities of smc6-74 mutants (Fig. 3D), as well as the mitotic defects conferred by HU treatment of smc6-74 cells (Table 4) and the severity of the mitotic defects of smc6-74 top2-191 cells (Table 3). Similar to swr1Δ, arp8Δ is synthetically lethal with top2-191 at 30°C (Fig. 3E), with significant mitotic defects (Table 3). However, arp8Δ cells are themselves lethal at 36°C (Fig. 3E) and display nuclear fragmentation and mitotic defects (Fig. 3F). This may contribute directly or indirectly to the genetic interaction with top2-191, and it should be noted that unlike Swr1 of SWR-C, Arp8 is a noncatalytic subunit of INO80. Thus, the severity of interaction with the top2-191 allele is likely to be a function of the precise molecular defect in INO80.
Histone H3 K56 acetylation suppresses the damage-induced mitotic defects of the smc6-74 mutation via H2A.Z.
It was recently demonstrated that the acetylation of histone H3 on K56 alters the activities for these chromatin remodelers in S. cerevisiae, stimulating an H2A.Z eviction activity for INO80 and SWR-C (48). This finding highlights multiple mechanisms by which these enzymes can lead to a net loss of H2A.Z, as this changes the specificity of SWR-C. We therefore asked whether H3 K56 acetylation modulates the chromosome segregation controls conferred by Smc5/6 and whether this affects the mitotic defects in smc6-74 cells after DNA damage or in combination with top2-191.
H3K56 is acetylated mostly during S phase, although it remains acetylated under conditions of replication stress (49). In both S. cerevisiae and S. pombe, H3K56 acetylation is catalyzed by Rtt109 (50) and deacetylated by Hst4 (51). We found that hst4Δ potently suppressed the DNA damage sensitivity of smc6-74 cells (Fig. 4A). Consistent with this suppression, deletion of rtt109 enhanced the sensitivity of smc6-74 mutant cells to DNA damage (Fig. 4B). In both cases, the phenotype correlated with the magnitude of mitotic defects (Table 4). As H3 K56 acetylation by Rtt109 stimulates H2A.Z turnover (48), we tested whether the enhanced MMS and HU sensitivities of smc6-74 rtt109Δ cells were suppressed by pht1Δ, and indeed this was the case (Fig. 4B).
FIG 4.

Histone H3 K56 acetylation suppresses Smc5/6 dysfunction. Cell spots in panels A to C are 10-fold serial dilutions, and plates were incubated at 30°C for 4 days. (A) Deletion of the H3K56 deacetylase hst4 suppresses the HU and MMS sensitivities of smc6-74 cells. (B) Deletion of H3K56 acetyltransferase rtt109 enhances the HU and MMS sensitivities of smc6-74 cells, and this is suppressed by pht1Δ. (C) The strains in the top two lines are on a wild-type background, and the lower six strains contain deletions of the indicated histone H3 and H4 genes. K56Q and K56R mutations in the remaining H3 gene (hht2) suppressed and enhanced the HU and MMS sensitivities of smc6-74 cells, respectively. (D) Western blot analysis of histone H3 K56 acetylation (K56Ac) and total H3 levels.
We then asked whether these observations could be recapitulated by mutations in H3 K56. To this end, we crossed smc6-74 mutants with strains where two out of three histone H3-H4 gene clusters (hht1, hhf1 and hht3, hhf3) have been deleted and the remaining histone H3 gene (hht2) contains nonacetylatable K56R or acetyl-mimicking K56Q mutations (51). On this background, reduced H3-H4 gene copy numbers partially suppressed the MMS and HU sensitivities of smc6-74 cells, while the hht2-K56Q mutation completely suppressed the sensitivities to these agents. Conversely, hht2-K56R enhanced the MMS and HU sensitivities of smc6-74 cells (Fig. 4C; Table 4), confirming the positive effect of K56 acetylation on the suppression of the damage-induced mitotic defects of smc6-74 cells. We did not observe major differences in K56 acetylation between wild-type and smc6-74 cells (Fig. 4D), and therefore Smc5/6 dysfunction is manifested under normal levels of K56 acetylation. However, Smc5/6 dysfunction is overcome when H3 K56 acetylation is enhanced by deletion of the deacetylase hst4, or by the acetyl-mimicking K56Q mutations.
H2A.Z does not modify the chromatin association of Smc5/6.
H2A.Z function is generally attributed to the regulation of gene expression, but transcriptional profiling of pht1Δ and pht1-4KR via microarrays did not detect significant changes in the mRNA levels of Smc5/6, Top2, or cohesin components or regulators (40). We tested this by quantitative reverse transcription-PCR for several genes in smc6-74, pht1Δ, pht1-36, and pht1-4KR cells (Fig. 5A). While a significant reduction of mRNA levels was observed in some cases, this included reduced expression of Cut1 in pht1Δ and pht1-4KR cells, which could exacerbate the mitotic defects. Moreover, expression was never reduced by >50%, and none of these genes was haplo-insufficient. However, we cannot rule out the combined effects of reduced expression of multiple genes.
FIG 5.

H2A.Z does not affect Smc5/6 chromatin association. (A) RT-qPCR assays of mRNA levels compared to wild type and normalized to cdc2 and expression levels in wild-type cells (100%; top dotted line). Data are means ± standard deviations (n = 4). All data points show no statistical significance (P < 0.05) compared to wild-type cells (Student's t test), except those with an asterisk, which indicates significance (P = 0.05 to 0.1). (B) Western blot showing levels of Nse4 and Smc5, both HA tagged at their C termini. (C) Smc5/6 ChIP assays with HA-tagged Nse4 were performed on asynchronously growing wild-type and pht1Δ cells, and occupancy at the indicated loci was measured by qPCR. STE1 is the subtelomeric region on chromosome 1 (33).
As nse4 mRNA levels were reduced in pht1Δ and pht1-4KR cells (but not in pht1-36 cells), we examined the Nse4 protein levels together with Smc5 by Western blotting. Notably, as with many multiprotein complexes, reduced expression of one Smc5/6 component in human cells reduces the levels of other Smc5/6 proteins (52). We observed no difference in Nse4 or Smc5 expression between wild-type and pht1Δ cells (Fig. 5B), suggesting that the nse4 mRNA differences are unlikely to be functionally significant. We therefore assayed the association of the Smc5/6 complex with chromatin in ChIP assays with hemagglutinin (HA)-tagged Nse4 (17, 53) (Fig. 5C). Again we saw no significant difference between asynchronously growing wild-type and pht1Δ cells. Thus, even though we saw reduced nse4 mRNA in pht1Δ cells, the Nse4 protein levels and chromatin recruitment were not affected. Therefore, the strong suppressive effects of pht1Δ on Smc5/6 dysfunction cannot be explained by gene expression changes or changes to Smc5/6 localization.
H2A.Z modulates cohesin levels on chromosome arms.
The overexpression of the cohesin protease separase suppresses smc6-74 top2-191 synthetic lethality and damage sensitivity as efficiently as does loss of H2A.Z (24). We therefore asked if H2A.Z affected the levels of cohesin on chromosomes by using ChIP assays and GFP-tagged Rad21. Steady-state levels of chromosome arm-associated cohesin were significantly reduced in pht1Δ cells at all loci tested (P < 0.01), whereas there was a more modest statistically insignificant effect at the centromeres (P = 0.4) (Fig. 6A) and at the centromere-adjacent tRNAMet and tRNALeu genes (data not shown).
FIG 6.
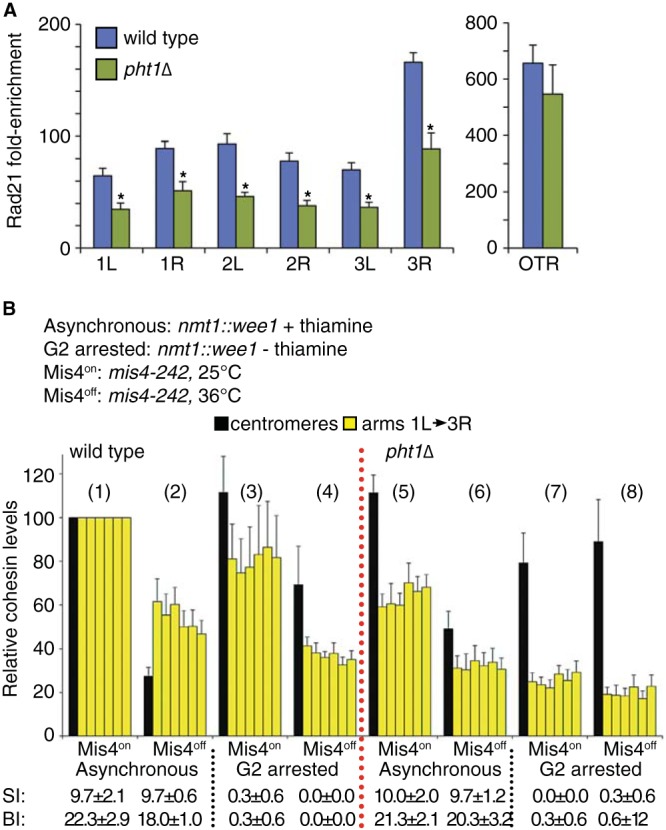
H2A.Z is required for wild-type cohesin levels on chromosome arms. (A) Cohesin ChIP assays using GFP-Rad21 were performed on wild-type and pht1Δ cells grown at 30°C. Data show the enrichment over untagged isogenic controls and are means ± standard errors of the means (SEM) for 5 biological replicates. Cohesin was detected on each chromosome arm (1L and 3R) and at the outer repeats (OTR) of the centromeres by qPCR. *, P < 0.01, Student's t test. The difference at the OTR was not significant (P = 0.4). (B) Wild-type (conditions 1 to 4) and pht1Δ (conditions 5 to 8) cells on a mis4-242 background were grown at 25°C. Cells also carried an integrated cassette expressing Wee1 from the nmt1 promoter and were grown either in the presence (promoter repressed, asynchronous) or absence (promoter derepressed, G2 arrested) of thiamine. Subsequently, half the culture was shifted to 36°C for 3 h to inactivate Mis4. Data were normalized to results with asynchronous wild-type cells and are means ± SEM (n = 4 to 5). Raw data are reported in Table 5. Below the graph are the septation index (SI) and binucleate index (BI) from 3 samples of 100 cells. Data are means ± standard deviations.
In S. pombe (and human cells), cohesin is removed from chromosomes for a very brief period postanaphase; in addition, pht1Δ cells show normal mitotic kinetics. Therefore, the effect of H2A.Z on chromosomal cohesin levels cannot be an effect of altered cell cycle regulation. We therefore asked whether the reduced cohesin levels on chromosomes were due to a defect in loading or retention. To this end, we used cells carrying a temperature-sensitive mutation in the cohesin loader, mis4-242, which is functional at 25°C (based on growth and cohesin ChIP) and inactivated at 36°C, where cohesin is not reloaded as cells pass through anaphase. However, we reproducibly observed reductions to 30 to 50% of steady-state levels of cohesin on postmitotic chromosomes assayed using Rad21-GFP ChIP, but this was not visible by microscopy in our hands. This likely reflected cohesin that encircled only one chromatid or perhaps was reloaded independently of Mis4 (24). Further, we collected both asynchronously growing cells (conditions 1, 2, 5, and 6) as well as cells arrested in G2 phase (conditions 3, 4, 7, and 8) by inducible Wee1 overexpression from the thiamine-repressible nmt1 promoter (54). The G2 arrest was confirmed by loss of septated and binucleated cells (Fig. 6B), as well as cell elongation and a 2C DNA content (data not shown). We then measured chromosomal cohesin levels by ChIP (Rad21-GFP) before (Mis4on, odd-numbered samples) and after (Mis4off, even-numbered samples) inactivation of Mis4 at 36°C in both wild-type (conditions 1 to 4) and pht1Δ (conditions 5 to 8) cells. The rationale behind this design was that if cohesin retention is defective, Mis4 inactivation in G2 should result in loss of cohesin without the normal mitotic removal, as it cannot be reloaded. For this experiment, the ChIP data were normalized against asynchronous cells grown at 25°C to enable comparison between different loci, and the raw data are shown in Table 5.
TABLE 5.
Normalizing enrichment of Rad21-GFP for the strains in Fig. 6B
| Locus | Fold enrichmenta |
|---|---|
| OTR | 108.81 ± 6.58 |
| 1L | 19.09 ± 2.26 |
| 1R | 22.04 ± 3.12 |
| 2L | 27.91 ± 2.50 |
| 2R | 19.44 ± 2.13 |
| 3L | 23.73 ± 2.51 |
| 3R | 22.63 ± 1.84 |
Data are means ± standard errors of the means (n = 4 to 8) and are the raw data for Fig. 6B (normalizing point, 25°C, with thiamine).
In wild-type cells, cohesin levels were maintained during a G2 arrest when Mis4 was active (compare conditions 1 to 3) but were reduced on chromosome arms following Mis4 inactivation in G2 (Fig. 6B, compare conditions 3 and 4). Hence, cohesin is reloaded in a Mis4-dependent manner in G2 and cohesin complexes on chromosome arms are more dynamic than those at centromeres. In pht1Δ cells, the reduced levels of chromosome arm cohesin (compare condition 1 to condition 5) were further reduced during the G2 arrest, regardless of Mis4 activity (Fig. 6B, compare condition 5 to conditions 7 and 8 and condition 3 to 7). Hence, cohesin retention (and possibly loading) in G2 is dependent on H2A.Z.
pht1Δ suppresses the aberrant retention of cohesin in smc6-74 top2-191 cells.
To test the physiological significance of the decrease in cohesin levels in pht1Δ cells, we assayed chromosomal cohesin levels in smc6-74 top2-191 cells in the presence and absence of H2A.Z. At 30°C, smc6-74 top2-191 cells have a well-characterized postanaphase retention of cohesin on chromosome arms, and the retained cohesin can be removed in cells that overexpress separase, even though endogenous separase functions predominantly at the centromeres. Again, we used a mis4-242 background to prevent cohesin reloading immediately following anaphase, as without this the transient and very brief loss of cohesin cannot be seen (17, 24). However, at 30°C, the restrictive temperature for smc6-74 top2-191 cells, mis4-242 inactivation is slower and requires a longer incubation (6 h) before cohesin loss is observed (24). We also included the actin poison latrunculin B, in order to prevent lethal cytokinesis and chromosome breakage following mitotic failure. Although a previous report indicated that there was a mitotic delay induced by latrunculin B (55), this experiment was performed in rich medium, and we have not observed this in minimal medium, which is used for all our ChIP assays (24).
As we observed before (24), cohesin was retained on chromosome arms after smc6-74 top2-191 cells passed through mitosis. In keeping with the genetic suppression of smc6-74 top2-191 by pht1Δ at 30°C, this postanaphase retention of cohesin was significantly diminished in the absence of H2A.Z (Fig. 7; these data were normalized to those for samples taken at 25°C for each strain, and the raw data are presented in Table 6). The reduction in cohesin retention conferred by pht1Δ was very similar to that seen with separase overexpression (24). Thus, as with separase overexpression, the absence of H2A.Z suppresses the mitotic defects resulting from Smc5/6 and Top2 dysfunction by lowering the levels of cohesin on chromosome arms that would otherwise block sister chromatid separation. These results confirm H2A.Z as a physiologically relevant regulator of cohesin dynamics.
FIG 7.
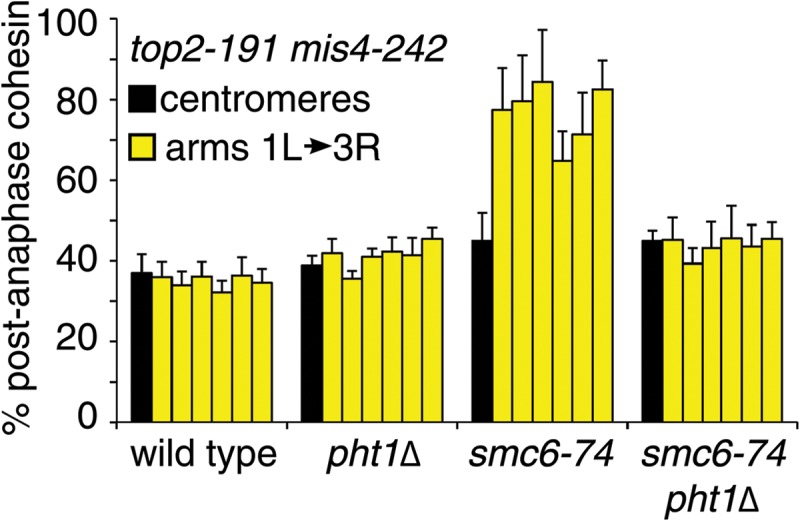
H2A.Z is required for aberrant cohesin retention in smc6-74 top2-191 cells. ChIP assays of cells with a top2-191 mis4-242 background were performed as in Fig. 6, except that latrunculin B was added to prevent lethal cytokinesis. The data show postmitotic levels normalized to results with asynchronous cultures, following a shift from 25°C to 30°C for 6 h. Data are means ± standard errors of the means (n = 4 to 8). Raw data are shown in Table 6.
TABLE 6.
Normalizing enrichment of Rad21-GFP for the strains in Fig. 7
| Genotype and chromosome arm | Fold enrichment |
|---|---|
| top2-191 mis4-242 rad21-GFP | |
| OTR | 214.31 ± 9.29 |
| 1L | 35.98 ± 1.75 |
| 1R | 31.15 ± 2.47 |
| 2L | 34.49 ± 1.99 |
| 2R | 34.62 ± 1.31 |
| 3L | 38.15 ± 2.67 |
| 3R | 33.91 ± 1.46 |
| pht1Δ top2-191 mis4-242 rad21-GPF | |
| OTR | 188.33 ± 11.74 |
| 1L | 29.3 ± 1.41 |
| 1R | 27.52 ± 1.26 |
| 2L | 27.04 ± 1.11 |
| 2R | 28.43 ± 1.12 |
| 3L | 30.35 ± 2.48 |
| 3R | 25.01 ± 1.92 |
| smc6-74 top2-191 mis4-242 rad21-GFP | |
| OTR | 176.45 ± 11.9 |
| 1L | 29.38 ± 1.78 |
| 1R | 23.14 ± 1.18 |
| 2L | 28.19 ± 1.93 |
| 2R | 25.61 ± 1.32 |
| 3L | 28.32 ± 2.44 |
| 3R | 24.43 ± 1.04 |
| smc6-74 pht1Δ top2-191 mis4-242 rad21-GFP | |
| OTR | 221.23 ± 8.96 |
| 1L | 30.13 ± 3.31 |
| 1R | 26.03 ± 1.67 |
| 2L | 28.55 ± 3.34 |
| 2R | 29.4 ± 3.91 |
| 3L | 31.41 ± 3.64 |
| 3R | 27.66 ± 2.94 |
Data are means ± standard errors of the means (n = 4 to 5) and are the raw data for Fig. 7 (normalizing point, 25°C).
DISCUSSION
Our previous findings showed not only that nse1-C216S cells are wild type for damage sensitivity but also that this disruption of the vRING domain prevents Smc5/6 complex accumulation at sites of DNA damage (17). Here we have shown that nse1-C216S is a separation-of-function mutant, and confers as strong mitotic defects when combined with top2-191 as do any of the repair-deficient mutants. Passage through mitosis requires Smc5/6 function, as mitotic failure is seen with null Smc5/6 alleles without extrinsic DNA damage. Therefore, in the case of the hypomorphs where mitosis fails after DNA damage or in combination with top2-191, it appears that these conditions potentiate a defect that manifests in the complete absence of Smc5/6 without additional challenges. To date, all the data points to cohesin retention as the cause of the mitotic defects of Smc5/6 mutants in S. pombe, which has also recently been observed in S. cerevisiae (26).
How does Smc5/6 regulate cohesin? To date, we can only speculate and perhaps rule out some possibilities. The Smc5/6 hypomorphs do not seem to affect cohesin expression, premitotic cohesin levels on chromosomes, or the recruitment of cohesin to DSBs (24). Therefore, any effect of Smc5/6 on cohesin appears to be subsequent to its loading. Conceivably, Smc5/6 could act directly on cohesin, although physical interactions have not been found. Further, Nse2-catalyzed SUMOylation is not essential for mitosis (16), and so this is also unlikely to be a cohesin regulator. Given the interactions with Top2 and H2A.Z, it is more likely the regulation of chromosome structure by Smc5/6 in turn influences the cohesin cycle in an indirect manner. Clearly, determining this regulatory cross talk is important, and independent demonstration of these phenomena in different species and through different experimental approaches will facilitate progress in this direction.
As a result of our suppressor screen, we clearly show that Smc5/6 dysfunction leading to cohesin retention and lethal mitoses is suppressed when the chromatin landscape is altered by H2A.Z depletion from chromosomes. We have demonstrated this occurs not only with H2A.Z loss but also by independent modulation of H2A.Z dynamics by chromatin remodelers and their regulation of H3 K56 acetylation. H2A.Z is acetylated by the essential Mst1 acetyltransferase (40). However, this enzyme also acetylates the canonical histones (56, 57), and so not surprisingly, smc6-74 is not suppressed by a conditional allele of mst1, mst1-L271P, at semipermissive temperatures (data not shown). Although modifications other than acetylation to the H2A.Z N-terminal lysines are possible, these are yet to be described. Further, the mechanism of suppression may involve yet to be identified additional proteins recruited to acetylated H2A.Z or modifications elsewhere on the protein, such as the C-terminal monoubiquitination described in human cells (58).
Although H2A.Z is frequently implicated as a regulator of gene expression, we cannot explain our phenotypes by functionally significant changes in cohesin or Smc5/6 gene expression. Rather, H2A.Z appears to have a direct effect in opposition to Smc5/6 to promote sister chromatid cohesin along chromosome arms but not at centromeres. Both mammalian (59) and S. pombe (38) cells lacking H2A.Z display chromosome segregation defects, and our findings suggest this is likely to be, at least in part, due to disruption of the cohesin cycle. Notably, the removal of arm cohesin is mechanistically different from that at the centromeres (12, 60, 61), with the latter requiring cohesin cleavage by separase (10).
We have shown that H2A.Z is required for the retention of cohesin on chromosome arms but not at the outer repeats of the centromeres. Consistent with this, an independent study of the effect of H2A.Z on chromosome architecture in S. pombe concluded no effect on centromere structure or function (40). The increase in mitotic defects conferred by pht1Δ in this study is also similar to ours (8-fold versus 6-fold, respectively). However, the magnitude of mitotic defects reported in that other study, 20 to 50% (40), was orders of magnitude over what we observed and greater than levels seen in many lethal mitotic mutants (Tables 1 and 2). Perhaps this reflects a difference in genetic background, culturing conditions, or assay sensitivity, but in any case it suggests the primary role for H2A.Z is at chromosome arms.
Several experiments have shown that H2A and H2A.Z modulate the interaction of condensin with chromosomes (40, 62). Recombinant N-terminal peptides from these histones interact with condensin from HeLa cell nuclear extracts, suggesting that H2A and H2A.Z may be “condensin receptors” on chromosomes. We have performed similar experiments using immobilized synthetic peptides of both acetylated and unacetylated H2A and H2A.Z as affinity reagents for cohesin, but we did not see a robust interaction. Thus, we cannot implicate acetylated H2A.Z as a cohesin “receptor” at this stage, although we also cannot rule out that such an activity occurs only in the case of intact nucleosomes or requires additional factors not present in our experiments. Given that H2A.Z is proposed to be a receptor for condensin, it is surprising that pht1Δ actually suppresses loss-of-function condensin defects in S. pombe (40). This suggests that the functional relationship between H2A.Z and condensin is nonlinear, and the same may be true for cohesin and Smc5/6. That is, these genes may act both up- and downstream of each other, and genetic interactions can depend on the nature of the alleles and the point at which they interact. For example, a defect in opening cohesin would have opposite effects if it were inflicted prior to or after cohesin loading (no cohesion versus no sister chromatid separation).
Although premature sister chromatid separation was recently reported in S. cerevisiae mutants lacking H2A.Z, this study concluded that this effect was in fact independent of cohesin dynamics (63). Mechanisms controlling cohesin dynamics in this organism appear to be exclusively driven by separase cleavage of Scc1 (13), and sister chromatid cohesion is resolved in several S. cerevisiae strains without loss of cohesin from chromosomes, presumably bound to one sister chromatid (64). Moreover, H2A.Z is required for DNA repair in S. cerevisiae (65) but not in S. pombe. Thus, the relationship between our findings and those regarding H2A.Z function in S. cerevisiae are difficult to compare. Nevertheless, our data firmly establish acetylated H2A.Z (and potential interactors) as an important regulator of cohesin dynamics, presumably as a local regulator of chromatin topology that mediates the complex interactions of the many proteins that ensure the cycle of events that control the accurate segregation of replicated chromatids.
Sister chromatid cohesion is established coincident with DNA replication, which also coincides with the kinetics of histone H3 K56 acetylation, a modification that is promoted by replication stress, and like Smc5/6 function, is required for replication stress resistance (50, 51). Importantly, H3 K56 acetylation modulates the activity of H2A.Z chromatin remodelers by favoring H2A.Z nucleosomes for exchange events (48). This is consistent with the suppression of rtt109Δ smc6-74 by pht1Δ, although it is possible (if not likely) that other aspects of the histone code will regulate the SMC complexes to ensure accurate chromosome segregation.
H2A.Z, and also the centromere-specific histone H3 variant CENP-A (66), are the most ancient of the histone variants. Metazoans have evolved several other histone H2A and H3 variants, and functional studies implicate these in processes such as X-inactivation, developmental fate, and pluripotency (67), which are critical for tissue patterning but not for cell proliferation per se. Increased genome integrity from the regulation of chromosome segregation at the centromeres (CENP-A) and arms (H2A.Z) may have selected for the emergence of the histone variants, which subsequently expanded with evolutionary complexity.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH NIGMS grant GM088162.
We are grateful to Michael Keogh, Antony Carr, Patrick Varga-Weisz, Rohinton Kamakaka, Johanne Murray, and Nancy Walworth for provision of strains, to the Mount Sinai Genomics Core facility for genomic sequence generation, and to Lekhana Bhandary for assistance with the early part of this study. We also acknowledge Nagma Shah for assistance with preparation of Fig. 4B.
Footnotes
Published ahead of print 31 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00193-14.
REFERENCES
- 1.Hirano T. 2006. At the heart of the chromosome: SMC proteins in action. Nat. Rev. Mol. Cell Biol. 7:311–322. 10.1038/nrm1909 [DOI] [PubMed] [Google Scholar]
- 2.Murray JM, Carr AM. 2008. Smc5/6: a link between DNA repair and unidirectional replication? Nat. Rev. Mol. Cell Biol. 9:177–182. 10.1038/nrm2309 [DOI] [PubMed] [Google Scholar]
- 3.Nasmyth K, Haering CH. 2005. The structure and function of SMC and kleisin complexes. Annu. Rev. Biochem. 74:595–648. 10.1146/annurev.biochem.74.082803.133219 [DOI] [PubMed] [Google Scholar]
- 4.Aono N, Sutani T, Tomonaga T, Mochida S, Yanagida M. 2002. Cnd2 has dual roles in mitotic condensation and interphase. Nature 417:197–202. 10.1038/417197a [DOI] [PubMed] [Google Scholar]
- 5.Birkenbihl RP, Subramani S. 1992. Cloning and characterization of rad21 an essential gene of Schizosaccharomyces pombe involved in DNA double-strand-break repair. Nucleic Acids Res. 20:6605–6611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McAleenan A, Clemente-Blanco A, Cordon-Preciado V, Sen N, Esteras M, Jarmuz A, Aragon L. 2013. Post-replicative repair involves separase-dependent removal of the kleisin subunit of cohesin. Nature 493:250–254. 10.1038/nature11630 [DOI] [PubMed] [Google Scholar]
- 7.Nagao K, Adachi Y, Yanagida M. 2004. Separase-mediated cleavage of cohesin at interphase is required for DNA repair. Nature 430:1044–1048. 10.1038/nature02803 [DOI] [PubMed] [Google Scholar]
- 8.Parelho V, Hadjur S, Spivakov M, Leleu M, Sauer S, Gregson HC, Jarmuz A, Canzonetta C, Webster Z, Nesterova T, Cobb BS, Yokomori K, Dillon N, Aragon L, Fisher AG, Merkenschlager M. 2008. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 132:422–433. 10.1016/j.cell.2008.01.011 [DOI] [PubMed] [Google Scholar]
- 9.Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, Tsutsumi S, Nagae G, Ishihara K, Mishiro T, Yahata K, Imamoto F, Aburatani H, Nakao M, Imamoto N, Maeshima K, Shirahige K, Peters JM. 2008. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 451:796–801. 10.1038/nature06634 [DOI] [PubMed] [Google Scholar]
- 10.Nasmyth K, Haering CH. 2009. Cohesin: its roles and mechanisms. Annu. Rev. Genet. 43:525–558. 10.1146/annurev-genet-102108-134233 [DOI] [PubMed] [Google Scholar]
- 11.Rudra S, Skibbens RV. 2013. Cohesin codes: interpreting chromatin architecture and the many facets of cohesin function. J. Cell Sci. 126:31–41. 10.1242/jcs.116566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hauf S, Roitinger E, Koch B, Dittrich CM, Mechtler K, Peters JM. 2005. Dissociation of cohesin from chromosome arms and loss of arm cohesion during early mitosis depends on phosphorylation of SA2. PLoS Biol. 3(3):e69. 10.1371/journal.pbio.0030069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uhlmann F, Lottspeich F, Nasmyth K. 1999. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature 400:37–42. 10.1038/21831 [DOI] [PubMed] [Google Scholar]
- 14.Palecek J, Vidot S, Feng M, Doherty AJ, Lehmann AR. 2006. The SMC5-6 DNA repair complex: bridging of the SMC5-6 heads by the Kleisin, NSE4, and non-Kleisin subunits. J. Biol. Chem. 281:36952–36959. 10.1074/jbc.M608004200 [DOI] [PubMed] [Google Scholar]
- 15.Lindroos HB, Strom L, Itoh T, Katou Y, Shirahige K, Sjogren C. 2006. Chromosomal association of the Smc5/6 complex reveals that it functions in differently regulated pathways. Mol. Cell 22:755–767. 10.1016/j.molcel.2006.05.014 [DOI] [PubMed] [Google Scholar]
- 16.Andrews EA, Palecek J, Sergeant J, Taylor E, Lehmann AR, Watts FZ. 2005. Nse2, a component of the Smc5-6 complex, is a SUMO ligase required for the response to DNA damage. Mol. Cell. Biol. 25:185–196. 10.1128/MCB.25.1.185-196.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tapia-Alveal C, O'Connell MJ. 2011. Nse1-dependent recruitment of Smc5/6 to lesion-containing loci contributes to the repair defects of mutant complexes. Mol. Biol. Cell 22:4669–4682. 10.1091/mbc.E11-03-0272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doyle JM, Gao J, Wang J, Yang M, Potts PR. 2010. MAGE-RING protein complexes comprise a family of E3 ubiquitin ligases. Mol. Cell 39:963–974. 10.1016/j.molcel.2010.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lehmann AR, Walicka M, Grittiths DJF, Murray JM, Watts FZ, McCready S, Carr AM. 1995. The rad18 gene of Schizosaccharomyces pombe defines a new subgroup of the SMC superfamily involved in DNA repair. Mol. Cell. Biol. 15:7067–7080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phipps J, Nasim A, Miller DR. 1985. Recovery, repair, and mutagenesis in Schizosaccharomyces pombe. Adv. Genet. 23:1–72 [DOI] [PubMed] [Google Scholar]
- 21.Birkenbihl RP, Subramani S. 1995. The rad21 gene product of Schizosaccharomyces pombe is a nuclear, cell cycle-regulated phosphoprotein. J. Biol. Chem. 270:7703–7711 [DOI] [PubMed] [Google Scholar]
- 22.Harvey SH, Sheedy DM, Cuddihy AR, O'Connell MJ. 2004. Coordination of DNA damage responses via the Smc5/Smc6 complex. Mol. Cell. Biol. 24:662–674. 10.1128/MCB.24.2.662-674.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verkade HM, Bugg SJ, Lindsay HD, Carr AM, O'Connell MJ. 1999. Rad18 is required for DNA repair and checkpoint responses in fission yeast. Mol. Biol. Cell 10:2905–2918. 10.1091/mbc.10.9.2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Outwin EA, Irmisch A, Murray JM, O'Connell MJ. 2009. Smc5-Smc6-dependent removal of cohesin from mitotic chromosomes. Mol. Cell. Biol. 29:4363–4375. 10.1128/MCB.00377-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tapia-Alveal C, Outwin EA, Trempolec N, Dziadkowiec D, Murray JM, O'Connell MJ. 2010. SMC complexes and topoisomerase II work together so that sister chromatids can work apart. Cell Cycle 9:2065–2070. 10.4161/cc.9.11.11734 [DOI] [PubMed] [Google Scholar]
- 26.Copsey A, Tang S, Jordan PW, Blitzblau HG, Newcombe S, Chan AC, Newnham L, Li Z, Gray S, Herbert AD, Arumugam P, Hochwagen A, Hunter N, Hoffmann E. 2013. Smc5/6 coordinates formation and resolution of joint molecules with chromosome morphology to ensure meiotic divisions. PLoS Genet. 9(12):e1004071. 10.1371/journal.pgen.1004071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pebernard S, Perry JJ, Tainer JA, Boddy MN. 2008. Nse1 RING-like domain supports functions of the Smc5-Smc6 holocomplex in genome stability. Mol. Biol. Cell 19:4099–4109. 10.1091/mbc.E08-02-0226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moreno S, Klar A, Nurse P. 1991. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 194:795–723 [DOI] [PubMed] [Google Scholar]
- 29.Bass KL, Murray JM, O'Connell MJ. 2012. Brc1-dependent recovery from replication stress. J. Cell Sci. 125:2753–2764. 10.1242/jcs.103119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calonge TM, Eshaghi M, Liu J, Ronai Z, O'Connell MJ. 2010. Transformation/transcription domain-associated protein (TRRAP)-mediated regulation of Wee1. Genetics 185:81–93. 10.1534/genetics.110.114769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Calonge TM, O'Connell MJ. 2006. Antagonism of Chk1 signaling in the G2 DNA damage checkpoint by dominant alleles of Cdr1. Genetics 174:113–123. 10.1534/genetics.106.060970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keogh MC, Kim JA, Downey M, Fillingham J, Chowdhury D, Harrison JC, Onishi M, Datta N, Galicia S, Emili A, Lieberman J, Shen X, Buratowski S, Haber JE, Durocher D, Greenblatt JF, Krogan NJ. 2006. A phosphatase complex that dephosphorylates γH2AX regulates DNA damage checkpoint recovery. Nature 439:497–501. 10.1038/nature04384 [DOI] [PubMed] [Google Scholar]
- 33.Tomita K, Cooper JP. 2008. Fission yeast Ccq1 is telomerase recruiter and local checkpoint controller. Genes Dev. 22:3461–3474. 10.1101/gad.498608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iben JR, Epstein JA, Bayfield MA, Bruinsma MW, Hasson S, Bacikova D, Ahmad D, Rockwell D, Kittler EL, Zapp ML, Maraia RJ. 2011. Comparative whole genome sequencing reveals phenotypic tRNA gene duplication in spontaneous Schizosaccharomyces pombe La mutants. Nucleic Acids Res. 39:4728–4742. 10.1093/nar/gkr066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wood V, Harris MA, McDowall MD, Rutherford K, Vaughan BW, Staines DM, Aslett M, Lock A, Bahler J, Kersey PJ, Oliver SG. 2012. PomBase: a comprehensive online resource for fission yeast. Nucleic Acids Res. 40:D695–D699. 10.1093/nar/gkr853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. 10.1186/gb-2009-10-e-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carr AM, Dorrington SM, Hindley J, Phear GA, Aves SJ, Nurse P. 1994. Analysis of a histone H2A variant from fission yeast: evidence for a role in chromosome stability. Mol. Gen. Genet. 245:628–635 [DOI] [PubMed] [Google Scholar]
- 39.Zlatanova J, Thakar A. 2008. H2A.Z: view from the top. Structure 16:166–179. 10.1016/j.str.2007.12.008 [DOI] [PubMed] [Google Scholar]
- 40.Kim HS, Vanoosthuyse V, Fillingham J, Roguev A, Watt S, Kislinger T, Treyer A, Carpenter LR, Bennett CS, Emili A, Greenblatt JF, Hardwick KG, Krogan NJ, Bahler J, Keogh MC. 2009. An acetylated form of histone H2A.Z regulates chromosome architecture in Schizosaccharomyces pombe. Nat. Struct. Mol. Biol. 16:1286–1293. 10.1038/nsmb.1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hou H, Wang Y, Kallgren SP, Thompson J, Yates JR, III, Jia S. 2010. Histone variant H2A.Z regulates centromere silencing and chromosome segregation in fission yeast. J. Biol. Chem. 285:1909–1918. 10.1074/jbc.M109.058487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krogan NJ, Keogh MC, Datta N, Sawa C, Ryan OW, Ding H, Haw RA, Pootoolal J, Tong A, Canadien V, Richards DP, Wu X, Emili A, Hughes TR, Buratowski S, Greenblatt JF. 2003. A Snf2 family ATPase complex required for recruitment of the histone H2A variant Htz1. Mol. Cell 12:1565–1576. 10.1016/S1097-2765(03)00497-0 [DOI] [PubMed] [Google Scholar]
- 43.Zofall M, Fischer T, Zhang K, Zhou M, Cui B, Veenstra TD, Grewal SI. 2009. Histone H2A.Z cooperates with RNAi and heterochromatin factors to suppress antisense RNAs. Nature 461:419–422. 10.1038/nature08321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Papamichos-Chronakis M, Watanabe S, Rando OJ, Peterson CL. 2011. Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell 144:200–213. 10.1016/j.cell.2010.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Attikum H, Fritsch O, Gasser SM. 2007. Distinct roles for SWR1 and INO80 chromatin remodeling complexes at chromosomal double-strand breaks. EMBO J. 26:4113–4125. 10.1038/sj.emboj.7601835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Attikum H, Fritsch O, Hohn B, Gasser SM. 2004. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell 119:777–788. 10.1016/j.cell.2004.11.033 [DOI] [PubMed] [Google Scholar]
- 47.Hogan CJ, Aligianni S, Durand-Dubief M, Persson J, Will WR, Webster J, Wheeler L, Mathews CK, Elderkin S, Oxley D, Ekwall K, Varga-Weisz PD. 2010. Fission yeast Iec1-ino80-mediated nucleosome eviction regulates nucleotide and phosphate metabolism. Mol. Cell. Biol. 30:657–674. 10.1128/MCB.01117-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Watanabe S, Radman-Livaja M, Rando OJ, Peterson CL. 2013. A histone acetylation switch regulates H2A.Z deposition by the SWR-C remodeling enzyme. Science 340:195–199. 10.1126/science.1229758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Downs JA. 2008. Histone H3 K56 acetylation, chromatin assembly, and the DNA damage checkpoint. DNA Repair (Amst.) 7:2020–2024. 10.1016/j.dnarep.2008.08.016 [DOI] [PubMed] [Google Scholar]
- 50.Xhemalce B, Miller KM, Driscoll R, Masumoto H, Jackson SP, Kouzarides T, Verreault A, Arcangioli B. 2007. Regulation of histone H3 lysine 56 acetylation in Schizosaccharomyces pombe. J. Biol. Chem. 282:15040–15047. 10.1074/jbc.M701197200 [DOI] [PubMed] [Google Scholar]
- 51.Haldar D, Kamakaka RT. 2008. Schizosaccharomyces pombe Hst4 functions in DNA damage response by regulating histone H3 K56 acetylation. Eukaryot. Cell 7:800–813. 10.1128/EC.00379-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taylor EM, Copsey AC, Hudson JJ, Vidot S, Lehmann AR. 2008. Identification of the proteins, including MAGEG1, that make up the human SMC5-6 protein complex. Mol. Cell. Biol. 28:1197–1206. 10.1128/MCB.00767-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pebernard S, Schaffer L, Campbell D, Head SR, Boddy MN. 2008. Localization of Smc5/6 to centromeres and telomeres requires heterochromatin and SUMO, respectively. EMBO J. 27:3011–3023. 10.1038/emboj.2008.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maundrell K. 1993. Thiamine-repressible expression vectors pREP and pRIP for fission yeast. Gene 123:127–130 [DOI] [PubMed] [Google Scholar]
- 55.Gachet Y, Tournier S, Millar JB, Hyams JS. 2004. Mechanism controlling perpendicular alignment of the spindle to the axis of cell division in fission yeast. EMBO J. 23:1289–1300. 10.1038/sj.emboj.7600156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gomez EB, Nugent RL, Laria S, Forsburg SL. 2008. Schizosaccharomyces pombe histone acetyltransferase MstI (KAT5) is an essential protein required for damage response and chromosome segregation. Genetics 179:757–771. 10.1534/genetics.107.085779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nugent RL, Johnsson A, Fleharty B, Gogol M, Xue-Franzen Y, Seidel C, Wright AP, Forsburg SL. 2010. Expression profiling of S. pombe acetyltransferase mutants identifies redundant pathways of gene regulation. BMC Genomics 11:59. 10.1186/1471-2164-11-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sarcinella E, Zuzarte PC, Lau PN, Draker R, Cheung P. 2007. Monoubiquitylation of H2A.Z distinguishes its association with euchromatin or facultative heterochromatin. Mol. Cell. Biol. 27:6457–6468. 10.1128/MCB.00241-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rangasamy D, Greaves I, Tremethick DJ. 2004. RNA interference demonstrates a novel role for H2A.Z in chromosome segregation. Nat. Struct. Mol. Biol. 11:650–655. 10.1038/nsmb786 [DOI] [PubMed] [Google Scholar]
- 60.Sumara I, Vorlaufer E, Stukenberg PT, Kelm O, Redemann N, Nigg EA, Peters JM. 2002. The dissociation of cohesin from chromosomes in prophase is regulated by Polo-like kinase. Mol. Cell 9:515–525. 10.1016/S1097-2765(02)00473-2 [DOI] [PubMed] [Google Scholar]
- 61.Waizenegger IC, Hauf S, Meinke A, Peters JM. 2000. Two distinct pathways remove mammalian cohesin from chromosome arms in prophase and from centromeres in anaphase. Cell 103:399–410. 10.1016/S0092-8674(00)00132-X [DOI] [PubMed] [Google Scholar]
- 62.Tada K, Susumu H, Sakuno T, Watanabe Y. 2011. Condensin association with histone H2A shapes mitotic chromosomes. Nature 474:477–483. 10.1038/nature10179 [DOI] [PubMed] [Google Scholar]
- 63.Sharma U, Stefanova D, Holmes SG. 2013. Histone variant H2A.Z functions in sister chromatid cohesion in Saccharomyces cerevisiae. Mol. Cell. Biol. 33:3473–3481. 10.1128/MCB.00162-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guacci V. 2007. Sister chromatid cohesion: the cohesin cleavage model does not ring true. Genes Cells 12:693–708. 10.1111/j.1365-2443.2007.01093.x [DOI] [PubMed] [Google Scholar]
- 65.Bandyopadhyay S, Mehta M, Kuo D, Sung MK, Chuang R, Jaehnig EJ, Bodenmiller B, Licon K, Copeland W, Shales M, Fiedler D, Dutkowski J, Guenole A, van Attikum H, Shokat KM, Kolodner RD, Huh WK, Aebersold R, Keogh MC, Krogan NJ, Ideker T. 2010. Rewiring of genetic networks in response to DNA damage. Science 330:1385–1389. 10.1126/science.1195618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ekwall K. 2007. Epigenetic control of centromere behavior. Annu. Rev. Genet. 41:63–81. 10.1146/annurev.genet.41.110306.130127 [DOI] [PubMed] [Google Scholar]
- 67.Vardabasso C, Hasson D, Ratnakumar K, Chung CY, Duarte LF, Bernstein E. 2013. Histone variants: emerging players in cancer biology. Cell. Mol. Life Sci. 71:379–404. 10.1007/s00018-013-1343-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Suto RK, Clarkson MJ, Tremethick DJ, Luger K. 2000. Crystal structure of a nucleosome core particle containing the variant histone H2A.Z. Nat. Struct. Biol. 7:1121–1124. 10.1038/81971 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.