

Abstract
The formation of protein-RNA granules is a part of both natural cellular function (P-bodies and nuclear HNRNPs) and the response to cellular stress (stress granules and ND10 bodies). To better understand the role of stress-induced granules in viral infection, we have studied the ability of cells to restrict poxvirus replication through the formation of antiviral granules (AVGs). Of cells infected with a wild-type poxvirus, a small number spontaneously formed AVGs. In these AVG-positive cells, viral gene expression was inhibited. The addition of compounds that altered RNA helicase activity, induced oxidative stress, or stimulated translation initiation factor phosphorylation significantly increased the number of AVG-positive cells. When AVGs formed, both viral translation and titers were decreased even when host translation persisted. Treatment with the antiviral compound isatin β-thiosemicarbazone (IBT), a compound that was used to treat smallpox infections, induced AVGs, suggesting a role for these structures in the pharmacological inhibition of poxvirus replication. These findings provide evidence that AVGs are an innate host response that can be exogenously stimulated to combat virus infection. Since small molecules are able to stimulate AVG formation, it is a potential target for new antiviral development.
INTRODUCTION
Poxviruses are a family of double-stranded DNA viruses that include the human pathogens smallpox, monkeypox, and cowpox viruses. The family also includes vaccinia virus (VACV), the vaccine strain that was used to eradicate smallpox from circulation in humans. These viruses are unique among DNA viruses in that their replication occurs entirely in the cytoplasm and uses viral biosynthetic machinery for DNA and RNA synthesis. Vaccinia virus, the prototype poxvirus, expresses over 200 unique proteins during the course of replication through an interdependent cascade of stage-specific transcription events (1–3). As soon as the viral core enters the cytoplasm, factors packaged within the virion begin transcribing viral mRNAs. Genome replication is subsequently initiated at discrete sites in the cytoplasm, called viral factories. These perinuclear DNA-filled factories are the site of all viral transcription and are where virus assembly is initiated. Viral factories become larger and more diffuse as intermediate and late transcription produce virus structural components that facilitate virion morphogenesis (4, 5).
In addition to expressing proteins that drive virus replication, poxviruses also express a large number of proteins that block the induction of host antiviral responses (6). Successful poxvirus replication also requires the evasion of cellular responses that are able to act immediately. An important example of this response is the recognition of double-stranded RNA (dsRNA), a potent pathogen-associated molecular pattern (PAMP) that is produced during poxvirus infection. Recognition of dsRNA by PKR promotes the phosphorylation of the α subunit of eukaryotic translation initiation factor 2 (eIF2α), inhibiting both host and viral translation (7). Vaccinia virus prevents PKR activation by sequestering dsRNA with the viral E3 protein (8–11). Viruses lacking E3 activate PKR, resulting in the inhibition of viral protein synthesis. This attenuates viral replication, highlighting the importance of eIF2α in facilitating poxvirus infection (9, 10, 12–14).
There are three other stress-activated eIF2α kinases that are capable of inducing a broad range of responses designed to protect the cell. PERK, HRI, and GCN2 respond to endoplasmic reticulum (ER) stress, oxidative stress, and nutrient deprivation, respectively (14–18). Collectively, the sensing and response pathways that lead to eIF2α inactivation are called the integrated stress response (ISR). The ISR encompasses an extensive transformation of global gene expression, switching from eIF2α-dependent general host translation to alternative eIF2α-independent stress expression (19–21).
The activation of the integrated stress response often promotes the formation of ribonucleoprotein aggregates, called stress granules (SGs), that form at seemingly random sites throughout the cytoplasm of stressed cells (22). They are composed of mRNA and RNA binding proteins (including G3BP1, CAPRIN1, and TIA1) and translation initiation components (including 40S ribosomal subunits, eIF3, and eIF4A/E/G) (23). It has been proposed that stress granules function as a dynamic microdomain in which mRNA is sorted between decay, storage, or polysome assembly (24, 25). The assembly of stress granules has been shown to be influenced by the rates of both translation initiation and termination; inhibitors of initiation cause decreased polysome formation and increased SG formation, whereas the prevention of polysome disassembly with drugs such as emetine and cycloheximide causes the disassembly of SGs (25, 26). Stress granule formation has been reported to play a role in restricting virus replication, and many viruses appear to have evolved mechanisms to circumvent the formation of these granules during infection (27).
In previous work, we found that cells infected with a mutant vaccinia virus that lacks the PKR antagonist protein E3 induce a host response to virus infection that includes the assembly of cytoplasmic granules. Importantly, these granules are distinct from stress granules in both composition and function. The granules, which we named antiviral granules (AVGs), show a defined localization, embedded across the surface and within cytoplasmic viral factories. They do not contain the 40S ribosomal subunit (which localizes to SGs) and, unlike SGs, do not disassemble when translation termination is prevented with polysome-stabilizing drugs such as cycloheximide and emetine (25, 28). We also found that attenuated vaccinia viruses that induce AVG formation grow to higher titers in cells where the AVG component TIA1 is deleted than in wild-type cells. This restoration demonstrates that AVGs are specifically required for the full inhibition of vaccinia virus replication.
Having found that AVG formation is an important part of restricting ΔE3L mutant vaccinia virus (ΔE3L-VACV), we hypothesized that AVGs might also be capable of inhibiting wild-type vaccinia virus (WT-VACV) replication. Here we report that AVGs are naturally induced spontaneously in WT-VACV infection at a low level and that their formation can be selectively enhanced.
MATERIALS AND METHODS
Cell lines.
U2OS cells were a kind gift from David Sabatini (Whitehead Institute for Biomedical Research). HeLa cells were obtained from the ATCC. All cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with glutamine and 10% fetal bovine serum (FBS) (D10).
Viruses, viral infections, and plaque assays.
The wild-type Western Reserve strain of vaccinia virus was obtained from Bernard Moss (NIAID, NIH) and grown in BHK21 cells. ΔE3L-VACV was created by using homologous recombination as previously described (28). Virus stock titers were determined in triplicate on Vero cells, and virus was stored in aliquots at −80°C.
For growth curve experiments, plaque assays were performed as previously described (28). Samples were serially diluted in triplicate by using D2 medium, added to confluent BSC-40 cells in 96-well plates, and grown at 37°C for 18 h. Plates were prepared for immunofluorescence as detailed below. Cells were stained with rabbit polyclonal antiserum raised against vaccinia virus A33R (catalog number NR-628; BEI Resources), followed by Alexa Fluor 568-conjugated donkey anti-rabbit secondary antibody (Invitrogen). Plaques were manually counted by using a Zeiss Axiovert 200M fluorescence microscope.
Fluorescent virus reporter assays using LREV (late mCherry- and early VENUS-expressing) virus were performed as previously described (29, 30). 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) cytotoxicity assays were performed as previously described (31, 32).
Immunofluorescence and drug treatments.
For immunofluorescence (IF) experiments, cells were seeded into a 24-well dish containing 12-mm clean glass no. 1.5 coverslips (catalog no. GG-12; Neuvitro Corp.). All cells were fixed in 4% paraformaldehyde (PFA; Electron Microscopy Sciences) at room temperature for 15 min. Coverslips were permeabilized with cold methanol for 5 min and blocked with fetal bovine serum (FBS) (5% FBS and 0.05% sodium azide in phosphate-buffered saline [PBS]) before continuing with IF staining. Primary and secondary antibodies were diluted in block and incubated with rocking for 1 h at room temperature. Coverslips were washed three times with PBS after both primary and secondary incubations and mounted by using ProLong Gold antifade with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen-Molecular Probes). Where noted, the final wash buffer also included 5 μM DRAQ5 far-red fluorescent DNA dye (catalog number 4084; Cell Signaling Technology).
Mouse monoclonal antibodies (MAbs) against G3BP1 (tt-y) and poly(A) binding protein (PABP) (10E10), goat polyclonal antibody against TIA1, and rabbit polyclonal serum against eIF4E were purchased from Santa Cruz Biotechnology (catalog numbers sc-81940, sc-32318, sc-1751, and sc-13963). Rabbit MAb against eIF3H (D9C1) was obtained from Cell Signaling Technology (catalog number 3413). Rabbit polyclonal serum against CAPRIN1 was purchased from Proteintech Group Inc. (catalog number 15112-1-AP). Alexa Fluor 488- and 568-conjugated donkey polyclonal secondary antibodies were obtained from Invitrogen.
Image scoring was performed by manually counting random fields of cells stained to visualize viral factories (DAPI regions distinct from the nucleus) and granules (TIA1, G3BP1, or CAPRIN1). RNP granules were scored as AVGs if their localization was associated with DNA-stained viral factories. At least 50 cells were scored for any individual data point shown on the charts (see Fig. 1, 3, and 7).
FIG 1.

Antiviral granules form in a small population of cells during wild-type vaccinia virus infection and suppress viral translation. HeLa and U2OS cells were infected with WT-VACV at an MOI of 10. Nascent proteins were metabolically labeled during the final 30 min of growth and fixed at 7 hpi (see Materials and Methods for details). Cells were stained with antibodies to G3BP1 (red) and with DAPI (blue). (A) Example field of view collected at a ×63 magnification, with a 200% magnification taken from the boxed region. Wild-type viral factories surrounded by diffuse cytoplasmic G3BP1 (arrowheads) or with surrounding G3BP1 granules (arrows) are marked. Bar = 25 μm. (B) Cells were stained as described above for panel A and additionally processed to conjugate nascent peptides to an Alexa Fluor 488 fluorophore (green). Examples of each type of factory observed are boxed and labeled (bar = 10 μm). (C) Channel-separated examples of each factory type: early, active, and repressed. (D) Factories were categorized for factory-localized enrichment of translation or G3BP1 labeling (early, 13.4% ± 5.2%; active, 78.1% ± 6.8%; repressed, 8.5% ± 1.9% [mean ± SEM]) (n = 149 cells from 3 biological replicates).
FIG 3.

Arsenite-induced AVGs recruit components similar to those recruited by ΔE3L-VACV-induced AVGs. HeLa cells were either mock infected (A to C) or infected with WT-VACV (D to I) or ΔE3L-VACV (J to L) at an MOI of 10. PBS (A to F and J to L) or 500 μM arsenite (G to I) was added at 6 hpi, and cells were fixed an hour later. Triplicate coverslips were stained with antibodies against either G3BP1 and eIF3H (A, D, G, and J), TIA1 and eIF4E (B, E, H, and K), or CAPRIN1 and PABP (C, F, I, and L) and stained with DAPI. Maximum-intensity projections from multiple z-slices. Boxed areas mark zoomed regions. Bar = 5 μm.
FIG 7.
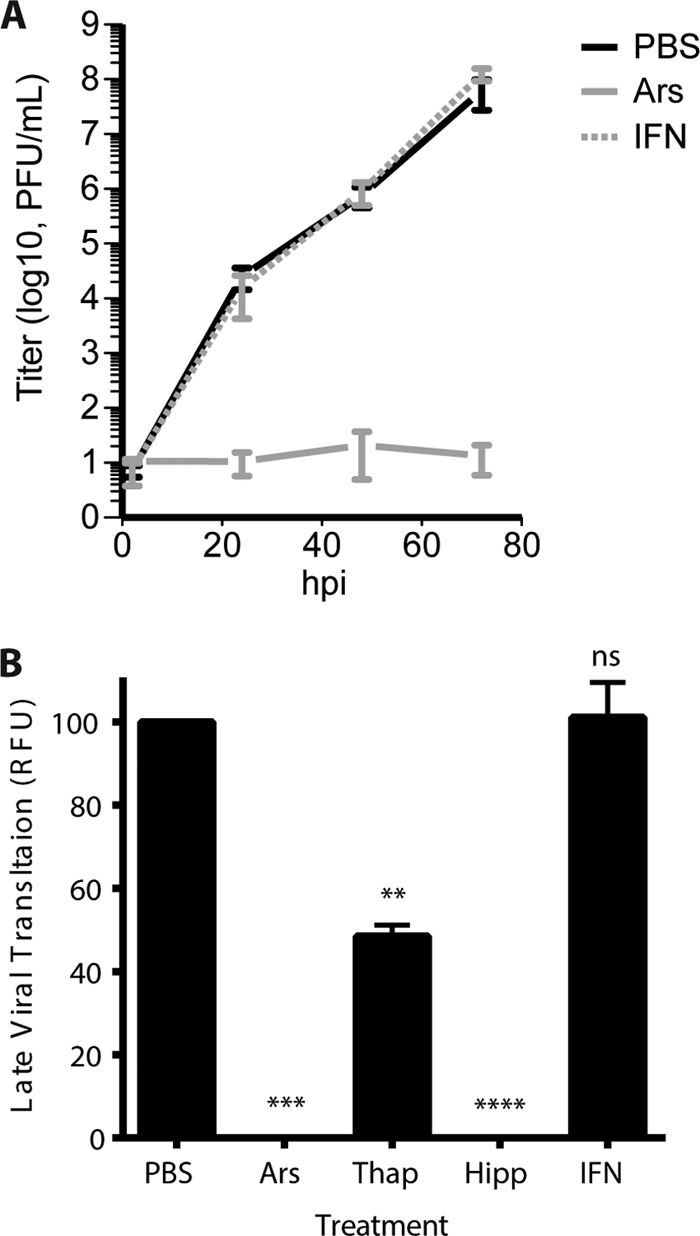
Arsenite-induced antiviral granules suppress wild-type vaccinia virus replication. (A) HeLa cells were infected with WT-VACV at an MOI of 0.1 and treated at 1 hpi in triplicate with PBS or 50 μM arsenite. In a separate set of wells, 200 U/ml beta 1 interferon was added 24 h before infection. Samples were harvested at 2, 24, 48, and 72 hpi, and viral titers were measured by a plaque assay on BSC-40 cells. The chart displays standard errors of the means for at least 2 independent experiments. (B) HeLa cells were infected with an LREV vaccinia virus expressing the mCherry reporter from the late promoter (G8) at an MOI of 1. Normalized late-stage expression after treatment with arsenite (−0.7% ± 1.0%), thapsigargin (48.5% ± 2.7%), and hippuristanol (−0.2% ± 0.5%) but not IFN (101.1% ± 8.4%) (P = 0.9999) showed a significant decrease (**, P < 0.05; ***, P < 0.005; ****, P < 0.0001).
For experiments using sodium arsenite (Ars) (catalog number 35000FLUKA; Sigma), thapsigargin (Thap) (catalog number 586005; Calbiochem), hippuristanol (Hipp) (a kind gift from J. Pelletier), and rohinitib (RHT) (also called rocaglate 8e [33]) (a kind gift from John Porco at the Center for Chemical Methodology and Library Development at Boston University), HeLa or U2OS cells were infected with WT-VACV at a multiplicity of infection (MOI) of 10, as described above. One hour prior to fixation (6 h postinfection [hpi]), the appropriate chemical was added as a dilution in PBS for a final concentration of 500 μM Ars, 1 μM Thap, 1 μM Hipp, or 1 μM RHT. For treatments longer than 1 h (e.g., growth assay, fluorescence reporter assay, MTT assay, and protein extract preparation), a reduced concentration of 50 μM arsenite was used to prevent cell death, as indicated in the figure legends. Similar levels of AVG induction were seen after 1 h of arsenite treatment with both 50 μM and 500 μM (data not shown). Poxvirus inhibitors (see Fig. 6E) were added at 1 hpi, fixed at 7 hpi, and processed for immunofluorescence as described above. Concentrations of 1 μg/ml 1-β-d-arabinofuranosylcytosine (AraC) (catalog number C6645; Sigma), 50 μM isatin β-thiosemicarbazone (IBT) (catalog number NC9075202; Fisher Scientific), 50 μg/ml rifampin (catalog number R3501; Sigma), and 5 μM ST-246 (a kind gift of Siga Labs, Corvallis, OR) were used as previously described (30, 34, 35). Type 1 interferon (IFN) was added 24 h prior to infection at 200 U/ml (human beta 1a interferon, catalog number 11415-1; PBL Interferon Source). After the inoculum was removed and cells were washed, additional IFN in D2 medium was replaced. All cells were fixed at 7 hpi unless otherwise noted.
FIG 6.

Arsenite-induced antiviral granules suppress wild-type vaccinia virus translation. (A) Example fields of view from cells grown as described in the legend of Fig. 2, with the addition of PBS (i) or 500 μM arsenite (ii) during the final hour of growth (6 to 7 hpi). Yellow arrows, factories with AVGs; white arrows, factories without granules (bar = 25 μm). (B) HeLa cells were infected as described above for panel A; treated with 0, 10, 50, or 500 μM arsenite; and fixed an hour later. Cells were stained with antibodies to G3BP1 and TIA1 and with DAPI, and AVGs were counted (white bars display mean numbers of infected cells with AVGs ± SEM) (0 μM, 5.2% ± 1.3%; 10 μM, 16.0% ± 2.8%; 50 μM, 78.0% ± 19.2%; 500 μM, 90.7% ± 3.0%) (n ≥ 50 for at least 3 biological replicates). An MTT cytotoxicity assay was performed on HeLa cells with similar treatments according to procedures described in Materials and Methods. Gray bars display the mean percentages ± SEM of retained cellular metabolism normalized to PBS values (0 μM, 100.0% ± 1.2%; 10 μM, 85.6% ± 4.3%; 50 μM, 52.7% ± 1.6%; 500 μM, 31.3% ± 1.3%) (from at least 4 biological replicates). A two-tailed unpaired t test of 50 and 500 μM arsenite treatments showed a statistically significant difference in cell toxicity (P < 0.0001) but not AVG induction (P = 0.2345). (C) Arsenite (50 μM) was added to HeLa cells, and cells were infected with WT-VACV at an MOI of 10 or mock treated. Whole-cell extracts made from cells at 0, 6, 8, or 24 hpi were blotted with a vaccinia virus virion polyclonal antibody (i), a vaccinia virus monoclonal antibody (ii), or a beta-actin loading control (iii). (Di) Autoradiogram of an SDS-PAGE gel showing [35S]Met-Cys incorporation into newly synthesized proteins (cells were grown as described above for panel C). (ii) Lanes at 24 hpi are grouped together for direct comparison. Molecular mass standards are indicated at the left in kilodaltons.
For fluorescence metabolic labeling, cells were prepared as described above, by treatment with either PBS or 500 μM Ars. One hour prior to fixation (6 hpi), medium was replaced with D2−Met/Cys depletion medium (DMEM with no methionine and no cysteine; Sigma Gibco). Twenty minutes prior to fixation, medium was changed again and replaced with D2−Met/Cys medium supplemented with 50 μM Click-iT l-azidohomoalanine (AHA) (or an l-homopropargylglycine [HPG] azide analog) (Invitrogen Molecular Probes). Following fixation (7 hpi), cells were permeabilized and incubated for 30 min at room temperature with 1× Click-iT cell reaction buffer and Alexa Fluor 488 detection reagent. Coverslips were further processed for IF as described above.
Polysome stability assays were performed similarly to previously described assays (25). HeLa cells were infected with WT-VACV at an MOI of 10 in D2 medium. Ars solution was added for a final concentration of 500 μM at 5.5 hpi. Thirty minutes later, 20 μg/ml cycloheximide (CHX; Sigma), 20 μg/ml puromycin (Puro; InvivoGen), or 0.05% dimethyl sulfoxide (DMSO) was added and incubated for an additional 2 h. Control wells were incubated individually with DMSO, CHX, or Puro alone for a final 2 h. All cells were fixed at 7 hpi.
Coverslips were viewed by using an Axioplan 200M microscope with a Plan-Apochromat 63×/1.4-numerical-aperture oil immersion objective and Axiovision software (Zeiss). Three-dimensional stacks were collected and deconvolved by using ImageJ software (version 1.47n, 1997 to 2013; W. S. Rasband, U.S. National Institutes of Health, Bethesda, MD, USA [http://imagej.nih.gov/ij/]) and the Parallel Iterative Deconvolution plug-in released by Piotr Wendykier (GNU General Public License). A theoretical point spread function was calculated for each image by using the Diffusion PSF 3D plug-in (R. Dougherty [http://www.optinav.com/imagej.html]). Images were also captured by using an LSM710-Live Duoscan confocal microscope (Zeiss).
Radiolabeling, gel electrophoresis, and Western blot analysis.
SDS-PAGE and Western blotting were performed as previously reported (28). HeLa cells grown in 12-well dishes were extracted with NP-40 lysis buffer, transferred onto a polyvinylidene difluoride (PVDF) membrane, and probed with antibodies according to the manufacturer's instructions (Li-Cor Biosciences). A polyclonal antibody recognizing vaccinia virus virion proteins was purchased from Virostat (catalog number 8101). A monoclonal antibody recognizing vaccinia virus virion protein was obtained from BEI (AB-VACC-MAB6, catalog number DD-42). Mouse monoclonal antibody against beta-actin was purchased from Santa Cruz Biotechnology (catalog number sc-47778). Rabbit polyclonal serum recognizing either total eIF2α or a phosphorylation site specifically at serine 52 of eIF2α was purchased from Cell Signaling Technologies or Assay Designs, respectively (catalog numbers 9722 and BML-SA405-0020, respectively). Goat anti-mouse IRDye 680RD and anti-rabbit IRDye 800CW secondary antibodies were diluted 1:2,000 in Li-Cor blocking buffer and incubated with a membrane for 1 h (Li-Cor Biosciences). All blots were scanned at a resolution of 169 μm. Quantification was performed by using ImageJ.
Pulse labeling of adherent HeLa cells with [35S]methionine was performed as previously described (36). Phosphorimages were analyzed by using a Personal Molecular Imager system and Quantity One software (Bio-Rad).
RESULTS
Poxvirus-infected cells can spontaneously form antiviral granules around viral factories.
To understand whether antiviral granule (AVG) structures form during normal virus infection, we examined cells infected with wild-type vaccinia virus (WT-VACV). HeLa cells were infected with WT-VACV and stained with antibodies against the AVG marker G3BP1 and DAPI to visualize cytoplasmic viral factories. We noted that G3BP1 staining was evenly diffuse throughout the cytoplasm in most infected cells (Fig. 1A, arrowheads). A small number of cells showed G3BP1 localization in distinct granules across the surface and embedded within the viral replication factory (Fig. 1A, arrows and magnified inset). Similar experiments were performed with other established markers of AVGs (CAPRIN1 and TIA1), and we found that factory-associated granules occurred in 4.8% (±1.4%) of infected HeLa cells (n = 622 cells from 7 biological replicates). This observation was confirmed by using U2OS cells, where factory-associated granules were seen in 5.8% (±0.6%) of infected cells (n = 615 cells from 6 replicates). Based on their similar morphology compared to previously described ΔE3L-induced AVGs (28), we hypothesized that these granules were antiviral in nature.
Cells that form antiviral granules suppress wild-type vaccinia virus translation.
For cells that have been infected with vaccinia virus, active translation has been suggested to be concentrated at viral cytoplasmic factories (37, 38). To determine whether translation was inhibited at viral factories in the subset of cells that form AVGs, we visualized active protein translation in individual vaccinia virus-infected cells. Infections were performed as described above, with the addition of the amino acid analog l-azidohomoalanine (AHA) 20 min prior to fixation. After fixation, proteins that had incorporated AHA were conjugated to a fluorescent probe by using a copper-catalyzed reaction, allowing us to label nascent peptides (39–42). Cells were also stained with an antibody against G3BP1 to mark factory-associated granules. Figure 1B shows a representative field of view from one such experiment, containing three types of viral factories: early, active, and repressed. Early factories were smaller and not enriched for G3BP1, and only minimal translation was observed (Fig. 1C). Active factories displayed areas of highly active protein translation, observed as enriched foci across the surface of viral factories, with no G3BP1 recruitment (Fig. 1C). Factories containing G3BP1 granules, despite having sizes and shapes similar to those of active factories, did not contain significant translation staining (Fig. 1C). When factories were scored for translation and G3BP1 enrichment, we saw a consistent trend that factories fell into one of the three categories (early [13.4% ± 3.4%], active [78.1% ± 8.1%], or repressed [8.5 ± 1.9%] [means ± standard errors of the means {SEM}]) (n = 149 cells from 3 biological replicates) (Fig. 1D). Importantly, we did not find any factories that displayed both active translation and G3BP1 recruitment, arguing that the host protein G3BP1 negatively regulates viral translation, which is in contrast to the previously proposed role in support of viral translation (38). This inverse correlation between viral factory-based translation and G3BP1 recruitment provides evidence that granules formed during wild-type vaccinia virus infection are capable of repressing viral translation.
AVG formation is enhanced in response to changes in translation initiation.
These results showed that a small subset of cells infected with vaccinia virus formed AVGs in response to poxvirus replication and blocked virus translation. Since AVG induction during ΔE3L-VACV infection was shown to require the phosphorylation of eIF2α (28), we hypothesized that the activation of independent environmental stress pathways would increase the incidence of AVG formation. We first investigated whether small-molecule stimulators of eIF2α phosphorylation, such as the oxidative stress inducer arsenite, would promote AVG formation during wild-type virus infection (Fig. 2A). Consistent with previous reports, phosphorylation of eIF2α was enhanced >4-fold in cells treated with 500 μM sodium arsenite for 1 h (Fig. 2B) (18). Arsenite treatment of infected HeLa cells led to a dramatic increase in the number of AVGs around wild-type vaccinia virus factories (Fig. 2Cii). AVGs were also formed when cells infected with WT-VACV were treated with the ER stress inducer thapsigargin (Thap) (1 μM), which stimulates eIF2α phosphorylation through activation of the PERK kinase (Fig. 2Ciii). Using these distinct inducers of the host stress response pathway, we were able to promote the formation of AVGs by enhancing eIF2α phosphorylation.
FIG 2.

Antiviral granule formation is enhanced during alterations of translation initiation. HeLa and U2OS cells were infected with WT-VACV at an MOI of 10. At 6 hpi, cells were treated with PBS, 500 μM arsenite (Ars), 1 μM thapsigargin (Thap), 1 μM hippuristanol (Hipp), or 1 μM rohinitib (RHT) and fixed an hour later at 7 hpi. Alternatively, cells were pretreated with 200 U/ml beta 1 interferon for 24 h before infection. Cells were stained with antibodies to G3BP1, TIA1, or CAPRIN1 and with DAPI. (A) Schematic describing parallel methods to alter translation initiation with small molecules. The activation of multiple stress kinases results in the phosphorylation of the alpha subunit of eIF2, preventing the formation of the ternary complex necessary for translation initiation. Alternatively, eIF4A is inhibited by hippuristanol and RHT in an eIF2α-independent manner. (B) Whole-cell extracts from mock-infected and infected cells treated with 500 μM Ars were separated by SDS-PAGE and blotted with an antibody against phosphorylated eIF2α (S51) (i) and total eIF2α (ii). A band densitometry comparison of the phosphorylated form normalized to total eIF2α is shown between gel images. (C) Example images for G3BP1 (red) (middle) and DAPI (blue) (right) labeling and merged images (left) for PBS (i)-, arsenite (ii)-, thapsigargin (iii)-, hippuristanol (iv)-, rohinitib (v)-, and beta 1 interferon (vi)-treated cells. N, cell nuclei; arrows, factories with AVGs; solid arrowheads, factories without granules; open arrowheads, stress granules (bar = 5 μm). (D) Quantification of infected cells with AVGs after treatment with arsenite (91.3% ± 1.9% [mean ± SEM]), thapsigargin (32.6% ± 7.4%), hippuristanol (77.9% ± 9.6%), and RHT (92.8% ± 1.6%) but not IFN (4.1% ± 0.7%) (P = 0.9997) showed a highly significant (****, P < 0.0001) increase compared to cells treated with PBS alone (5.3% ± 0.8%). All experiments included >50 cells from at least 2 biological replicates. ns, not significant. (E) HeLa cells were infected with WT-VACV, and 500 μM arsenite was added 1 h before fixation at 4 to 8 hpi. (i) Cells were stained as described above for panel C (bar = 5 μm). (ii and iii) Magnified dashed boxes containing viral factories with AVGs are shown in orthogonal views as a maximum-intensity projection, including the top view (x and y, bar = 5 μm) (ii) and side view (z, bar = 2 μm) (ii). Borders of DAPI-stained factories are denoted with dashed lines.
The stimulation of AVGs was not dependent solely on eIF2α phosphorylation. Treatment of WT-VACV-infected cells with the eIF4A inhibitors hippuristanol (Hipp) (1 μM) and rohinitib (RHT) (1 μM) also induced AVGs at VACV factories (Fig. 2Civ and v) (21, 33, 43). This suggested that various stresses which alter normal cellular translation initiation lead to AVG formation. Importantly, the induction of AVGs was not a universal response to all forms of cell stress. When cells were treated with 200 U/ml type 1 interferon (IFN) for 24 h prior to infection to induce antiviral genes, they did not induce AVGs in WT-VACV-infected cells, nor did the treatment appear to block the ability of vaccinia virus to enter cells and set up active replication centers (Fig. 2Cvi). To determine the extent of granule formation in infected cells, we quantified the frequency of AVG-positive cells after treatment of both HeLa and U2OS cells for 1 h before fixation at 7 hpi (Fig. 2D). All treatments except IFN induced AVGs in the majority of WT-VACV factories (Ars, 91.2% ± 1.9%; Thap, 32.6% ± 7.3%; Hipp, 77.9% ± 9.6%; RHT, 92.1%; IFN, 4.8% [means ± SEM of infected cells with virus-associated granules]) (44). AVGs were seen across nearly all factories with arsenite, hippuristanol, and RHT treatments, while thapsigargin had a lesser, although highly significant, effect, consistent with its reported lesser effect on HeLa cells. These treatments also induced similar numbers of SGs in both infected and uninfected cells, as previously established (44). These experiments demonstrated that AVG formation in virus-infected cells could be enhanced by a variety of compounds that alter rates of translation initiation through either eIF2α-dependent or -independent mechanisms.
AVGs are formed around and within cytoplasmic viral factories and can be induced during all stages of factory development.
AVGs become apparent during ΔE3L-VACV infections, surrounding and embedded within factories as soon as a viral factory is produced (28). To determine if there was any effect of time of infection or factory size on the ability to chemically induce AVGs during infection with WT-VACV, we treated cells with 500 μM sodium arsenite for 1 h in cells that had been infected for increasing amounts of time. As previously described, cells began to show viral DNA replication and evidence of factory formation at between 2 and 4 hpi (45). AVGs were readily seen to be associated with DAPI-stained cytoplasmic viral factories at all times after factory formation (Fig. 2E). AVGs induced during early times of infection appeared to be restrained to the periphery of 1- to 3-μm-diameter factories (4 to 5 hpi) (Fig. 2E; see also Movies S1 and S2 in the supplemental material). This may be due to the presence of an ER-derived membrane, which has been shown to be present until virion assembly begins at around 6 hpi (46). AVGs formed later in infection (6 to 8 hpi) were observed across the surface and embedded throughout the volume of larger cytoplasmic factories (3- to 8-μm diameter), consistent with those previously observed at similar times of infection with ΔE3L-VACV (6 to 8 hpi) (Fig. 2E; see also Movies S3 to S5 in the supplemental material) (28). These findings show that stress-induced AVGs are capable of forming at all stages of WT-VACV factory development and are found adjacent to and within the factory itself.
AVG structures formed in wild-type-infected cells are multiprotein complexes that are insensitive to cycloheximide dissolution.
To verify that the induced AVGs that we observed during WT-VACV infection were similar to those formed during ΔE3L-VACV infections and not simply SGs that are also induced under these circumstances, we examined their composition and stability. The colocalization of three mRNA binding proteins (G3BP1, TIA1, and CAPRIN1) and three translation initiation proteins (eIF3H, eIF4E, and PABP) with AVGs was determined by using immunofluorescence (Fig. 3). As expected, each of these proteins showed a diffuse labeling pattern in the cytoplasm of mock-infected cells and WT-VACV-infected and PBS-treated cells (Fig. 3A to F). Consistent with its role in pre-mRNA metabolism, TIA1 was also seen in the nucleus under similar conditions (Fig. 3B and E) (22, 47). The addition of 500 μM arsenite to wild-type-infected HeLa cells caused a rapid enrichment of all AVG components into dense granules localized coincident with DAPI-stained factories (Fig. 3G to I). Staining for multiple components simultaneously showed a large proportion of overlapping signals, showing colocalization into similar granule structures (Fig. 3G to I).
Cycloheximide has been shown to cause dissociation of stress granules but not AVGs (25, 28). Infected cells treated with 20 μg/ml cycloheximide in the presence of 500 μM arsenite retained factory-associated AVGs, whereas non-factory-associated granules, presumably stress granules, were lost (96% of infected cells with AVGs; n = 51) (Fig. 4D). Similar results were obtained by using emetine (D. K. Rozelle and J. H. Connor, unpublished data). Next, we showed that 20 μg/ml puromycin after 500 μM arsenite treatment caused no change in AVG or stress granule prevalence, indicating that dispersion of stress granule components requires polysome disassembly and not merely ribosome inhibition (97% and 96% of cells retained arsenite-induced AVGs [n = 40] and SGs [n = 52], respectively) (Fig. 4F and L). The initial experiments established that AVGs are able to form during wild-type vaccinia virus infections.
FIG 4.

Arsenite-induced stress granules but not antiviral granules disassemble during elongation inhibition. HeLa cells were infected with WT-VACV at an MOI of 10 or mock infected and left untreated (A and G), treated with 0.5 mM arsenite for 30 min (B and H), treated with 10 μg/ml cycloheximide for 1 h (C and I), treated with arsenite for 90 min with the addition of cycloheximide after 30 min (D and J), treated with 20 μg/ml puromycin for 1 h (E and K), or treated with arsenite for 90 min with the addition of puromycin after 30 min (F and L). Cells were fixed at 7 hpi and stained with antibodies to CAPRIN1 (red) and with DAPI (blue). Fields of view were collected at a ×63 magnification. Arrows mark viral factories with AVGs, white arrowheads mark factories without AVGs, and open arrowheads mark SGs. Bar = 5 μm. Quantification of infected cells showing factory-associated CAPRIN1 granules (A to F) or of all cells with stress granules (G to L) is shown in each panel (n = 100 each).
AVGs sequester viral mRNAs.
To understand how AVGs might repress viral translation, we determined whether the subcellular localization of viral mRNAs was altered when AVGs were formed. To accomplish this, we used fluorescence in situ hybridization (RNA-FISH) to specifically label viral mRNAs. RNA-FISH probes recognizing the vaccinia virus F9L gene were seen spread throughout the cytoplasm of productively infected HeLa cells at 7 hpi (Fig. 5i). When infected cells were treated with 500 μM arsenite (as in Fig. 2), both G3BP1 staining and F9L mRNA probes became localized to granules (Fig. 5ii). We saw a similar shift of both G3BP1 protein and viral F9L mRNA after treatment with hippuristanol (Fig. 5iii).
FIG 5.

Antiviral granules are formed around viral mRNA. HeLa cells were infected with WT-VACV at an MOI of 10. At 6 hpi, cells were treated with PBS, 500 μM arsenite (Ars), or 1 μM hippuristanol (Hipp) and fixed an hour later. Eighteen custom fluorescent oligonucleotides targeting the entire open reading frame of vaccinia virus F9L mRNA were hybridized prior to staining with antibodies to G3BP1 and with DAPI. Shown are example fields of view with individual channels for DAPI (blue), G3BP1 (red), and F9L mRNA (green) and merged images for PBS (i), Ars (ii), and Hipp (iii) treatments. N, cell nuclei; arrows, factories with AVGs; solid arrowheads, factories without granules; open arrowheads, SGs (bar = 5 μm). Charts display channel intensities of DAPI (blue), G3BP1 (red), and F9L mRNA (green) normalized for each channel between all images. The regions analyzed are marked with white bars on the merged images. The background level is indicated on each chart with a dashed line (0.2 RFU).
To determine if the G3BP1 protein aggregates and viral F9L mRNA foci were localized in a similar structure, we analyzed their colocalization. Individual AVGs were identified as regions where levels of staining for G3BP1 protein (Fig. 5, green line) and viral factory DNA (blue) were above the normalized background level. Background levels were 0.2 relative fluorescence units (RFU) using PBS-treated cells infected with WT-VACV (Fig. 5i). In infected cells treated with either 500 μM arsenite or 1 μM hippuristanol, the level of F9L mRNA staining (red) was above background levels at all locations identified as AVGs (Fig. 5ii and iii). To quantify the degree of mRNA enrichment at AVGs, we compared the mean intensity of the F9L RNA probe signal at these AVG regions with that of nongranule cytoplasmic regions. Infected cells treated with arsenite showed an enrichment in the F9L mRNA intensity of 159.1% ± 8.8% at AVGs relative to that in the cytoplasm, and cells treated with hippuristanol showed a similar 168.3% ± 12.5% enrichment (n ≥ 13 cells from 2 biological replicates). This result shows that AVGs contain components capable of enriching viral mRNAs from the cytoplasm. Levels of enrichment were found to be similar at SGs (data not shown), suggesting that SGs could also sequester viral RNAs.
Induced AVGs suppress translation of viral but not stress-induced host mRNAs.
Since induced AVGs form in almost all cells following the addition of arsenite, we hypothesized that they have the ability to significantly inhibit vaccinia virus replication. To determine the effect of AVGs on viral gene expression, we examined protein synthesis following arsenite treatment. Microscopic images of nascent peptides in virus-infected HeLa cells showed that active translation was localized primarily to AVG-free factories in infected cells (Fig. 6Ai, white arrows). In contrast, in cells treated with 500 μM arsenite, viral factories surrounded by AVGs were devoid of translation, indicating that arsenite-promoted AVG formation prevented viral translation (Fig. 6Aii, yellow arrows).
Our investigations until this point were performed by using 500 μM arsenite to induce cytoplasmic granules, consistent with previous reports (44, 48–50). While this concentration of arsenite consistently produces a robust stress response and assembly of granules, we wanted to determine if AVGs were able to form at lower concentrations. By adding a range of arsenite concentrations for 1 h to HeLa cells infected with WT-VACV, we determined that similar numbers of AVGs were induced in infected cells treated with 50 μM arsenite (78.0% ± 19.2%) and 500 μM arsenite (90.7% ± 3.0%; P = 0.2345), compared with the reduced response seen after 10 μM arsenite treatment (16.0% ± 2.8%) (Fig. 6B, white bars). When the cytotoxicity imposed upon cells by treatment with 500 μM arsenite (31.3% ± 1.3% of control cell metabolism) was compared to that imposed by treatment with 50 μM (52.7% ± 1.6%), we found a statistically significant increase in host metabolic capacity (P < 0.0001) (Fig. 6B, gray bars). This showed us that 50 μM arsenite was the lowest concentration at which AVGs still formed in the majority of cells. By using this reduced amount of arsenite, especially during prolonged experiments, such as the growth assays described below, we were able to minimize the pleiotropic effects of arsenite while retaining most of its AVG-inducing potency.
To verify that the AVG-induced translation inhibition shown in Fig. 6A led to a decrease in viral protein accumulation, we probed whole-cell extracts with vaccinia virus-specific antibodies. Two antibodies raised against vaccinia virus virions (polyclonal and monoclonal) were used to compare the inhibition of viral protein production during AVG production. The polyclonal serum detected increasing amounts of viral protein at molecular masses of 80, 50, 30, and 25 kDa at between 6 and 24 hpi in WT-VACV-infected cells treated with PBS (Fig. 6Ci, lanes 5 to 8). In contrast, infected cells treated with 50 μM arsenite had negligible protein production, even at 24 hpi (Fig. 6Civ, lanes 9 to 12). A similar expression pattern was seen by using an alternative monoclonal antibody recognizing an individual 35-kDa vaccinia virus protein (Fig. 6Cii).
To determine whether this inhibition of viral protein synthesis could be explained by a general inhibition of all cellular translation, we metabolically labeled cells with [35S]methionine-cysteine. Radiolabeled proteins made in vaccinia virus-infected cells showed a characteristic shift from host expression to WT-VACV expression at between 6 and 24 hpi (Fig. 6Di, lanes 4 to 6) (51). Cells infected in the presence of 50 μM arsenite showed none of the bands associated with viral protein production at either 6 or 24 hpi (Fig. 6Di, lanes 7 to 9). Instead of viral proteins, we observed a translation pattern which resembled that seen during a cellular stress response (Fig. 6Di) (52). When lanes at 24 hpi are compared side by side (Fig. 6Dii), it is apparent that conditions promoting AVG formation allow the translation of a subset of host proteins but inhibit viral protein synthesis. Therefore, AVG formation is associated with an inhibition of viral protein translation but not with a complete inhibition of host mRNA translation.
Arsenite-induced AVGs suppress wild-type vaccinia virus replication.
Since stress-induced AVGs resulted in decreased viral protein production, we hypothesized that AVGs would prevent the formation of mature virions and reduce viral titers. To quantify viral replication, HeLa cells were infected with WT-VACV and grown in medium containing PBS, 50 μM arsenite, or 200 U/ml interferon (IFN was added 24 h prior to infection; all other treatments were added at 1 hpi). Virus titers in cells treated with PBS displayed the expected exponential growth rate for wild-type vaccinia virus, reaching a maximal titer of 6.2 × 107 PFU/ml by 72 hpi (Fig. 7A). Virus grown in cells treated with arsenite showed a complete absence of viral replication, with no significant increase in titers by 72 hpi (Fig. 7A). Growth of virus in IFN-treated cells was indistinguishable from that with PBS-alone treatments, reaching similar titers at all time points (Fig. 7A).
These results were confirmed by using a modified WT-VACV that expresses a fluorescent reporter from a late viral promoter. Cells treated with PBS or IFN as described above showed strong fluorescence expression at 16 hpi (100% ± 1.3% and 92.7% ± 1.0% [means ± SEM] for normalized PBS- and IFN-treated cells, respectively). In contrast, cells infected in the presence of 50 μM arsenite showed a fluorescent signal indistinguishable from the background signal (0.04% ± 0.1% relative to PBS) (Fig. 7B). Similar results were obtained when cells were grown in the presence of 1 μM hippuristanol (0.5% ± 0.3% relative to PBS) (Fig. 7B). Thapsigargin (1 μM), which induced AVGs in an intermediate number of infected cells (Fig. 2D), showed a corresponding reduction in viral growth, as predicted (48.8% ± 1.7% relative to PBS) (Fig. 6B). These data support the idea that vaccinia virus replication detected by a plaque assay and a reporter virus is inversely proportional to AVG formation (Fig. 2D). Viral growth was abrogated by both arsenite and hippuristanol, which induced the largest number of AVGs, supporting our hypothesis that AVG formation limits the replication of wild-type VACV.
The smallpox virus antiviral IBT induces AVGs, while other poxvirus antivirals do not.
Following our finding that compounds that modulate translation initiation by perturbing cellular homeostasis could inhibit poxvirus replication, we were interested in whether known poxvirus inhibitors also induced the formation of AVGs. To investigate this, we treated infected cells with several known poxvirus inhibitors: 1 μg/ml arabinofuranosylcytosine (AraC) (a CTP analog that inhibits viral DNA replication), 50 μM isatin β-thiosemicarbazone (IBT) (inhibits normal viral transcription termination), 50 μg/ml rifampin (virion assembly inhibitor), and 5 μM ST-246 (virion assembly inhibitor) (53–56). While a majority of the compounds did not significantly induce AVG formation, IBT enhanced AVG formation even better than arsenite (AraC, 1.3% ± 1.4%; IBT, 94.5% ± 2.2%; rifampin, 6.8% ± 3.8%; ST-246, 3.4% ± 1.0% [mean ± SEM for infected cells with AVGs]) (n ≥ 50 with at least 2 biological replicates) (Fig. 8A and C). Immunofluorescence analysis of IBT-treated WT-VACV-infected HeLa cells showed that G3BP1, TIA1, and CAPRIN1 (Fig. 8A and data not shown) all locate to granules associated with cytoplasmic factories. Hybridization with fluorescent probes recognizing F9L viral mRNA (as shown in Fig. 5) showed a similar enrichment at AVGs of 157.3% compared to the level in the cytoplasm (n = 8). Intriguingly, no cytoplasmic SGs were apparent with IBT treatment, suggesting a more specific method of AVG induction than with arsenite and hippuristanol, both of which promote AVG and SG formation simultaneously.
FIG 8.

The smallpox virus antiviral IBT induces antiviral granules. (A) HeLa and U2OS cells were infected with WT-VACV at an MOI of 10 in the presence of 0.05% DMSO, 1 μg/ml 1-β-d-arabinofuranosylcytosine (AraC), 50 μM isatin β-thiosemicarbazone (IBT), 50 μg/ml rifampin (Rifamp), or 5 μM ST-246 and fixed at 7 hpi. Coverslips were stained with antibodies to G3BP1 (red), hybridized with F9L mRNA probes (green) and DAPI (blue) as described in the legend of Fig. 5. (A) Example field of view for cells treated with IBT, showing the localization of G3BP1 and F9L mRNA to AVGs at cytoplasmic viral factories. (B) Cells were grown as described above for panel A, with the addition of 10 μg/ml cycloheximide during the final hour of growth (6 to 7 hpi). Quantification of infected cells showing factory-associated G3BP1/CAPRIN1 granules is shown in the merged panel (91%; n > 50 from each of 2 biological replicates). (C) Chart showing quantification of infected cells with AVGs after treatment with DMSO (0.0%), AraC (1.4% ± 1.4%), IBT (94.5% ± 2.2%), rifampin (6.8% ± 3.8%), and ST-246 (3.4% ± 1.0%). Only IBT treatment showed a significant incidence of AVG formation compared to treatment with DMSO alone (****, P < 0.0001). All experiments included >50 cells from at least 2 biological replicates. (D) HeLa cells were treated with 0.05% DMSO, 500 μM arsenite, 50 μM arsenite, or 50 μM IBT for 24 h before the MTT assay was performed. Absorbance values normalized to DMSO values (relative light units [RLU]) showed a statistically significant increase in cell metabolism for treatment with 50 μM IBT (85.0% ± 3.7%) compared to treatment with either 500 μM arsenite (30.2% ± 1.0%; P = 0.0004) or 50 μM arsenite (50.7% ± 4.5%; P = 0.013). (E and F) Viral growth was determined after the addition of 50 μM IBT by a plaque assay (log10 PFU/ml) (E) and a fluorescence reporter assay (0.17% ± 1.4% RFU; P < 0.0001) (F).
Arsenic-based therapies have a long history of use as potent therapeutic agents (57, 58). Despite their accepted usage for several severe diseases, arsenic therapies remain highly toxic. To compare the toxicity of our in vitro arsenite and IBT treatments, we performed an MTT assay. Ideally, an antiviral treatment would have sufficient specificity to limit virus replication at a concentration below that which causes cellular damage. Despite optimizing our arsenite treatment by using the lowest concentration possible, cells treated with 50 μM arsenite retained only 50.7% of the metabolic capacity of PBS-treated control cells, indicating that the antiviral effect of arsenite occurred at concentrations at which cellular damage also occurred (Fig. 6B). In comparison, cells treated with IBT retained 83% of their metabolic capability (Fig. 7C). This significant reduction in cellular cytotoxicity paired with an equivalent potency for viral inhibition in the absence of SG induction shows that IBT is a substantially more specific antiviral treatment than arsenite.
IBT is an antipoxvirus drug thought to inhibit postreplicative transcription termination, resulting in excessive dsRNA production (54). To verify that AVG enrichment occurred under conditions that inhibit viral replication, we grew WT-VACV in the presence of 50 μM IBT. Plaque assays showed inhibition similar to that with arsenite treatment (Fig. 8E). Late-stage fluorescence reporter virus expression corroborated this finding, yielding only background fluorescence signals compared with PBS treatment (<0.1% relative to PBS) (Fig. 8F).
DISCUSSION
The experiments presented above show that the disruption of poxvirus replication by various means triggers the formation of host protein dense antiviral granules (AVGs). AVGs are differentiated from other ribonucleoprotein granules based on their localization to cytoplasmic viral factories, their composition (lacking eIF3B and 40S ribosome subunits), and their stability in the presence of translation elongation inhibitors. AVGs contain many of the same components as canonical stress granules, such as mRNA binding proteins (TIA1, G3BP1, and CAPRIN1), translation initiation components (eIF4E, eIF3H, and PABP), translation regulators (FXR1, FXR2, and FMRP), as well as other proteins (28, 59).
While neither SGs nor AVGs have been shown to contain large ribosomal subunits, consistent with their arrest before the formation of the 80S complex, stress granules accumulate 40S ribosomes, while AVGs fail to recruit small subunits. In previous work, AVGs have been shown to be required for the suppression of vaccinia virus replication. ΔE3L-VACV grown in mouse embryonic fibroblast (MEF) cells lacking the core AVG component TIA1 showed a rescue of viral titers despite similar levels of eIF2α phosphorylation in both knockout and WT MEF cells (28). This leads us to propose that AVGs are not temporary holding and sorting sites of preinitiated mRNAs (the proposed function for SGs) but exist solely to restrict viral translation.
Our experiments show that viral mRNA is enriched at AVGs (and SGs) and that these granules lack ribosomal components necessary for translation. This supports a mechanism in which viral mRNA in AVGs is sequestered from the translation-competent pool of mRNAs. Translation modulation (through ISR stimulation or eIF4A modulation) triggers an antiviral survey of the cell, during which mRNAs that are not in active polysomes are identified as nonself transcripts. In our model, we do not propose that viral mRNAs are uniquely trafficked to AVGs, but rather, we propose that AVGs are formed where concentrations of mRNAs that are not associated with ribosomes are located. Thus, it is possible for newly synthesized viral mRNAs to be sequestered into AVGs following cell stress, while mRNAs that were actively being translated are shuttled into SGs. This could explain the presence of viral mRNAs in both structures following arsenite and hippuristanol treatments (Fig. 5).
Previous descriptions of the vaccinia virus cytoplasmic replication factory have proposed tight coordination between transcription and translation apparatuses at vaccinia virus factories (38). Our experiments make use of recent advances in click chemistry to draw a clear distinction between factories that support active translation and those that are repressed by AVGs (Fig. 1 and 6). Our analysis does provide some support that factory-associated translation sites do exist at the surface of viral factories. Our data, however, clearly distinguish between actively translating foci at viral factories (which lack G3BP1, CAPRIN1, and TIA1) and AVG foci, where translation is repressed. We did not find support for previous work suggesting that G3BP and Caprin1 are recruited to active translation sites (38). Instead, our results suggest that these proteins are recruited only when poxvirus translation is inhibited.
The fact that both viral and environmental stressors can induce the formation of AVGs in poxvirus-infected cells suggests that cells are perpetually balanced between normal translation and stress-responsive translation, consistent with the model shown in Fig. 9. This transition is controlled by the phosphorylation of eIF2α in response to canonical stress-sensing kinases (PERK, PKR, HRI, or GCN2) (14–18). Activation of stress pathways leads to an extensive transformation in global gene expression, preventing viral and host housekeeping mRNAs from recruiting 60S ribosomal subunits while at the same time promoting the transcription and translation of stress response genes (19). Inhibition of translation initiation leads to polysome disassembly and the formation of cytoplasmic granules through the binding of RNA binding proteins (TIA1, G3BP1, and CAPRIN1) to nontranslating mRNAs. Stress granules formed in this manner have been proposed to serve as mRNA triage and sorting compartments (24). When cells undergo the transition from normal to stressed translation during infection with vaccinia virus, viral RNA levels are enriched at both SGs and AVGs. While data supporting sorting and triage roles for AVGs are lacking, we predict that this is a potential mechanism by which AVGs could exert a specific antiviral effect on viral messages.
FIG 9.
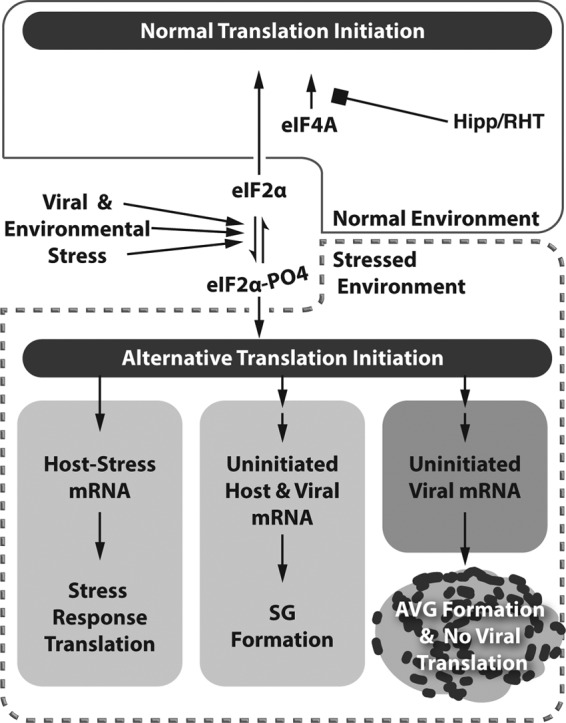
Model of stress-induced AVG formation. Our results show that both viral and environmental stressors are capable of inducing AVGs. Phosphorylation of eIF2α can be accomplished by activating canonical stress sensors (PERK, PKR, HRI, or GCN2). This leads to an extensive transformation of global gene expression (stressed environment marked by a dashed box), preventing viral and host housekeeping mRNAs from recruiting 60S ribosome components while at the same time promoting the transcription and translation of stress response genes. An alternative alteration of translation initiation through the use of eIF4A inhibitors also leads to an increased amount of non-polysome-associated mRNA (both constitutive host mRNA and viral mRNA), which becomes bound by mRNA binding proteins such as G3BP1, TIA1, and CAPRIN1. During vaccinia virus infection, the formation of AVG-type granules through this process causes the aggregation of cytoplasmic viral factory-associated AVGs that are capable of viral translation and replication repression.
The sensitivity of poxviruses to cytoplasmic granule formation is contrary to the behavior of other viruses in the face of homeostatic disruption. Flaviviruses (Japanese encephalitis virus [JEV]), togaviruses (Semliki forest virus [SFV] and chikungunya virus [CHIKV]), and picornaviruses (poliovirus [PV]) all produce proteins that directly antagonize granule formation. In these viruses, SG formation is prevented through the direct sequestration of host proteins. JEV core protein directly binds CAPRIN1, preventing it from recognizing viral mRNA. Preventing this interaction allows SG formation and impairs viral propagation (48). Similarly, nsP3 proteins from SFV and CHIKV have been shown to sequester G3BP1 in viral replication complexes and to prevent SG formation (60, 61). Polioviruses produce a protease that cleaves G3BP1, resulting in the formation of compositionally unique RNA granules that are unable to repress viral replication (62, 63). Influenza A virus takes an indirect approach to avoid SG formation by suppressing upstream signaling. Virally encoded NS1 protein sequesters immunogenic dsRNA and blocks PKR activation, much like the role that the E3 protein plays in blocking AVG formation during vaccinia virus infection (64, 65).
AVGs are a natural host defense that we have now shown is induced by various natural stresses, including the ISR. Vaccinia virus has numerous immunomodulatory proteins that are capable of preventing the recognition of and response to normal replication. We have shown that these blockages can be overcome by therapeutically targeting alternative pathways. Significant research has already been performed on integrated stress pathways. The fact that we are able to induce an AVG response using numerous cell and virus stress agents suggests that there are multiple targets to activate AVG formation. Initial attempts at inducing AVGs with arsenite during infections with monkeypox virus have also confirmed that AVGs are not a vaccinia virus-specific effect and likely act in a similar manner for all poxviruses (C. M. Filone, D. K. Rozelle, and J. H. Connor, unpublished data). Additionally, because AVGs are formed by the aggregation of host proteins, there is likely a higher barrier to the development of drug-resistant mutants. An additional benefit of targeting such a conserved pathway is the likelihood of broad antiviral activity. With the monumental financial and technical burdens of developing novel antiviral therapeutics, future development will favor broadly applicable drug targets such as AVGs.
Supplementary Material
ACKNOWLEDGMENTS
We thank Doug Grosenbach, (Siga Technologies, Corvallis, OR) for providing ST-246 and Jerry Pelletier (McGill University, Montreal, Quebec, Canada) for the kind gift of hippuristanol. We thank John Porco and Neil Lajkiewicz (Department of Chemistry, Boston University) for generously providing a sample of the rocaglate rohinitib (RHT).
We appreciate David Center of the Clinical and Translational Science Institute (CTSI) for providing funds through NIH grant UL1-TR000157 for use at the Boston University Cellular Imaging Core. D.K.R. was supported by an NIH training grant in immunology to Boston University (grant 5T32AI 7309). C.M.F. was additionally supported by the NIH Program in Inflammatory Disorders at Boston University (grant T32 AI 89673-3). This work was supported by NIH grants RO1AI1096159-01 and K22AI-064606 (to J.H.C.).
Footnotes
Published ahead of print 24 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01630-13.
REFERENCES
- 1.Smith GL, Benfield CTO, Maluquer de Motes C, Mazzon M, Ember SWJ, Ferguson BJ, Sumner RP. 2013. Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. J. Gen. Virol. 94:2367–2392. 10.1099/vir.0.055921-0 [DOI] [PubMed] [Google Scholar]
- 2.Moss B. 2007. Poxviridae: the viruses and their replication, p 2906–2945 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 3.Broyles SS. 2003. Vaccinia virus transcription. J. Gen. Virol. 84:2293–2303. 10.1099/vir.0.18942-0 [DOI] [PubMed] [Google Scholar]
- 4.Yang Z, Bruno DP, Martens CA, Porcella SF, Moss B. 2010. Simultaneous high-resolution analysis of vaccinia virus and host cell transcriptomes by deep RNA sequencing. Proc. Natl. Acad. Sci. U. S. A. 107:11513–11518. 10.1073/pnas.1006594107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang Z, Reynolds SE, Martens CA, Bruno DP, Porcella SF, Moss B. 2011. Expression profiling of the intermediate and late stages of poxvirus replication. J. Virol. 85:9899–9908. 10.1128/JVI.05446-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith GL, Symons JA, Khanna A, Vanderplasschen A, Alcamí A. 1997. Vaccinia virus immune evasion. Immunol. Rev. 159:137–154. 10.1111/j.1600-065X.1997.tb01012.x [DOI] [PubMed] [Google Scholar]
- 7.Farrell PJ, Balkow K, Hunt T, Jackson RJ, Trachsel H. 1977. Phosphorylation of initiation factor elF-2 and the control of reticulocyte protein synthesis. Cell 11:187–200. 10.1016/0092-8674(77)90330-0 [DOI] [PubMed] [Google Scholar]
- 8.Watson JC, Chang HW, Jacobs BL. 1991. Characterization of a vaccinia virus-encoded double-stranded RNA-binding protein that may be involved in inhibition of the double-stranded RNA-dependent protein kinase. Virology 185:206–216. 10.1016/0042-6822(91)90768-7 [DOI] [PubMed] [Google Scholar]
- 9.Chang HW, Watson JC, Jacobs BL. 1992. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc. Natl. Acad. Sci. U. S. A. 89:4825–4829. 10.1073/pnas.89.11.4825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Myskiw C, Arsenio J, van Bruggen R, Deschambault Y, Cao J. 2009. Vaccinia virus E3 suppresses expression of diverse cytokines through inhibition of the PKR, NF-kappaB, and IRF3 pathways. J. Virol. 83:6757–6768. 10.1128/JVI.02570-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kwon J-A, Rich A. 2005. Biological function of the vaccinia virus Z-DNA-binding protein E3L: gene transactivation and antiapoptotic activity in HeLa cells. Proc. Natl. Acad. Sci. U. S. A. 102:12759–12764. 10.1073/pnas.0506011102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Langland JO, Jacobs BL. 2002. The role of the PKR-inhibitory genes, E3L and K3L, in determining vaccinia virus host range. Virology 299:133–141. 10.1006/viro.2002.1479 [DOI] [PubMed] [Google Scholar]
- 13.Bonnet MC, Weil R, Dam E, Hovanessian AG, Meurs EF. 2000. PKR stimulates NF-kappaB irrespective of its kinase function by interacting with the IkappaB kinase complex. Mol. Cell. Biol. 20:4532–4542. 10.1128/MCB.20.13.4532-4542.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. 2000. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6:1099–1108. 10.1016/S1097-2765(00)00108-8 [DOI] [PubMed] [Google Scholar]
- 15.Srivastava SP, Kumar KU, Kaufman RJ. 1998. Phosphorylation of eukaryotic translation initiation factor 2 mediates apoptosis in response to activation of the double-stranded RNA-dependent protein kinase. J. Biol. Chem. 273:2416–2423. 10.1074/jbc.273.4.2416 [DOI] [PubMed] [Google Scholar]
- 16.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. 2000. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 5:897–904. 10.1016/S1097-2765(00)80330-5 [DOI] [PubMed] [Google Scholar]
- 17.Wek SA, Zhu S, Wek RC. 1995. The histidyl-tRNA synthetase-related sequence in the eIF-2 alpha protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol. Cell. Biol. 15:4497–4506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McEwen E, Kedersha N, Song B, Scheuner D, Gilks N, Han A, Chen J-J, Anderson P, Kaufman RJ. 2005. Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J. Biol. Chem. 280:16925–16933. 10.1074/jbc.M412882200 [DOI] [PubMed] [Google Scholar]
- 19.Ron D. 2002. Translational control in the endoplasmic reticulum stress response. J. Clin. Invest. 110:1383–1388. 10.1172/JCI16784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawai T, Fan J, Mazan-Mamczarz K, Gorospe M. 2004. Global mRNA stabilization preferentially linked to translational repression during the endoplasmic reticulum stress response. Mol. Cell. Biol. 24:6773–6787. 10.1128/MCB.24.15.6773-6787.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wek RC, Jiang H-Y, Anthony TG. 2006. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 34:7–11. 10.1042/BST0340007 [DOI] [PubMed] [Google Scholar]
- 22.Kedersha NL, Gupta M, Li W, Miller I, Anderson P. 1999. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J. Cell Biol. 147:1431–1442. 10.1083/jcb.147.7.1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kedersha N, Anderson P. 2009. Regulation of translation by stress granules and processing bodies. Prog. Mol. Biol. Transl. Sci. 90:155–185. 10.1016/S1877-1173(09)90004-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson P, Kedersha N. 2008. Stress granules: the Tao of RNA triage. Trends Biochem. Sci. 33:141–150. 10.1016/j.tibs.2007.12.003 [DOI] [PubMed] [Google Scholar]
- 25.Kedersha N, Cho MR, Li W, Yacono PW, Chen S, Gilks N, Golan DE, Anderson P. 2000. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J. Cell Biol. 151:1257–1268. 10.1083/jcb.151.6.1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mazroui R, Sukarieh R, Bordeleau M-E, Kaufman RJ, Northcote P, Tanaka J, Gallouzi I, Pelletier J. 2006. Inhibition of ribosome recruitment induces stress granule formation independently of eukaryotic initiation factor 2alpha phosphorylation. Mol. Biol. Cell 17:4212–4219. 10.1091/mbc.E06-04-0318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lloyd RE. 2012. How do viruses interact with stress-associated RNA granules? PLoS Pathog. 8:e1002741. 10.1371/journal.ppat.1002741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simpson-Holley M, Kedersha N, Dower K, Rubins KH, Anderson P, Hensley LE, Connor JH. 2011. Formation of antiviral cytoplasmic granules during orthopoxvirus infection. J. Virol. 85:1581–1593. 10.1128/JVI.02247-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dower K, Rubins KH, Hensley LE, Connor JH. 2011. Development of vaccinia reporter viruses for rapid, high content analysis of viral function at all stages of gene expression. Antiviral Res. 91:72–80. 10.1016/j.antiviral.2011.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dower K, Filone CM, Hodges EN, Bjornson ZB, Rubins KH, Brown LE, Schaus S, Hensley LE, Connor JH. 2012. Identification of a pyridopyrimidinone inhibitor of orthopoxviruses from a diversity-oriented synthesis library. J. Virol. 86:2632–2640. 10.1128/JVI.05416-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Meerloo J, Kaspers GJL, Cloos J. 2011. Cell sensitivity assays: the MTT assay. Methods Mol. Biol. 731:237–245. 10.1007/978-1-61779-080-5_20 [DOI] [PubMed] [Google Scholar]
- 32.Van de Loosdrecht AA, Beelen RH, Ossenkoppele GJ, Broekhoven MG, Langenhuijsen MM. 1994. A tetrazolium-based colorimetric MTT assay to quantitate human monocyte mediated cytotoxicity against leukemic cells from cell lines and patients with acute myeloid leukemia. J. Immunol. Methods 174:311–320. 10.1016/0022-1759(94)90034-5 [DOI] [PubMed] [Google Scholar]
- 33.Santagata S, Mendillo ML, Tang Y, Subramanian A, Perley CC, Roche SP, Wong B, Narayan R, Kwon H, Koeva M, Amon A, Golub TR, Porco JA, Jr, Whitesell L, Lindquist S. 2013. Tight coordination of protein translation and HSF1 activation supports the anabolic malignant state. Science 341:1238303. 10.1126/science.1238303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sodeik B, Griffiths G, Ericsson M, Moss B, Doms RW. 1994. Assembly of vaccinia virus: effects of rifampin on the intracellular distribution of viral protein p65. J. Virol. 68:1103–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grosenbach DW, Jordan R, Hruby DE. 2011. Development of the small-molecule antiviral ST-246 as a smallpox therapeutic. Future Virol. 6:653–671. 10.2217/fvl.11.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bonifacino J. 2002. Protein labeling and immunoprecipitation. Curr. Protoc. Cell Biol. 15:7.0.1–7.0.3. 10.1002/0471143030.cb0700s15 [DOI] [Google Scholar]
- 37.David A, Dolan BP, Hickman HD, Knowlton JJ, Clavarino G, Pierre P, Bennink JR, Yewdell JW. 2012. Nuclear translation visualized by ribosome-bound nascent chain puromycylation. J. Cell Biol. 197:45–57. 10.1083/jcb.201112145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katsafanas GC, Moss B. 2007. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe 2:221–228. 10.1016/j.chom.2007.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Breinbauer R, Köhn M. 2003. Azide-alkyne coupling: a powerful reaction for bioconjugate chemistry. Chembiochem 4:1147–1149. 10.1002/cbic.200300705 [DOI] [PubMed] [Google Scholar]
- 40.Wang Q, Chan TR, Hilgraf R, Fokin VV, Sharpless KB, Finn MG. 2003. Bioconjugation by copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc. 125:3192–3193. 10.1021/ja021381e [DOI] [PubMed] [Google Scholar]
- 41.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. 2002. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 41:2596–2599. [DOI] [PubMed] [Google Scholar]
- 42.Kolb HC, Finn MG, Sharpless KB. 2001. Click chemistry: diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. Engl. 40:2004–2021 http://www.renyi.hu/∼stipsicz/Mezo_DECEMBER/click/click.pdf [DOI] [PubMed] [Google Scholar]
- 43.Bordeleau M-E, Mori A, Oberer M, Lindqvist L, Chard LS, Higa T, Belsham GJ, Wagner G, Tanaka J, Pelletier J. 2006. Functional characterization of IRESes by an inhibitor of the RNA helicase eIF4A. Nat. Chem. Biol. 2:213–220. 10.1038/nchembio776 [DOI] [PubMed] [Google Scholar]
- 44.Kedersha N, Anderson P. 2007. Mammalian stress granules and processing bodies. Methods Enzymol. 431:61–81. 10.1016/S0076-6879(07)31005-7 [DOI] [PubMed] [Google Scholar]
- 45.Dales S, Siminovitch L. 1961. The development of vaccinia virus in Earle's L strain cells as examined by electron microscopy. J. Biophys. Biochem. Cytol. 10:475–503. 10.1083/jcb.10.4.475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tolonen N, Doglio L, Schleich S, Krijnse Locker J. 2001. Vaccinia virus DNA replication occurs in endoplasmic reticulum-enclosed cytoplasmic mini-nuclei. Mol. Biol. Cell 12:2031–2046. 10.1091/mbc.12.7.2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dember LM, Kim ND, Liu KQ, Anderson P. 1996. Individual RNA recognition motifs of TIA-1 and TIAR have different RNA binding specificities. J. Biol. Chem. 271:2783–2788. 10.1074/jbc.271.5.2783 [DOI] [PubMed] [Google Scholar]
- 48.Katoh H, Okamoto T, Fukuhara T, Kambara H, Morita E, Mori Y, Kamitani W, Matsuura Y. 2013. Japanese encephalitis virus core protein inhibits stress granule formation through an interaction with caprin-1 and facilitates viral propagation. J. Virol. 87:489–502. 10.1128/JVI.02186-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith JA, Schmechel SC, Raghavan A, Abelson M, Reilly C, Katze MG, Kaufman RJ, Bohjanen PR, Schiff LA. 2006. Reovirus induces and benefits from an integrated cellular stress response. J. Virol. 80:2019–2033. 10.1128/JVI.80.4.2019-2033.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.White JP, Cardenas AM, Marissen WE, Lloyd RE. 2007. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2:295–305. 10.1016/j.chom.2007.08.006 [DOI] [PubMed] [Google Scholar]
- 51.White SD, Jacobs BL. 2012. The amino terminus of the vaccinia virus E3 protein is necessary to inhibit the interferon response. J. Virol. 86:5895–5904. 10.1128/JVI.06889-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Novoa I, Zhang Y, Zeng H, Jungreis R, Harding HP, Ron D. 2003. Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 22:1180–1187. 10.1093/emboj/cdg112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kufe DW, Munroe D, Herrick D, Egan E, Spriggs D. 1984. Effects of 1-beta-D-arabinofuranosylcytosine incorporation on eukaryotic DNA template function. Mol. Pharmacol. 26:128–134 [PubMed] [Google Scholar]
- 54.Prins C, Cresawn SG, Condit RC. 2004. An isatin-beta-thiosemicarbazone-resistant vaccinia virus containing a mutation in the second largest subunit of the viral RNA polymerase is defective in transcription elongation. J. Biol. Chem. 279:44858–44871. 10.1074/jbc.M408167200 [DOI] [PubMed] [Google Scholar]
- 55.Moss B, Rosenblum EN, Katz E, Grimley PM. 1969. Rifampicin: a specific inhibitor of vaccinia virus assembly. Nature 224:1280–1284. 10.1038/2241280a0 [DOI] [PubMed] [Google Scholar]
- 56.Yang G, Pevear DC, Davies MH, Collett MS, Bailey T, Rippen S, Barone L, Burns C, Rhodes G, Tohan S, Huggins JW, Baker RO, Buller RLM, Touchette E, Waller K, Schriewer J, Neyts J, DeClercq E, Jones K, Hruby D, Jordan R. 2005. An orally bioavailable antipoxvirus compound (ST-246) inhibits extracellular virus formation and protects mice from lethal orthopoxvirus challenge. J. Virol. 79:13139–13149. 10.1128/JVI.79.20.13139-13149.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Antman KH. 2001. Introduction: the history of arsenic trioxide in cancer therapy. Oncologist 6(Suppl 2):1–2. 10.1634/theoncologist.6-suppl_2-1 [DOI] [PubMed] [Google Scholar]
- 58.Cutler EG, Bradford EH. 1878. Action of iron, cod-liver oil, and arsenic on the globular richness of the blood. Am. J. Med. Sci. 75:74–84 [Google Scholar]
- 59.Anderson P, Kedersha N. 2006. RNA granules. J. Cell Biol. 172:803–808. 10.1083/jcb.200512082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Panas MD, Varjak M, Lulla A, Eng KE, Merits A, Karlsson Hedestam GB, McInerney GM. 2012. Sequestration of G3BP coupled with efficient translation inhibits stress granules in Semliki Forest virus infection. Mol. Biol. Cell 23:4701–4712. 10.1091/mbc.E12-08-0619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fros JJ, Domeradzka NE, Baggen J, Geertsema C, Flipse J, Vlak JM, Pijlman GP. 2012. Chikungunya virus nsP3 blocks stress granule assembly by recruitment of G3BP into cytoplasmic foci. J. Virol. 86:10873–10879. 10.1128/JVI.01506-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.White JP, Lloyd RE. 2011. Poliovirus unlinks TIA1 aggregation and mRNA stress granule formation. J. Virol. 85:12442–12454. 10.1128/JVI.05888-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Piotrowska J, Hansen SJ, Park N, Jamka K, Sarnow P, Gustin KE. 2010. Stable formation of compositionally unique stress granules in virus-infected cells. J. Virol. 84:3654–3665. 10.1128/JVI.01320-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mok BW-Y, Song W, Wang P, Tai H, Chen Y, Zheng M, Wen X, Lau S-Y, Wu WL, Matsumoto K, Yuen K-Y, Chen H. 2012. The NS1 protein of influenza A virus interacts with cellular processing bodies and stress granules through RNA-associated protein 55 (RAP55) during virus infection. J. Virol. 86:12695–12707. 10.1128/JVI.00647-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Onomoto K, Jogi M, Yoo J-S, Narita R, Morimoto S, Takemura A, Sambhara S, Kawaguchi A, Osari S, Nagata K, Matsumiya T, Namiki H, Yoneyama M, Fujita T. 2012. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS One 7:e43031. 10.1371/journal.pone.0043031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Filone CM, Caballero IS, Dower K, Mendillo ML, Cowley GS, Santagata S, Rozelle DK, Yen J, Rubins KH, Hacohen N, Root DE, Hensley LE, Connor J. 2014. The master regulator of the cellular stress response (HSF1) is critical for orthopoxvirus infection. PLoS Pathog. 10:e1003904. 10.1371/journal.ppat.1003904 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.