

ABSTRACT
Kaposi's sarcoma-associated herpesvirus (KSHV) has been shown to be recognized by two families of pattern recognition receptors (PRRs), Toll-like receptors (TLRs) and NOD-like receptors (NLRs). Here we show that MAVS and RIG-I (retinoic acid-inducible gene 1), an RLR family member, also have a role in suppressing KSHV replication and production. In the context of primary infection, we show that in cells with depleted levels of MAVS or RIG-I, KSHV transcription is increased, while beta interferon (IFN-β) induction is attenuated. We also observed that MAVS and RIG-I are critical during the process of reactivation. Depletion of MAVS and RIG-I prior to reactivation led to increased viral load and production of infectious virus. Finally, MAVS depletion in latent KSHV-infected B cells leads to increased viral gene transcription. Overall, this study suggests a role for MAVS and RIG-I signaling during different stages of the KSHV life cycle.
IMPORTANCE We show that RIG-I and its adaptor protein, MAVS, can sense KSHV infection and that these proteins can suppress KSHV replication following primary infection and/or viral reactivation.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV) was isolated and identified from a Kaposi's sarcoma (KS) lesion in 1994 by Chang et al. (1). KSHV, also known as human herpesvirus 8 (HHV-8), is classified as a member of the gammaherpesvirus subfamily within the Herpesviridae family. KSHV is the etiological agent of KS and is the primary cause of two B cell proliferative cancers, primary effusion lymphoma (PEL) and a variant of multicentric Castleman's disease (MCD) (1, 2). The host cell range of KSHV includes B cells, endothelial cells, epithelial cells, monocytes, hematopoietic progenitor cells, and dendritic cells (3–7). KSHV exists primarily in a latent state in which the viral genome is tethered to host chromosomes via the viral protein latency associated nuclear antigen (LANA). In a latent infection, KSHV exists in a dormant state, in which only a small subset of viral genes are transcribed, and is able to persist for the lifetime of the host. KSHV undergoes lytic replication, typically during the first 72 to 96 h following primary infection (8) but also during periods of reactivation, which is necessary for genome maintenance in the host.
KSHV contains 84 open reading frames (ORFs), and more than 15 viral proteins have been identified as modulators of host immune responses (9). Modulation of the host immune response is critical for the life cycle of KSHV; without subversion of the host response, it is likely that the virus would be eliminated prior to the establishment of latency. Innate immune signaling is primarily initiated by receptors/sensors belonging to one of three families: Toll-like receptors (TLRs), RIG-I like receptors (RLRs) (reviewed in reference 10), and nucleotide-binding domain leucine-rich repeat-containing receptors (NLRs) (reviewed in reference 11). Our laboratory has previously shown that proteins from two of these families play a significant role during the life cycle of KSHV. We have demonstrated a role for TLR activation during KSHV primary infection and reactivation (12–14). We have also shown that NLR signaling is important during KSHV infection (15). Each of these signaling pathways can lead to the secretion of type I interferon (IFN), which is one of the critical defenses against invading pathogens. It is presumed that interferon activation aids in the establishment of an antiviral state.
The RLR and NLR families of receptors are cytosolic receptors, while TLRs are membrane-bound receptors. As mentioned above, we have shown a role for TLR and NLR signaling during KSHV infection; however, there is little information on the role of RLR signaling during KSHV infection. RIG-I, the founding member of the RLR family, is a cytosolic sensor of double-stranded RNA (dsRNA) or single-stranded RNA (ssRNA), typically of viral origin (16, 17), and plays a significant role in the induction of type I interferon responses following viral infection. Recognition of viral RNA by RIG-I initiates a signaling cascade that results in production of type I IFN and proinflammatory cytokines. One of the key components of this signaling cascade is mitochondrial antiviral signaling protein (MAVS). MAVS is a membrane-bound adaptor protein which transmits signals from RIG-I to downstream signaling molecules, most notably TBK-1 and the IκB kinase (IKK) family of proteins (18). These, in turn, signal through the transcription factors NF-κB, IRF3, and IRF7 to produce inflammatory cytokines, including IFN-β (reviewed in reference 19).
ORF64, the KSHV-encoded deubiquitinase, was shown to inhibit RIG-I signaling via prevention of ubiquitination of RIG-I, which is critical for its activity (20, 21). Epstein-Barr virus (EBV) and herpes simplex virus (HSV) have both been shown to be recognized by RIG-I during infection (22–27). Based on our previous observations that members of the TLR and NLR families play critical roles during KSHV infection, we wanted to investigate whether loss of MAVS or RIG-I would have an impact on interferon production following KSHV infection. We found that both the adaptor protein MAVS and the cytosolic sensor RIG-I play a role in type I interferon (IFN-β) production following KSHV infection.
MATERIALS AND METHODS
Cell culture.
293 cells were maintained in Dulbecco modified Eagle medium (DMEM; Cellgro) containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (Pen-Str). iSLK cells harboring latent rKSHV.219 (a kind gift from D. Ganem) were maintained in DMEM supplemented with 10% FBS, 1% Pen-Str, G418 (250 μg/ml), hygromycin (400 μg/ml), and puromycin (10 μg/ml). Cells were maintained at 37°C in 5% carbon dioxide. Sf9 cells were maintained at 30°C in Sf900 II serum-free medium (Gibco) supplemented with 500 μl of Pen-Str and 500 μl of amphotericin B (Sigma). B cells were maintained in RPMI medium supplemented with 20% FBS, 1% Pen-Str, 1% l-glutamine, and 0.05 mM β-mercaptoethanol.
Virus production and purification.
Virus was propagated and purified as described previously (13). Briefly, baculovirus ORF50, a gift from Jeff Vieira (28), was propagated in Sf9 cells for 72 h. Vero cells stably expressing rKSHV.219 were then infected with baculovirus ORF50 in DMEM containing 2% FBS, 1% Pen-Str, and 2 mM sodium butyrate to initiate reactivation. After 72 to 96 h, supernatant was harvested and cell debris was pelleted and filtered through a sterile 0.45-μm filter. To concentrate the virus, 30 ml of supernatant was layered over a 5-ml cushion of 20% sucrose (optical grade; Sigma), and the virus was pelleted by ultracentrifugation in an SW32 rotor (Beckman Coulter) for 2.5 h at 4°C. Supernatant was decanted, and virus pellets were resuspended in endotoxin-free phosphate-buffered saline (PBS) in 1% of the original volume. UV inactivation of KSHV was performed as previously described (13).
Viral transcription.
To quantify viral transcription in infected cells, RNA was isolated from cells using the RNeasy Plus minikit (Qiagen) per the manufacturer's instructions. In order to generate cDNA, 1 to 2 μg of RNA was reverse transcribed using Superscript III reverse transcriptase (Invitrogen) and oligo(dT) primers (Invitrogen). Quantitative real-time PCR (qPCR) was performed for specific viral genes, using the cDNA as a template in a SYBR green PCR master mix (Bio-Rad). β-Actin was used as the endogenous control for all of the qPCRs.
Depletion of MAVS or RIG-I.
Mission short hairpin RNA (shRNA) constructs targeted to MAVS and RIG-I were purchased from Sigma. Transient transfection was first performed using Lipofectamine 2000 (Invitrogen) per the manufacturer's instructions to test which constructs successfully knocked down MAVS and RIG-I. Optimal knockdown was achieved for MAVS using a combination of two plasmids, while only one plasmid was needed to achieve optimal knockdown of RIG-I. In order to create stable cells knocked down for either MAVS or RIG-I, along with a scrambled control, lentivirus expressing each of the shRNAs was made via transfection of 293T cells. Briefly, 5 × 106 293T cells were plated in 10-cm dishes and 5 μg of DNA (2.5 μg of each plasmid for MAVS) was transfected along with 15 μg of ViraPower packaging mix (Invitrogen) in antibiotic-free medium. Transfection medium was replaced with complete medium 4 to 6 h posttransfection. Forty-eight hours posttransfection, the medium was harvested and filtered through a 0.45-μm filter and stored at −80°C prior to use. The supernatant was used to infect 293 cells, and cells were selected via addition of puromycin. Once a week, cells were harvested and tested for knockdown until maximum knockdown was achieved. Cells were then stably maintained under selection using puromycin at a concentration of 1 μg/ml. To achieve knockdown of MAVS in BCBL1 cells, 1 × 106 cells were plated and transduced with 1.5 ml of lentivirus, prepared as described above. Cells were centrifuged with the lentivirus at 2,500 rpm for 90 min at 30°C. Following transduction, 1 ml of complete medium was added to the cells and the cells were incubated at 37°C for 72 or 96 h, after which they were harvested, pelleted, washed, and flash frozen prior to RNA isolation.
Transfection and reactivation of iSLK cells.
iSLK cells were maintained as described above and were transfected using X-tremeGENE HP (Roche) per the manufacturer's instructions. Four micrograms of DNA of each construct (shSCR, shMAVS [2 μg of each plasmid], and shRIG-I) was transfected into iSLK cells; at 24 h posttransfection, the medium was changed to DMEM containing 1% Pen-Str, 10% FBS, and 0.2 μg/ml of doxycycline for reactivation (29). At 48 h postreactivation, cells were collected and RNA was harvested via the RNeasy Plus minikit for analysis of levels of viral transcripts.
Generation of iSLK-BAC16 stable cells.
The DH10B-BAC16 construct was kindly provided by Jae Jung (30). 293T cells were transfected with DH10B-BAC16 DNA and selected with hygromycin (100 μg/ml). To create the iSLK-BAC16 cells, 293T-BAC16 stable cells were cocultured with naive iSLK cells in DMEM containing 2% FBS at a ratio of 1:1. Tetradecanoyl phorbol acetate (TPA; 25 ng/ml) and sodium butyrate (0.5 mM) were added to induce reactivation of the 293T-BAC16 cells. At 72 h postreactivation, selection was begun using hygromycin (1200 μg/ml), puromycin (1 μg/ml), and G418 (250 μg/ml); it was continued until 100% of the iSLK cells were green fluorescent protein (GFP) positive. A total of 1 × 106 iSLK-BAC16 cells were then plated and reactivated with doxycycline (3 μg/ml) for 72 h. The supernatant was collected and filtered through a 0.45-μm filter. Naive iSLK cells were infected with the filtered supernatant via spinoculation as described previously (13, 14). KSHV-infected iSLK cells were cultured and maintained in DMEM supplemented with 10% FBS, 1% Pen-Str, hygromycin (1,200 μg/ml), puromycin (1 μg/ml), and G418 (250 μg/ml).
Immunofluorescence assay.
iSLK-BAC16 cells were cultured as described previously (30) and plated in 2-well Lab-Tek chamber slides (Sigma). At 80% confluence, cells were reactivated with doxycycline for 48 h as described previously (29). At 48 h, the medium was removed and cells were fixed in 4% paraformaldehyde (PFA) for 15 min at room temperature, followed by two washes with ice-cold PBS. Cells were permeabilized with PBS containing 0.5% Triton X-100 (PBST), washed, and blocked with 1% bovine serum albumin (BSA) in PBST. The samples were incubated with mouse monoclonal antibody J2 to dsRNA (English and Scientific Consulting) at 1:100 overnight at 4°C, washed, and incubated with goat anti-mouse IgG tetramethyl rhodamine isocyanate (TRITC) in 1% BSA. Samples were washed and mounted using VECTASHIELD (Vector Laboratories Inc.). Microscopy was performed using a Nikon Eclipse Ti.
qPCR viral array.
An improved qPCR array was designed to specifically amplify KSHV mRNAs following the strategy of our earlier work (31–33). One hundred ninety-two primer pairs were included (K. Tamburro and D. P. Dittmer, unpublished data). Between 1 and 3 primer pairs for each annotated KSHV open reading frame (ORF), targeting both the 3′ and 5′ ends of each gene where applicable, were designed based on the KSHV BC1 isolate (locus KSU75698). Reference genes for cellular transcripts were included for normalization. Primers were designed using Primer3 (34), and specificity was confirmed using BLAST. All primers produce a 200-bp product and share similar melting temperatures. This results in amplification reactions with similar efficiencies and thus allows us to directly compare the expression levels among different mRNAs (35). qPCRs were plated in 384-well plates using the Tecan Freedom Evo liquid handling robot and cycled and analyzed using Roche LightCycler 480, as previously described (36). A detailed, step-by-step protocol is available at http://www.med.unc.edu/vironomics/protocols.
RESULTS
Knockdown of MAVS and RIG-I leads to increased infection efficiency.
We established stable 293 cells depleted of either MAVS (shMAVS) or RIG-I (shRIG-I) using lentivirus expressing shRNAs that targeted these genes. Stable cells were created using puromycin selection and maintained as a pool of cells. Stable 293 cells expressing a scrambled control (shSCR) shRNA were also generated. Significant knockdown was achieved with both shMAVS (Fig. 1A) and shRIG-I (Fig. 1B) compared to the shSCR control cell line. In order to determine whether inhibition of these genes alters KSHV infection or replication, we infected cells as described previously (13, 14) with a recombinant KSHV expressing GFP (rKSHV.219) (28) and monitored the cells via GFP expression, as a marker of infection. We found that there was increased infection as indicated by GFP positivity at both 48 h (Fig. 2A) and 72 h (Fig. 2B) postinfection in MAVS- or RIG-I-depleted cells compared to the control cells. These infections were performed over 10 times, with representative images being shown. This suggests that MAVS and RIG-I suppress KSHV replication postinfection. To ensure that MAVS or RIG-I depletion did not impact the efficiency of KSHV adsorption or attachment to host cells, we infected cells as described above and harvested cells at 2 h postinfection (30 min postspinoculation). DNA was isolated using the DNeasy (Qiagen) isolation kit per the manufacturer's instructions. Purified DNA was analyzed via qPCR for quantitation of viral genomes. As shown in Fig. 2C, there was no significant difference in viral genomes present between infected shSCR control cells and either the shMAVS or the shRIG-I knockdown cells. Previous studies have shown that analysis of viral DNA content at 1 to 2 h post-KSHV infection can be an indicator of the efficiency of infection (37). This suggests that the differences we observe in Fig. 2A and B are due to loss of MAVS or RIG-I and not due to more efficient infection of the depleted cells.
FIG 1.
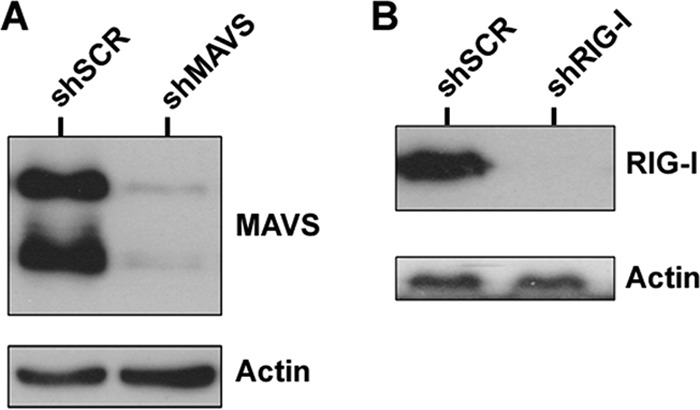
Western blot confirming knockdown of MAVS and RIG-I in 293 cells. (A) MAVS depletion compared to the scrambled control. (B) RIG-I depletion compared to the scrambled control. Actin was used as a loading control.
FIG 2.

KSHV infection of control and knockdown cell lines was monitored for GFP expression by fluorescence microscopy for 48 or 72 h. Control cells and MAVS-depleted and RIG-I-depleted stable knockdown 293 cells were infected for 48 h (A) or 72 h (B). GFP expression was significantly increased in both MAVS-depleted and RIG-I-depleted cells compared to the scrambled control. (C) Quantitation of viral genomes at 2 h postinfection (30 min postspinoculation). There was no significant difference in viral genomes detected between the shMAVS and shRIG-I cells compared to shSCR cells at this time point.
Inhibition of MAVS and RIGI-I leads to increased viral transcription.
We next determined whether the viral gene transcription profile of KSHV was affected in MAVS- and RIG-I-depleted cell lines following primary infection. We isolated poly(A) RNA from KSHV-infected cells and performed qPCR analysis using a primer collection, which spans the entire viral genome. As shown in Fig. 3, we observed broad increases in KSHV transcription in MAVS- and RIG-I-depleted cells. The viral mRNAs were largely undetectable at 0 h, as indicated by the blue color on the heat map; multiple viral mRNAs were detectable as indicated by the red color on the heat map. To construct the heat map as a visual representation of viral mRNA abundance, we proceeded as follows. We determined the abundance of each KSHV mRNA by individual qPCRs, yielding the cycle threshold value (CT) as a logarithmic measure of abundance. For each sample, we first normalized mRNA abundance based on the median of 11 housekeeping mRNAs to yield ΔCT. Next, we excluded all mRNAs which did not change significantly (>3 standard deviations) across our time points. This eliminated approximately one-third of all mRNAs. We next adjusted for variation in individual primer efficiency. Finally, we introduced a hard threshold of detection below which all values were set equal. This cutoff is based on our experience that variation at the limit of detection reflects technical variation of no biological significance. We observed a large number of mRNAs that were increased. This is as opposed to only a small, specific set of genes being induced, e.g., the viral interleukin 6 (vIL-6) in response to IFN-γ as published by Chatterjee et al. (38). Genome-wide increases in transcription would be consistent with the virus entering the lytic cycle, since we observed increased transcription of immediate early, early, and late genes. By 48 h, KSHV replication is expected to have initiated. Please note that our mRNA measurements were performed on poly(A)-selected RNA to avoid confounding the mRNA measurements by any potential DNA contamination.
FIG 3.

Global viral gene transcription was assessed using a viral qPCR array. Poly(A) RNA was isolated from scrambled control, MAVS-depleted, or RIG-I-depleted 293 cells infected with wild-type KSHV at 0 h and 48 h postinfection. Global viral gene transcription was increased in both MAVS- and RIG-I-depleted cells compared to the control. Shown is a heat map representation of relative expression levels of significantly changed mRNAs. Red indicates the presence and blue the absence of a particular mRNA. The names of the primers, indicating the gene name and primer position on the genome, are shown below the heat map. Primers were clustered using centroid clustering and Manhattan distance as metrics. A dendrogram of the result is shown above the heat map.
All in all, these data evidence for the first time that RIG-I and MAVS affect KSHV transcription. We cannot yet explain why the two of them would yield different KSHV signatures and what the underlying mechanism may be. Increased viral gene transcription in cells depleted of MAVS or RIG-I provides further evidence that these innate immune sensors are inhibitory to KSHV replication and support the above observation that KSHV infection in the absence of MAVS and RIG-I is more efficient.
KSHV infection of MAVS- or RIG-I-depleted cells resulted in decreased IFN-β production.
One of the primary functions of pattern recognition receptors (PRRs) is to initiate the type I IFN response, production and secretion of IFN-α and IFN-β, leading to the establishment of an antiviral state. Both MAVS and RIG-I are involved in signaling cascades that result in activation of the type I IFN response (reviewed in references 19 and 39). We wanted to determine whether KSHV infection of MAVS- or RIG-I-depleted cells would result in changes in IFN-β message production. We have previously shown that type I IFN can be induced via TLR signaling in multiple cell types following KSHV infection (13, 14) and that depletion of cellular sensors significantly affected the production of type I IFN in the context of a KSHV infection.
Figure 4A shows that infection with wild-type KSHV resulted in an approximately 8-fold increase in IFN-β message levels in control cells (shSCR) 24 h postinfection, while in MAVS (shMAVS)-depleted cells, IFN-β message levels were reduced approximately 16-fold compared to those in control cells. In RIG-I (shRIG-I)-depleted cells, the difference was not as dramatic, but there was an approximate 4-fold decrease compared to the level in control cells. Three biological replicates of these experiments were performed, and qPCR analysis was done in triplicate for each sample per experimental condition. To evaluate whether intact viral DNA is necessary to trigger these signaling pathways, we also infected cells with UV-inactivated virions. UV treatment was performed as described previously (13). In each of the three cells lines, infection with UV-treated virions resulted in little to no activation of IFN-β transcription (Fig. 4). These results indicate that intact viral DNA is necessary upon entry in order to trigger these signaling pathways. At 48 h postinfection, the amount of IFN-β message was increased 2.7-fold in infected control cells over the value for mock-infected control cells. The IFN-β message in the MAVS- and RIG-I-depleted cells remained significantly reduced (Fig. 4B).
FIG 4.
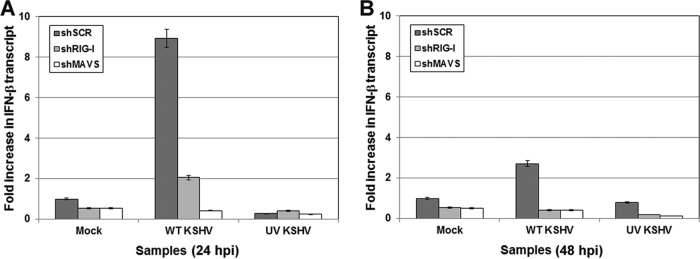
IFN-β transcription was monitored following infection of control, MAVS-depleted, or RIG-I-depleted 293 cells at 24 h postinfection with either wild-type KSHV or UV-inactivated KSHV. Both MAVS-depleted and RIG-I-depleted 293 cells showed decreased amounts of IFN-β transcripts compared to that in the control at 24 (A) and 48 (B) h postinfection. MAVS or RIG-I knockdown in 293 cells infected with UV-inactivated KSHV resulted in no induction of IFN-β transcription, indicating a requirement for infectious virus particles for stimulation of type I IFN.
We also tested whether ganciclovir treatment during KSHV infection would affect IFN-β production. Ganciclovir is known to inhibit late gene transcription, but immediate early and early gene transcription is not inhibited by this drug. At 24 and 48 h post-KSHV infection, ganciclovir treatment had no impact on the induction of IFN-β message in either the MAVS- or the RIG-I-depleted cells relative to control cells (data not shown). This suggests that RIG-I and MAVS sense viral transcripts made prior to late gene expression.
Inhibition of MAVS and RIG-I results in increased viral load and production following reactivation.
The default state for KSHV in vivo is latency. The virus is thought to sporadically reactivate from latency and enter the replicative cycle. We next investigated whether depletion of MAVS or RIG-I affected reactivation of KSHV from latency. For these studies, iSLK cells were used; iSLK is a line of epithelial origin harboring the rKSHV.219 clone under selection (see Materials and Methods for conditions) (29).
iSLK cells were transiently transfected with pLKO.1 shMAVS and shRIG-I shRNA constructs for 48 h, followed by a 48-h period of reactivation. Briefly, 3 μg of DNA was transfected into iSLK cells, and cells were incubated for 48 h in transfection medium. The medium was removed and replaced with complete medium plus doxycycline to induce reactivation. At 48 h posttransfection, cells were harvested and analyzed for knockdown of MAVS and RIG-I by Western blotting. shRNA knockdown was not as effective in these cells as in 293 cells; however, knockdown was sufficient to see decreased protein levels for both MAVS (shMAVS) and RIG-I (shRIG-I) (Fig. 5A) compared to the control (shSCR). To assess the effect of knockdown on reactivation, RNA was isolated from cells; cDNA was synthesized and analyzed by qPCR for lytic viral gene transcription, using vIL-6 as a readout. As shown in Fig. 5B, cells depleted of MAVS or RIG-I showed a 2- to 3-fold increase in vIL-6 levels compared to the control upon reactivation. We also observed increased levels of RFP (an indicator of reactivation) following reactivation in both MAVS- and RIG-I-depleted cells compared to that in control cells (Fig. 5C). As shown in Fig. 5D, we quantified the fluorescence (RFP) intensity from images of reactivated iSLK cells depleted of MAVS or RIG-I and compared the mean intensities to that of cells transfected with the control. These data are presented as the fold change in average mean intensity over three biological replicates. Depletion of either MAVS or RIG-I showed increased mean RFP intensity compared to the control in the context of iSLK reactivation. However, depletion of MAVS or RIG-I did not induce reactivation from latent iSLK cells (data not shown).
FIG 5.

Effect of MAVS and RIG-I depletion on KSHV reactivation. iSLK cells latently infected with KSHV.r219 were transfected with scrambled control, MAVS, or RIG-I shRNA constructs for 48 h. (A) Knockdown was confirmed by Western blotting. (B) At 48 h postreactivation viral transcript levels were monitored by qPCR. MAVS depletion resulted in a 7-fold increase and RIG-I depletion resulted in a 3.5-fold increase in viral genomes. (C) Reactivation was monitored by RFP expression using fluorescence microscopy. Both MAVS-depleted and RIG-I-depleted cells showed increased RFP compared to that in the scrambled control cells at 48 h postreactivation. (D) Mean RFP intensity was quantified using NIS Elements (Nikon imaging software) on the Nikon Eclipse Ti. Data are shown as the average mean intensity over three independent experiments. Both shMAVS and shRIG-I cells showed increased RFP intensity over that in shSCR cells following reactivation.
To confirm that knockdown of MAVS or RIG-I leads to increased viral load and virus production from latently infected iSLK cells, we infected naive 293 cells with supernatants harvested from iSLK cells 72 h postreactivation. Naive 293 cells were infected for 72 h, and infection was monitored by quantitating GFP-positive cells. As shown in Fig. 6A, there was a significant increase in GFP-positive cells in both MAVS- and RIG-I-depleted cells compared to the control. At 72 h postinfection, RNA was harvested from 293 cells to measure viral transcription. As shown in Fig. 6B, naive 293 cells infected with supernatants from reactivated iSLK cells depleted of MAVS or RIG-I showed increased GFP expression compared to that of 293 cells infected with supernatants from iSLK control cells. We also observed increased viral transcription in 293 cells infected with supernatants; vIL6 message levels were increased 3- to 4-fold in 293 cells infected with supernatants from MAVS- and RIG-I-depleted iSLK cells (Fig. 6C). These combined results point to a possibly significant role for MAVS and RIG-I in the context of reactivation.
FIG 6.

Infectious virus production following reactivation of latent KSHV from scrambled control and MAVS-depleted or RIG-I-depleted cells. (A) GFP-positive cells were quantitated by counting multiple fields. Naive 293 cells infected with supernatants from reactivated cells depleted for either MAVS or RIG-I showed a significant increase in the number of GFP-positive cells compared to control cells. (B) Microscopy images of infected cells described for panel A. (C) qPCR confirms that 293 cells infected with supernatant from reactivated iSLK cells, depleted for MAVS or RIG-I, show increased viral transcript levels compared to 293 cells infected with supernatants from the scrambled control.
Detection of dsRNA following reactivation of KSHV.
dsRNA is a common intermediate in both positive- and negative-sense RNA virus infections. These intermediates are much less common in DNA virus infections, and the presence of dsRNA has never been shown in KSHV-infected cells, although it has been shown in herpes simplex virus (HSV)-infected cells (25). In this study, we used iSLK cells latently infected with KSHV-BAC16 (which expresses only GFP) for detection of dsRNA species in the context of reactivation (30).
iSLK-BAC16 cells were reactivated for 48 h with doxycycline, and immunofluorescence assays were performed for detection of dsRNA. As shown in Fig. 7, unreactivated iSLK-BAC16 cells exhibited no specific staining for dsRNA; however, in reactivated cells, we were able to detect specific dsRNA signal, as indicated by the red fluorescence visible in the cytoplasm of infected cells. This suggests a possible mechanism for activation of RIG-I and MAVS in the context of reactivation. In KSHV-infected cells with intact RIG-I- and MAVS-mediated signaling pathways, these RNA intermediates could possibly be detected by RIG-I and the signal may be potentiated through MAVS leading to IFN-β and inflammatory cytokine production. Presumably, the expression of IFN-β and inflammatory cytokines would inhibit virus production following reactivation, making viral spread less efficient.
FIG 7.

Detection of dsRNA following reactivation of iSLK cells latently infected with KSHV. iSLK cells latently infected with KSHV-BAC16 were reactivated for 48 h with doxycycline. Immunofluorescence assay for dsRNA was performed. dsRNA, indicated by red fluorescence visible in the cytoplasm, was detected in the cytoplasm of reactivated cells (+Dox) but not in unreactivated (−Dox) cells.
Depletion of MAVS in latently infected B cells results in increased viral gene transcription.
B cells are one of the primary reservoirs of KSHV infection in the host. We wanted to determine whether inhibition of MAVS or RIG-I in a biologically relevant cell type would affect KSHV viral gene transcription.
In order to determine whether MAVS depletion played a role during viral latency in KSHV-infected B cells, we transduced BCBL1 cells, which are KSHV-positive, EBV-negative B cells, with lentivirus expressing shRNA to MAVS or RIG-I (or an SCR control). Briefly, 1 × 106 BCBL1 cells were transduced with each respective lentivirus and the cells were incubated for 72 to 96 h to determine whether MAVS or RIG-I knockdown alone, in the absence of reactivation, resulted in increased viral gene transcription. At 72 and 96 h after lentiviral infection, cells were harvested and washed, and RNA was isolated as described in Materials and Methods. qPCR was performed for ORF57, a readout for viral gene transcription, and for MAVS or RIG-I to confirm inhibition of the target gene transcript. As shown in Fig. 8, depletion of MAVS in BCBL1 cells leads to increased transcription of ORF57 at both 72 (Fig. 8A) and 96 (Fig. 8B) h. At 72 h, ORF57 transcription is increased over 4.5-fold, and at 96 h, the increase is approximately 9-fold. Fold changes were normalized to the housekeeping β-actin gene and calculated using the 2−ΔΔCT method where ΔΔCT = CT for target gene − CT for β-actin. We also confirmed that MAVS transcript levels were decreased 8- to 10-fold at each respective time point. We also performed qPCR for vIL6 to confirm these results (data not shown). These data suggest that in the latent setting, MAVS can negatively regulate viral lytic gene transcription, preventing the virus from reactivating in the absence of a direct stimulus. Depletion of RIG-I in this latent system resulted in no significant changes compared to the control (data not shown).
FIG 8.
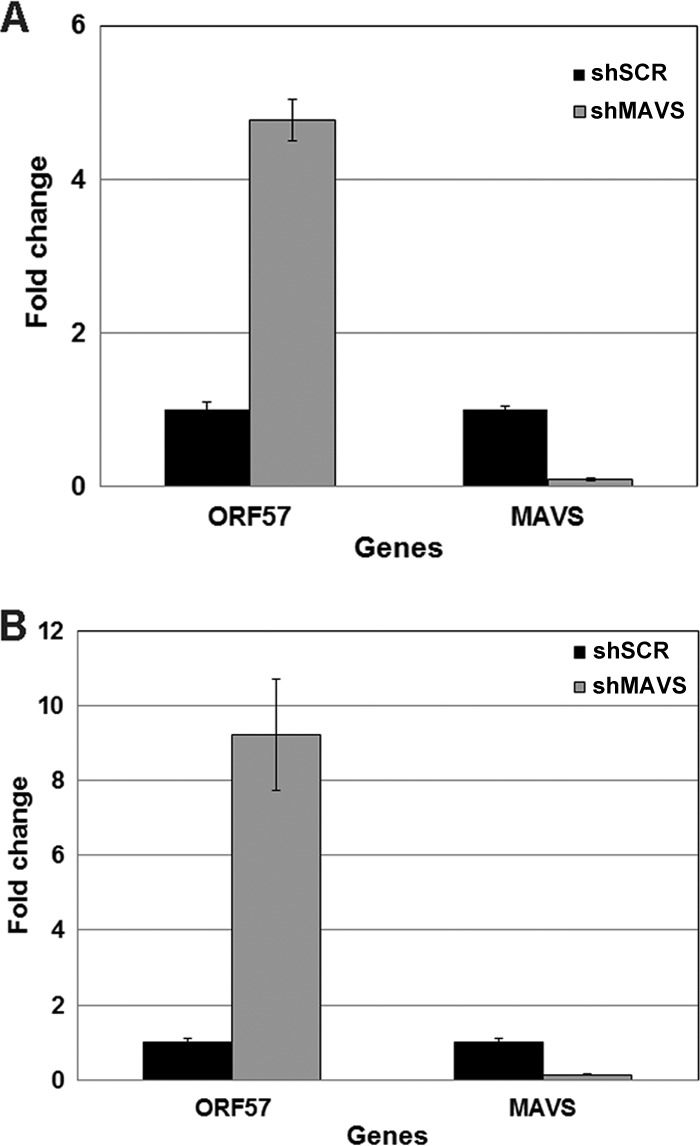
Latently infected B cells (BCBL1) were transduced with scrambled control (shSCR) or shMAVS lentiviruses. BCBL1 cells were incubated for 72 or 96 h with the lentiviruses, and expression of ORF57 mRNA transcript was measured. MAVS-depleted cells showed increased ORF57 viral gene transcription at both 72 (A) and 96 (B) h compared to the scrambled control. MAVS depletion was confirmed via qPCR. Fold changes were normalized to the housekeeping β-actin gene and calculated using the 2-ΔΔCT method where ΔΔCT = CT for target gene − CT for β-actin.
DISCUSSION
Pathogen infection typically results in the production of proinflammatory cytokines and type I IFN. The IFN response is one of the primary defenses utilized by the host innate immune system to control virus infection. A majority of immune cells in the body contain one or more sensors that monitor for pathogen invasion as part of the innate immune response. These sensors are classified as pattern recognition receptors (PRRs), because they recognize and bind specific patterns present on the surface of incoming pathogens (19). Members of the TLR, NLR, and RLR protein families are all classified as PRRs and have all been found to play a role in herpesvirus infection (reviewed in reference 40). Specifically, in the context of a KSHV infection, our laboratory has previously established a role for both TLR and NLR activation during the KSHV life cycle. However, there are no current reports identifying a role for MAVS during KSHV infection and only one report indicating that RIG-I (an RLR family member) may be important during KSHV infection, as the virus specifically targets RIG-I to inactivate it (21).
KSHV establishes a lifelong latent infection in the human host. To accomplish this, manipulation of the host immune response is required. We investigated whether two critical components of the innate immune response to viral infection, MAVS and RIG-I, were involved in hindering KSHV infection. Here we report the first evidence that MAVS has a role in limiting KSHV replication and virus production and that RIG-I also has an inhibitory function in the context of KSHV primary infection and during reactivation from latency. Using cell lines stably knocked down for either MAVS or RIG-I, we have shown that in cells depleted of these innate immune proteins, de novo KSHV infection results in increased global transcription of the viral genome. This is an indication that normal expression of these sensors inhibits KSHV transcription following primary infection. Although KSHV contains over 80 open reading frames whose transcripts are thought to be capped and polyadenylated, whole-genome transcriptome analysis has revealed that there are many additional long and short noncoding RNAs that are transcribed off the KSHV genome in addition to the viral microRNAs (41, 42). We surmise that some of these transcripts may be sensed by the RIG-I/MAVS pathway. Further evidence supporting the notion that expression of MAVS and RIG-I is inhibitory for KSHV infection includes the observation of reduced IFN-β transcription following KSHV infection of MAVS- and RIG-I-depleted cell lines, suggesting that these proteins recognize KSHV and initiate their signaling cascades in response to infection with this virus. Additionally, infection with UV-inactivated virus showed that intact viral DNA is required for normal activation of these signaling cascades and increased IFN-β transcription.
Inhibition of KSHV viral gene transcription by MAVS and RIG-I is not limited to primary infection. We report here that subsequent to reactivation, viral gene transcription is increased in cells deficient in MAVS and RIG-I compared to that in control cells. We also demonstrated that this increase in viral gene transcription reflected an increase in infectious virion production. Supernatants from reactivated control cells and MAVS- and RIG-I-deficient cells were used to infect naive 293 cells. Increased GFP expression along with increased viral load was observed in cells infected with supernatants from MAVS- and RIG-I-deficient cells. This further supports the notion that MAVS and RIG-I expression serves to inhibit KSHV reactivation.
The mechanism by which RIG-I senses KSHV following infection is unclear; however, it has been published that RNA polymerase III can mediate the recognition of dsDNA through RNA intermediates which, in turn, can be recognized by RIG-I (22, 24). This is a potential mechanism through which RIG-I could respond to KSHV infection, via RNA intermediates. Moreover, RNase L may also have a role in sensing RNA during KSHV infection or reactivation. RNase L is an endoribonuclease which, upon activation by phosphorylated 2′,5′-linked oligoadenylate [(2′-5′)p3A3], cleaves RNA that can initiate type I IFN production (43). This pathway has been shown to be active in the context of HSV-2 infection, and RNase L can cooperate with RIG-I and MDA-5 to induce expression of IFN-β during virus infection (25). The apparent suppression of KSHV replication and production by RIG-I and MAVS may aid KSHV in establishing a latent infection. The KSHV-encoded deubiquitinase, a tegument lytic protein, may counter this response since it can inactivate RIG-I through deubiquination (21). Collectively, our data suggest that MAVS and RIG-I sense KSHV during different phases of its life cycle and that depletion of MAVS from KSHV-infected B cells allows for viral reactivation.
ACKNOWLEDGMENTS
We thank Don Ganem for the iSLK cells and Jae Jung for the BAC16 construct. We also thank Jeff Vieira for the gift of the rKSHV.219 virus.
This work was supported by NIH grants AI107810, AI109965, and DE018281 to B.D. and DE018304 to D.P.D. J.A.W. was supported by grant 1F32AI078735. S.R.J. was supported by grants 5T32-CA09156 and 5T32-AI007151 and P.C. by grant CA109232. B.D. is a Leukemia & Lymphoma Society Scholar and Burroughs Wellcome Fund Investigator in Infectious Disease.
Footnotes
Published ahead of print 12 March 2014
REFERENCES
- 1.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869. 10.1126/science.7997879 [DOI] [PubMed] [Google Scholar]
- 2.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186–1191. 10.1056/NEJM199505043321802 [DOI] [PubMed] [Google Scholar]
- 3.Blasig C, Zietz C, Haar B, Neipel F, Esser S, Brockmeyer NH, Tschachler E, Colombini S, Ensoli B, Sturzl M. 1997. Monocytes in Kaposi's sarcoma lesions are productively infected by human herpesvirus 8. J. Virol. 71:7963–7968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boshoff C, Schulz TF, Kennedy MM, Graham AK, Fisher C, Thomas A, McGee JO, Weiss RA, O'Leary JJ. 1995. Kaposi's sarcoma-associated herpesvirus infects endothelial and spindle cells. Nat. Med. 1:1274–1278. 10.1038/nm1295-1274 [DOI] [PubMed] [Google Scholar]
- 5.Monini P, Colombini S, Sturzl M, Goletti D, Cafaro A, Sgadari C, Butto S, Franco M, Leone P, Fais S, Melucci-Vigo G, Chiozzini C, Carlini F, Ascherl G, Cornali E, Zietz C, Ramazzotti E, Ensoli F, Andreoni M, Pezzotti P, Rezza G, Yarchoan R, Gallo RC, Ensoli B. 1999. Reactivation and persistence of human herpesvirus-8 infection in B cells and monocytes by Th-1 cytokines increased in Kaposi's sarcoma. Blood 93:4044–4058 [PubMed] [Google Scholar]
- 6.Rettig MB, Ma HJ, Vescio RA, Pold M, Schiller G, Belson D, Savage A, Nishikubo C, Wu C, Fraser J, Said JW, Berenson JR. 1997. Kaposi's sarcoma-associated herpesvirus infection of bone marrow dendritic cells from multiple myeloma patients. Science 276:1851–1854. 10.1126/science.276.5320.1851 [DOI] [PubMed] [Google Scholar]
- 7.Wu W, Vieira J, Fiore N, Banerjee P, Sieburg M, Rochford R, Harrington W, Jr, Feuer G. 2006. KSHV/HHV-8 infection of human hematopoietic progenitor (CD34+) cells: persistence of infection during hematopoiesis in vitro and in vivo. Blood 108:141–151. 10.1182/blood-2005-04-1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krishnan HH, Naranatt PP, Smith MS, Zeng L, Bloomer C, Chandran B. 2004. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 78:3601–3620. 10.1128/JVI.78.7.3601-3620.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee HR, Brulois K, Wong L, Jung JU. 2012. Modulation of immune system by kaposi's sarcoma-associated herpesvirus: lessons from viral evasion strategies. Front. Microbiol. 3:44. 10.3389/fmicb.2012.00044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Creagh EM, O'Neill LA. 2006. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 27:352–357. 10.1016/j.it.2006.06.003 [DOI] [PubMed] [Google Scholar]
- 11.Ye Z, Ting JP. 2008. NLR, the nucleotide-binding domain leucine-rich repeat containing gene family. Curr. Opin. Immunol. 20:3–9. 10.1016/j.coi.2008.01.003 [DOI] [PubMed] [Google Scholar]
- 12.Gregory SM, West JA, Dillon PJ, Hilscher C, Dittmer DP, Damania B. 2009. Toll-like receptor signaling controls reactivation of KSHV from latency. Proc. Natl. Acad. Sci. U. S. A. 106:11725–11730. 10.1073/pnas.0905316106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.West J, Damania B. 2008. Upregulation of the TLR3 pathway by Kaposi's sarcoma-associated herpesvirus. J. Virol. 82:5440–5449. 10.1128/JVI.02590-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.West JA, Gregory SM, Sivaraman V, Su L, Damania B. 2011. Activation of plasmacytoid dendritic cells by Kaposi's sarcoma-associated herpesvirus. J. Virol. 85:895–904. 10.1128/JVI.01007-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gregory SM, Davis BK, West JA, Taxman DJ, Matsuzawa S, Reed JC, Ting JP, Damania B. 2011. Discovery of a viral NLR homolog that inhibits the inflammasome. Science 331:330–334. 10.1126/science.1199478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G. 2006. 5′-Triphosphate RNA is the ligand for RIG-I. Science 314:994–997. 10.1126/science.1132505 [DOI] [PubMed] [Google Scholar]
- 17.Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314:997–1001. 10.1126/science.1132998 [DOI] [PubMed] [Google Scholar]
- 18.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4:491–496. 10.1038/ni921 [DOI] [PubMed] [Google Scholar]
- 19.Akira S, Uematsu S, Takeuchi O. 2006. Pathogen recognition and innate immunity. Cell 124:783–801. 10.1016/j.cell.2006.02.015 [DOI] [PubMed] [Google Scholar]
- 20.Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. 2007. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446:916–920. 10.1038/nature05732 [DOI] [PubMed] [Google Scholar]
- 21.Inn KS, Lee SH, Rathbun JY, Wong LY, Toth Z, Machida K, Ou JH, Jung JU. 2011. Inhibition of RIG-I-mediated signaling by Kaposi's sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J. Virol. 85:10899–10904. 10.1128/JVI.00690-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V. 2009. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 10:1065–1072. 10.1038/ni.1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng G, Zhong J, Chung J, Chisari FV. 2007. Double-stranded DNA and double-stranded RNA induce a common antiviral signaling pathway in human cells. Proc. Natl. Acad. Sci. U. S. A. 104:9035–9040. 10.1073/pnas.0703285104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chiu YH, Macmillan JB, Chen ZJ. 2009. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138:576–591. 10.1016/j.cell.2009.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rasmussen SB, Jensen SB, Nielsen C, Quartin E, Kato H, Chen ZJ, Silverman RH, Akira S, Paludan SR. 2009. Herpes simplex virus infection is sensed by both Toll-like receptors and retinoic acid-inducible gene-like receptors, which synergize to induce type I interferon production. J. Gen. Virol. 90:74–78. 10.1099/vir.0.005389-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Samanta M, Iwakiri D, Kanda T, Imaizumi T, Takada K. 2006. EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. EMBO J. 25:4207–4214. 10.1038/sj.emboj.7601314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Samanta M, Iwakiri D, Takada K. 2008. Epstein-Barr virus-encoded small RNA induces IL-10 through RIG-I-mediated IRF-3 signaling. Oncogene 27:4150–4160. 10.1038/onc.2008.75 [DOI] [PubMed] [Google Scholar]
- 28.Vieira J, O'Hearn PM. 2004. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology 325:225–240. 10.1016/j.virol.2004.03.049 [DOI] [PubMed] [Google Scholar]
- 29.Myoung J, Ganem D. 2011. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J. Virol. Methods 174:12–21. 10.1016/j.jviromet.2011.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, Lee SH, Lee HR, Myoung J, Ganem D, Oh TK, Kim JF, Gao SJ, Jung JU. 2012. Construction and manipulation of a new Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome clone. J. Virol. 86:9708–9720. 10.1128/JVI.01019-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dittmer DP. 2011. Restricted Kaposi's sarcoma (KS) herpesvirus transcription in KS lesions from patients on successful antiretroviral therapy. mBio 2:e00138–00111. 10.1128/mBio.00138-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Papin J, Vahrson W, Hines-Boykin R, Dittmer DP. 2005. Real-time quantitative PCR analysis of viral transcription. Methods Mol. Biol. 292:449–480. 10.1385/1-59259-848-X:449 [DOI] [PubMed] [Google Scholar]
- 33.Dittmer DP. 2003. Transcription profile of Kaposi's sarcoma-associated herpesvirus in primary Kaposi's sarcoma lesions as determined by real-time PCR arrays. Cancer Res. 63:2010–2015 [PubMed] [Google Scholar]
- 34.Rozen S, Skaletsky H. 2000. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132:365–386. 10.1385/1-59259-192-2:365 [DOI] [PubMed] [Google Scholar]
- 35.Lock EF, Ziemiecke R, Marron J, Dittmer DP. 2010. Efficiency clustering for low-density microarrays and its application to QPCR. BMC Bioinformatics 11:386. 10.1186/1471-2105-11-386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chugh P, Tamburro K, Dittmer DP. 3 December 2010. Profiling of pre-micro RNAs and microRNAs using quantitative real-time PCR (qPCR) arrays. J. Vis Exp.. 10.3791/2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raghu H, Sharma-Walia N, Veettil MV, Sadagopan S, Chandran B. 2009. Kaposi's sarcoma-associated herpesvirus utilizes an actin polymerization-dependent macropinocytic pathway to enter human dermal microvascular endothelial and human umbilical vein endothelial cells. J. Virol. 83:4895–4911. 10.1128/JVI.02498-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chatterjee M, Osborne J, Bestetti G, Chang Y, Moore PS. 2002. Viral IL-6-induced cell proliferation and immune evasion of interferon activity. Science 298:1432–1435. 10.1126/science.1074883 [DOI] [PubMed] [Google Scholar]
- 39.Medzhitov R. 2007. Recognition of microorganisms and activation of the immune response. Nature 449:819–826. 10.1038/nature06246 [DOI] [PubMed] [Google Scholar]
- 40.Paludan SR, Bowie AG, Horan KA, Fitzgerald KA. 2011. Recognition of herpesviruses by the innate immune system. Nat. Rev. Immunol. 11:143–154. 10.1038/nri2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chandriani S, Xu Y, Ganem D. 2010. The lytic transcriptome of Kaposi's sarcoma-associated herpesvirus reveals extensive transcription of noncoding regions, including regions antisense to important genes. J. Virol. 84:7934–7942. 10.1128/JVI.00645-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dresang LR, Teuton JR, Feng H, Jacobs JM, Camp DG, II, Purvine SO, Gritsenko MA, Li Z, Smith RD, Sugden B, Moore PS, Chang Y. 2011. Coupled transcriptome and proteome analysis of human lymphotropic tumor viruses: insights on the detection and discovery of viral genes. BMC Genomics 12:625. 10.1186/1471-2164-12-625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Malathi K, Dong B, Gale M, Jr, Silverman RH. 2007. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 448:816–819. 10.1038/nature06042 [DOI] [PMC free article] [PubMed] [Google Scholar]