

ABSTRACT
Reactivation of human cytomegalovirus (HCMV) is a significant cause of disease and death in immunocompromised patients, underscoring the need to understand how latency is controlled. Here we demonstrate that HCMV has evolved to utilize cellular microRNAs (miRNAs) in cells that promote latency to regulate expression of a viral protein critical for viral reactivation. Our data reveal that hsa-miR-200 miRNA family members target the UL122 (immediate early protein 2) 3′ untranslated region, resulting in repression of this viral protein. Utilizing recombinant viruses that mutate the miRNA-binding site compared to the sequence of the wild-type virus results in lytic rather than latent infections in ex vivo infections of primary CD34+ cells. Cells permissive for lytic replication demonstrate low levels of these miRNAs. We propose that cellular miRNA regulation of HCMV is critical for maintenance of viral latency.
IMPORTANCE Human cytomegalovirus (HCMV) is a herpesvirus that infects a majority of the population. Once acquired, individuals harbor the virus for life, where the virus remains, for the most part, in a quiet or latent state. Under weakened immune conditions, the virus can reactivate, which can cause severe disease and often death. We have found that members of a family of small RNAs, termed microRNAs, encoded by human myeloid progenitor cells are capable of repressing a key viral protein, thus enabling the virus to ensure a quiet/latent state. As these progenitor cells mature further down the myeloid lineage toward cells that support active viral replication, the levels of these microRNAs decrease. Together, our data suggest that host cell microRNA regulation of HCMV is important for the quiet/latent state of this pathogen.
INTRODUCTION
Human cytomegalovirus (HCMV) is a ubiquitous pathogen that resides latently in the host for life. During times of immunological stress, the virus can reactivate to cause severe disease and often mortality (1). In adults, primary infection rarely causes HCMV-related complications. Rather, it is lytic reactivation of the virus from its latent state that results in various states of morbidity (2). Thus, understanding the mechanisms involved in viral latency and reactivation is of great significance.
During latency, the herpesviruses have silenced genomes and express only a limited number of viral transcripts. Of the more than 200 genes encoded by the HCMV genome, for example, only 5 are expressed during latent infection (3–7). Transcriptional silencing of the HCMV major immediate early promoter (MIEP) is a key step in establishing HCMV latency (8–14). This promoter regulates the expression of UL122 and UL123 (encoding immediate early [IE] protein 2 [IE2] and IE1, respectively), which ultimately increase expression from the MIEP as well as the early and late viral lytic genes in the HCMV lytic transcriptional cascade (reviewed in reference 15). Thus, suppressing this promoter silences the lytic HCMV genome.
Repressive chromatin marks likely represent the key mechanism by which HCMV silences its genome to establish and maintain a latent infection. In experimental ex vivo and in vivo latency systems, deposition of repressive chromatin marks on the viral genome indeed promotes HCMV latency via the action of histone deacetylases and histone methyltransferases (16). The MIEP, which contains binding sites for a variety of transcription factors, is also modulated by chromatinization. Additionally, in naturally latently infected individuals, the MIEP is associated with repressed chromatin (12), suggesting that this mechanism of viral genomic silencing is utilized to maintain a successful in vivo HCMV latent infection. However, it is unlikely that chromatinization is the only means by which HCMV maintains a latent infection. We hypothesize that to complement transcriptional repression due to chromatinization, HCMV has evolved a secondary backup mechanism which employs microRNAs (miRNAs) to impede viral translation.
The alpha- and gammaherpesviruses encode miRNAs that play important roles in the regulation of viral latency (17); however, to date, a direct role for miRNAs in the control of HCMV latency has not been reported. miRNAs are small RNAs (21 to 23 nucleotides in length) that target specific transcripts via a partial complementarity to binding sites typically within the 3′ untranslated region (UTR) of the target gene. Once bound to the target transcript, miRNAs either repress translation of the target protein or, alternatively, result in RNA instability. Herpesvirus-encoded miRNAs target both cellular and viral transcripts. For example, HCMV encodes 14 miRNAs (18–20), and we and others have shown that at least one of these miRNAs, hcmv-miR-UL112-1, targets both viral (21, 22) and cellular (23, 24) transcripts in the context of lytic infection. The present work was motivated by our identification of a family of cellular miRNAs that we predicted target the 3′ UTR of UL122. As discussed above, silencing of the MIEP is a major step in establishing and maintaining a latent infection, and thus, we hypothesized that miRNA targeting of the IE region is perhaps a critical secondary mechanism toward controlling latency.
We have recently described an in vitro system for the study of HCMV latency and reactivation (25). Our system utilizes the CD34+ progenitor Kasumi-3 cell line, which supports latent infection and reactivation, producing infectious viral progeny. This system affords us the ability to interrogate key questions about latency, including the regulation of the IE region by cellular miRNAs. In the study described here, we used our latency model system to demonstrate that the cellular hsa-miR-200 cluster of miRNAs specifically targets the 3′ UTR of HCMV IE2, encoded by UL122. We show that the targeting of this viral IE protein is key in regulating viral latency in vitro. Furthermore, we have found that these findings are consistent with those obtained in infected ex vivo primary CD34+ cells. Together, we demonstrate that HCMV utilizes cellular miRNAs to repress expression of an essential viral transcript as a means to facilitate successful silencing of lytic replication.
MATERIALS AND METHODS
In silico analysis.
Candidate host miRNAs were identified using a computational algorithm (22, 26) that calculates the probability that a specific miRNA binding to a defined 3′ UTR would occur by chance in a randomly generated pool of 3′ UTRs with the same mononucleotide and dinucleotide content as the real 3′ UTR. The smaller that such a probability is, the more likely it is that the miRNA-3′ UTR pair is functional. The UL122 3′ UTR was screened against human miRNAs to identify candidate miRNAs that are most likely to target this gene. Minimum free energies and binding hybridization of candidate miRNAs were calculated by use of the BiBiServe RNA hybrid algorithm (27).
Plasmids and lentiviral constructs.
To generate the pMirGloIE2wt plasmid, the following oligonucleotides corresponding to the predicted miRNA-binding site within the 3′ UTR of UL122 were annealed: top oligonucleotide 5′-TCGAGTAGGATCCATAGTGATTCCCCGTGACAGTATTAAT-3′ and bottom oligonucleotide 5′-CTAGATTAATACTGTCACGGGGAATCACTATGGATCCTAC-3′. The resulting product was ligated into the XhoI and XbaI restriction enzyme sites of the pMirGlo vector (Promega). A corresponding construct containing a 4-nucleotide substitution within the predicted seed sequence was generated by ligating two oligonucleotides, top oligonucleotide 5′-TCGAGTAGGATCCATAGTGATTCCCCGTGAGTCATAATAT-3′ and bottom oligonucleotide 5′-CTAGATATTATGACTCACGGGGAATCACTATGGATCCTA C-3′, and cloned, as described above, into pMirGlo to generate pMirGloIE2mut. The miRNA 200 (miR-200)-regulated positive control contained the 3′ UTR sequence of SIP1/ZEB2, which is regulated by the miR-200 family members (28), and the retroviral constructs which expressed the miR-200 family members were a kind gift of Yibin Kang (Princeton University). These retroviruses contain the human genomic fragments termed cluster 1 (C1), expressing hsa-miR-200b, -200a, and -429, or cluster 2 (C2), expressing hsa-miR-200c and -141, which were cloned into the pMSCV-puro and pMSCV-hygro retroviral vectors, respectively (29).
Luciferase reporter assay.
Transient transfections were performed using the FuGENE 6 transfection reagent (Roche Applied Science) according to the manufacturer's protocols. pCMV-U6 with no miRNA-coding insert was added to the transfections to ensure that equal amounts of total DNA were used for each transfection. After 48 h at 37°C, cell lysates were prepared using passive lysis solution (Promega) and assayed using a Dual-Glo luciferase assay system (Promega) according to the manufacturer's protocols. Luciferase units were assayed using a Veritas microplate luminometer (Promega). Firefly luciferase units were normalized to Renilla luciferase units to control for transfection and lysis variations.
Viruses and cells.
HCMV bacterial artificial chromosome (BAC)-derived strains FIX and TB40/E (clone 4) were used in these studies. We previously engineered these strains to express enhanced green fluorescent protein (eGFP) (22, 25). FixBACgfp and TB40/Egfp were used as the wild-type viruses in these studies, where indicated, and were also utilized either to generate the FIXgfpIE2cisΔ mutant using galK BAC recombineering protocols (30) or to generate the TB40/EgfpIE2cisΔ mutant using I-sce I-based recombineering protocols (31). The recombineering protocols and virus production protocols are described below.
Kasumi-3 cells (ATCC) were maintained in RPMI 1640 (ATCC; catalog no. 30-2001) supplemented with 20% fetal bovine serum (FBS), 100 U/ml each penicillin and streptomycin, and 100 μg/ml gentamicin. The Kasumi-3 cells used for viral infections were maintained in X-VIVO 15 medium (Lonza) supplemented with 100 U each of penicillin and streptomycin for 48 h prior to infection. Primary newborn human fibroblasts (NUFF-1 cells; GlobalStem) or primary human embryonic lung fibroblasts (MRC5 cells) were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% FBS, 2 mM l-glutamine, 0.1 mM nonessential amino acids, and 100 U/ml each penicillin and streptomycin. The murine breast cancer cell line 4T07 was maintained in DMEM supplemented with 10% newborn calf serum and 100 U/ml each penicillin and streptomycin. Primary CD34+ cells were isolated from cord blood by magnetic bead separation and cultured as previously described (32). Monocytes isolated from peripheral blood were a kind gift from Christine McDonald (Cleveland Clinic). Monocytes were maintained in culture in RPMI 1640 supplemented with 10% human serum. Macrophages were derived from a portion of the monocyte culture by treating them with 100 ng/ml each of macrophage colony-stimulating factor and interleukin-3 for 72 h (33). All cells, except where indicated, were maintained in a 37°C incubator with 5% CO2.
Generation of stable cell lines.
Retroviruses containing the genomic fragments expressing the miR-200 family members or their empty counterparts (pMSCV-puro and pMSCV-hygro) were transfected into Phoenix cells by lipid-based methods using the FuGENE 6 transfection reagent (Roche Applied Science) per the manufacturer's protocols. Supernatants were collected, clarified by filtering through a 0.45-μm-pore-size syringe filter, and concentrated by ultracentrifugation at 82,705 × g at 4°C for 90 min. Pellets were resuspended and used to transduce both fibroblasts and 4T07 cells in the presence of Polybrene (4 μg/ml) overnight at 37°C. On the following day, the medium was changed and transduced cells were selected by the addition of either puromycin (1 μg/ml) or hygromycin (100 μg/ml).
Viral recombineering and virus production.
To generate the mutations within the FixBAC strain, we transformed Escherichia coli SW105 with FixBACgfp and inserted the galK open reading frame (ORF) into the IE2 3′ UTR using the following primers: forward primer 5′-GATATATAAAAAAAAGCTACTTTTATTAAACAGCCTTCTCACCACACGTTAcctgttgacaattaatcatcggca-3′ and reverse primer 5′-AAAACTGGAAAGAGAGACATGGACTCTTGTACATAGTGATTCCCCGTGACAtcagcactgtcctgctcctt-3′, where uppercase letters correspond to the HCMV sequence and lowercase letters correspond to the sequence of the pGalK vector. GalK-positive clones were reverted by a second round of homologous recombination using the following annealed primers, resulting in the deletion of the miRNA-binding site: top primer 5′-AAAAAGCTACTTTTATTAAACAGCCTTCTCACCACACGTTAGGGGTGTCACGGGGAATCACTATGTACAAGAGTCCATGTCTCTCT-3′ and bottom primer 5′-AGAGAGACATGGACTCTTGTACATAGTGATTCCCCGTGACACCCCTAACGTGTGGTGAGAAGGCTGTTTAATAAAAGTAGCTTTTT-3′, where the bold letters denote the mutations within the seed sequence. All mutations within the recombinant viruses were validated by sequencing.
To generate the mutations within the clinical TB40/e BAC strain, we transformed E. coli GS1783 with TB40/Egfp and inserted the kanamycin-I-sce I cassette containing the mutation using the following primers: forward primer 5′-AGATGTATAAAAAAAGCTACTTTTATTAAACAGCATTCTCACCACACGTTAtcgatttattcaacaaagccacg-3′ and reverse primer 5′-GAAAAACTGGAAAGAGACATGGACTCTTGTACATAGTGATTCCCCGTGACAcgcgtatatctggcccgtacatcg-3′, where the lowercase characters correspond to the pEPKanS vector. Positive clones were then selected for by culture on kanamycin-chloramphenicol-LB agar plates overnight at 32°C, and then insertion of the cassette was confirmed by PCR. Positive clones were then selected and grown overnight in LB-chloramphenicol at 32°C overnight, after which the cells were diluted and grown to an optical density at 600 nm of 0.5 to 0.7. The insertion cassette containing the kanamycin-I-sce I fragment was then excised by 1.0% l-arabinose, resulting in a seamless insertion of the mutated sequence. Finally, clones were selected for on kanamycin-chloramphenicol-arabinose-LB plates, confirmed by PCR amplification, and then analyzed by genomic sequencing. To generate the repair virus TB40/EgfpIE2cisΔrep, we used the same technique described above with the following primers for the initial insertion: forward primer 5′-AGTAAGTGAAAAACTGGAAAGAGACATGGACTCTTGTACATAGTGATTCCCCGTGACAGTATTcgcgtatatctggcccgtacatcg-3′ and reverse primer 5′-AAAGCTACTTTTATTAAACAGCATTCTCACCACACGTTAATACTGTCACGGGGAATCACTAtcgatttattcaacaaagccacg-3′, where the lowercase characters correspond to the pEPKanS vector. All viral stocks were grown on primary fibroblasts, and after a 100% cytopathic effect was achieved, virus was harvested by ultracentrifugation through a 20% sorbitol cushion. Viral pellets were resuspended in X-VIVO 15 medium (Lonza) supplemented with 1.5% bovine serum albumin. Aliquots were flash frozen in liquid nitrogen and stored at −80°C until further use. Stock titers were assessed by 50% tissue culture infective dose assays on primary fibroblasts.
Infection of fibroblasts, Kasumi-3 cells, and CD34+ hematopoietic progenitor cells.
Fibroblasts (2.5 × 105) were infected at a multiplicity of 0.5 PFU/cell for 1 h at 37°C. Following adsorption, the inoculum was removed and the cells were washed three times with phosphate-buffered saline (PBS), after which the cultures were replenished with fresh medium. Viral supernatants were collected at 4 days postinfection (dpi), and viral titers were determined by immunofluorescence for IE1, as previously described (34). In brief, collected supernatants were serially diluted and used to infect primary MRC5 cells for 24 h. Each dilution was analyzed in triplicate. Cells were then fixed in ice-cold methanol for 20 min at −20°C and next stained with a monoclonal antibody that detects IE1 (clone 1B12 [35]) and a fluorescently conjugated antibody (Molecular Probes). Five random fields were quantified, as described previously (34).
Kasumi-3 cells were infected at a multiplicity of 1.0 PFU/cell by centrifugal enhancement at 1,000 × g for 30 min at room temperature in X-VIVO 15 medium (Lonza). On the next day, the cells were treated with trypsin to remove any viral particles that had not entered the cell. Infected cells were cushioned onto Ficoll-Paque (GE Healthcare) to remove debris and the viral inoculum, after which the cells were washed two times with PBS. Cells were then replated in X-VIVO 15 medium (Lonza) and harvested as described above.
Primary CD34+ cells were allowed to recover for 5 h postisolation (as described above). Cells were then infected at a multiplicity of 2.0 PFU/cell in X-VIVO 15 medium (Lonza) by low-speed centrifugation at 450 × g for 20 min at room temperature. The infected cells were then returned to culture overnight, and on the following day, the cells were washed with excess X-VIVO 15 medium (Lonza) to remove the viral inoculum. Cells were then replated and harvested as described above.
Detection of miRNAs, RNA, DNA, and protein.
To determine miRNA expression, total RNA was isolated from cell pellets using a mirVana miRNA isolation kit (Ambion), according to the manufacturer's procedure for total RNA isolation. This protocol allows total RNA isolation yet preserves the miRNA population. In brief, cell pellets were first lysed and disrupted, after which the lysates were extracted once with acid phenol-chloroform. Samples were then purified further over glass fiber filters, and finally, the filtrates were washed and eluted to yield total RNA. DNA was removed from the samples utilizing a DNA-free reagent kit (Ambion). Expression of the hsa-miR-200 cluster was then detected utilizing modified TaqMan-based stem-loop reverse transcription (RT)-quantitative PCR (qPCR) (36). RNA (10 ng) was reverse transcribed using a TaqMan microRNA reverse transcription kit (Applied Biosystems), according to the manufacturer's instructions, using stem-loop primers for either hsa-miR-200b or hsa-miR-200c (Applied Biosystems). Next, a 1:15 dilution of the product from the RT step was used for TaqMan qPCR with 1.5 mM forward primer, 0.7 mM reverse primer, 0.2 mM TaqMan probe, and 1× Universal TaqMan PCR master mix (Applied Biosystems). The primers and probe used in each reaction corresponded to either hsa-miR-200b or hsa-miR-200c, in accordance with the appropriate stem-loop primer. Each sample was analyzed in triplicate, and in each case, the level was normalized by quantifying the levels of human U44 small nuclear RNA using an RNU44 TaqMan control assay (Applied Biosystems).
RNA was isolated from infected Kasumi-3 cells at 7 dpi utilizing the Tri Reagent (Sigma) protocol, according to the instructions of the manufacturer. cDNA was generated from equal concentrations of RNA (500 ng) using TaqMan RT reagents (Roche), as previously described (22). Next, equal volumes of cDNA were used to quantify transcripts by RT-qPCR using a SYBR green PCR mix (Applied Biosystems) according to the manufacturer's protocol. Samples were analyzed in triplicate, and in each case, the level was normalized to that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH; forward primer 5′-ACCCACTCCTCCACCTTTGAC-3′ and reverse primer 5′-CTGTTGCTGTAGCCAAATTCGT-3′). Viral transcripts were assessed utilizing primers directed at UL123 (forward primer 5′-GCCTTCCCTAAGACCACCAAT-3′ and reverse primer 5′-ATTTTCTGGGCATAAGCCATAATC-3′).
Extracellular viral genomes were quantified by qPCR as previously described (25). In brief, samples were collected over a time course of 5 days. For each time point, cells were pelleted by centrifugation, and the supernatants were collected and stored at −80°C. Equal volumes of all samples were prepared by sodium dodecyl sulfate (SDS; 0.8%)-proteinase K (40 μg/ml) treatment overnight at 37°C in 400 mM NaCl, 10 mM Tris, pH 8.0, and 10 mM EDTA. DNA was extracted on the following day by phenol-chloroform-isoamyl alcohol purification followed by ethanol precipitation. Pellets were resuspended in an equal volume and analyzed by qPCR in triplicate using the UL123 primers described above.
For protein analysis, cells were infected at a multiplicity of 1.0 PFU/cell and harvested at the indicated time points. Proteins were then denatured and equal concentrations of each sample (30 μg) were loaded for separation by SDS-PAGE. Proteins were transferred onto polyvinylidene difluoride by semidry transfer and detected with the following antibodies: anti-IE2 (clone 3A9 [37]) diluted 1:100, antitubulin (Sigma) diluted 1:5,000, and horseradish peroxidase-conjugated goat antimouse secondary antibody (Jackson Laboratory) diluted 1:10,000.
RESULTS
The hsa-miR-200 cluster specifically targets the HCMV UL122 3′ UTR.
We have previously published our computational analysis algorithm that predicts the targets of both cellular and viral miRNAs (22, 26). Our analysis indicates that three of the five hsa-miR-200 miRNA family members are likely to target the 3′ UTR of UL122 (Fig. 1), a surprising finding as the 3′ UTR of UL122 is relatively short (∼90 nucleotides). The hsa-miR-200 family is comprised of five miRNAs, which can be subdivided into two groups on the basis of either chromosomal location or sequence (29). hsa-miR-200b, -200a, and -429 map to human chromosome 1 (cluster 1 [C1]), while hsa-miR-200c and -141 map to human chromosome 12 (cluster 2 [C2]). Our prediction analysis revealed that hsa-miR-200b, -200c, and -429, which contain a common seed sequence, display 100% seed complementarity to a conserved target site within the IE2 3′ UTR of either 8 nucleotides (hsa-miR-200b and hsa-miR-200c) or 10 nucleotides (hsa-miR-429) (Fig. 1). Moreover, this target site is conserved within all sequenced strains of HCMV and thus is very unlikely to have appeared randomly, suggesting that this site is functionally important. All three miRNAs are attractive candidates for binding to UL122, to prevent its translation (Fig. 1). To test if this hsa-miR-200 cluster indeed targets the UL122 3′ UTR, we utilized luciferase reporter assays. We transfected cells with either nontargeting miRNAs or a synthetic oligonucleotide corresponding to the hsa-miR-200b miRNA. We rationalized that any observed changes in protein expression due to hsa-miR-200b would be indicative of the changes seen in response to the other family members, as hsa-miR-200b has the weakest predicted binding to the 3′ UTR of UL122 among the miR-200 family members (Fig. 1). Each of the aforementioned transfected cell cultures was cotransfected with constructs expressing luciferase with either the wild-type UL122 3′ UTR or a mutant UL122 3′ UTR, as well as negative and positive controls. The mutant UL122 3′ UTR contains four mutated nucleotides within the miRNA seed sequence, such that the miRNA cannot bind. The positive control contains the 3′ UTR of ZEB2, a published target of the hsa-miR-200 family members (28). We found that only the wild-type UL122 3′ UTR showed repression when cotransfected with the hsa-miR-200 overexpression constructs, whereas a nonspecific miRNA did not show similar repressed luciferase expression (Fig. 2). Importantly, we found that the overexpression of hsa-miR-200b failed to repress the mutant UL122 3′ UTR (Fig. 2). Taken together, these findings suggest that the 3′ UTR of UL122 is specifically regulated by hsa-miR-200b within the seed sequence predicted by our algorithm.
FIG 1.
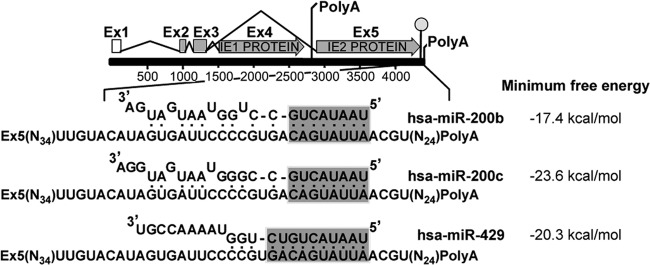
In silico analysis predicts that several cellular hsa-miR-200 family members bind the UL122 3′ UTR. In silico analysis predicts that three members of the cellular hsa-miR-200 family of miRNAs are likely to bind the 3′ UTR of UL122 with perfect sequence complementarity in the seed region (shaded). A schematic of the UL122/UL123 region of HCMV, with the 3′ UTR sequence of UL122 (IE2) magnified, is shown. Predicted hybridizations between the 3′ UTR of UL122 and hsa-miR-200b, hsa-miR-200c, or hsa-miR-429 are shown along with the calculated free energy of the interaction. Ex, exon.
FIG 2.
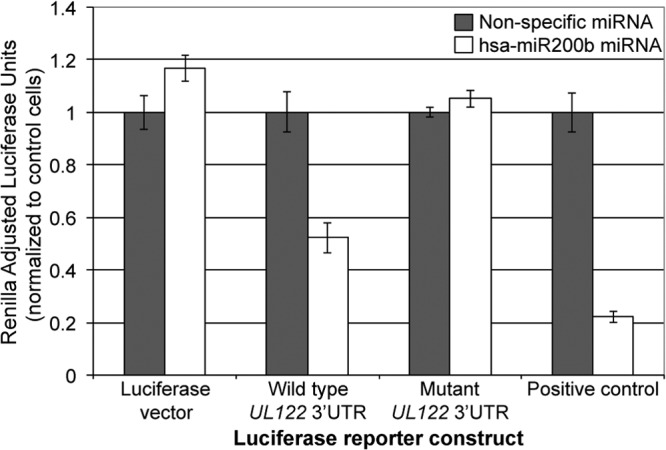
The wild-type 3′ UTR of HCMV UL122 is repressed by overexpression of hsa-miR-200b. 4T07 cells were transfected with either a control synthetic miRNA construct (gray bars) or a synthetic miRNA construct corresponding to miR-200b (white bars). These cells were then cotransfected with firefly luciferase constructs expressing either the wild-type UL122 3′ UTR, a mutant UL122 3′ UTR, a positive control (ZEB2 3′ UTR), or a negative control (empty luciferase vector). All samples were analyzed in triplicate, and the levels were adjusted to those for Renilla luciferase.
Overexpression of the hsa-miR-200 cluster in fibroblasts results in decreased viral titers.
To determine if the targeting of hsa-miR-200 has an effect on viral infection, we constructed viral mutants in the BAC-derived clinical strain FixBACgfp using bacterial recombineering. We mutated the 3′ UTR of IE2 (FixBACgfpIE2cisΔ) in a manner similar to that used for the luciferase reporter constructs used earlier, such that the predicted hsa-miR-200 family members' seed binding sequence was mutated. Fibroblasts are highly permissive for HCMV lytic replication and thus serve as an excellent cell type with which to investigate the role of miRNA targeting of IE2. To this end, we stably transduced primary fibroblasts with lentiviral constructs that overexpress the C1 hsa-miR-200 cluster or a control lentivirus. We observed an increase in hsa-miR-200b expression within the C1-transduced cells compared to the level of expression in cells transduced with an empty lentiviral construct (Fig. 3A). We then infected these cells with either wild-type virus or the FixBACgfpIE2cisΔ mutant. We found that wild-type infection resulted in decreased virus production in the hsa-miR-200-overexpressing fibroblasts compared to the level of production in the control fibroblasts (Fig. 3B). However, the FixBACgfpIE2cisΔ mutant displayed a similar viral growth phenotype in both control fibroblasts and the hsa-miR-200-overexpressing cells (Fig. 3C). We noted that the total titer of the FixBACgfpIE2cisΔ virus was lower than that of the wild-type virus in the control cells, suggesting that the conserved sequences in the UL122 (IE2) 3′ UTR found in all sequenced strains are likely important during lytic replication. To determine if the levels of IE2 are modulated in the context of a viral infection, we performed immunoblot analysis on these infected cells. Consistent with the results presented above, we found that hsa-miR-200-overexpressing fibroblasts infected with wild-type virus, which contains an intact miRNA-binding site, showed decreased IE2 levels compared to those in the FixBACgfpIE2cisΔ mutant virus (Fig. 3D). Taken together, these findings suggest that in the context of lytic viral infection, hsa-miR-200 family members inhibit viral replication via the 3′ UTR of UL122, resulting in decreased levels of viral IE2.
FIG 3.
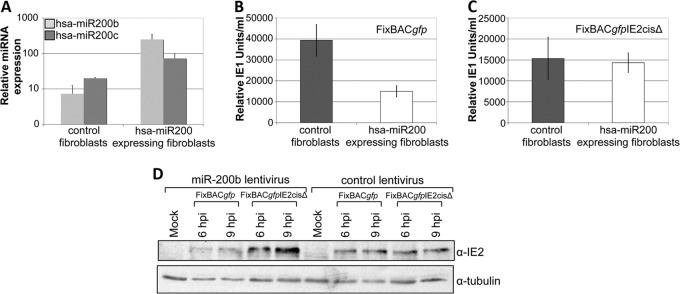
Cellular hsa-miR-200 inhibits wild-type infection but not infection with an UL122 (IE2) 3′ UTR mutant virus. (A) Primary human embryonic lung fibroblasts (MRC5 cells) were stably transduced with either a control retrovirus or one that overexpressed the C1 cluster of the hsa-miR-200 family. In each cell type, levels of hsa-miR-200b (light gray bars) or hsa-miR-200c (dark gray bars) were assessed by qPCR. Samples were normalized to those of cellular RNU44 and analyzed in triplicate. (B, C) MRC5 cells stably transduced with a C1-expressing lentivirus (white bars) or an empty control (gray bars) were then infected with either wild-type FixBACgfp virus (B) or FixBACgfpIE2cisΔ virus (C) at a multiplicity of 0.5 PFU/cell for 4 days. The titer of cell-free virus was then determined by a modified immunofluorescence assay for IE1. Samples were analyzed in triplicate. (D) MRC5 cells transduced with either the C1-expressing or control lentivirus were infected with either wild-type FixBACgfp virus or FixBACgfpIE2cisΔ virus at a multiplicity of 1 PFU/cell. Cell lysates were harvested at the indicated time points (hpi, hours postinfection), and IE2 levels were assessed using a monoclonal antibody (clone 3A9). α-Tubulin was used as a control.
IE2cisΔ infection of Kasumi-3 cells favors lytic replication during experimental in vitro latency.
Our data suggest that the hsa-miR-200 cluster targets the 3′ UTR of UL122 as a means by which to suppress lytic replication. Thus, we hypothesized that this cellular miRNA family might function during latency to maintain the latent rather than the lytic state of the virus. To investigate the function of these miRNAs during latency, we generated a mutation within the 3′ UTR of UL122 (IE2) within the TB40/E strain of HCMV, a strain that we have used in our previous studies of HCMV latency (25). Using our Kasumi-3 cell model for in vitro HCMV latent infection, we asked if infection with the TB40/EgfpIE2cisΔ mutant could maintain a latent infection. We infected Kasumi-3 cells with either wild-type or recombinant viruses. Interestingly, we found that when we infected Kasumi-3 cells with the TB40/EgfpIE2cisΔ mutant, viral IE gene expression was not silenced like it was in the wild-type infections at 7 dpi (Fig. 4A). Consistent with this finding, the TB40/EgfpIE2cisΔ mutant infection resulted in the production of extracellular virions at a level which was significantly increased over that in wild-type infections (Fig. 4B). To ensure that these observations were not due to off-site mutations in the recombineered TB40/EgfpIE2cisΔ mutant, we generated a repair recombinant derived from the mutant BAC parent, TB40/EgfpIE2cisΔrep. As demonstrated in Fig. 4, infection of Kasumi-3 cells with the TB40/EgfpIE2cisΔrep virus resulted in phenotypes consistent with those obtained with the wild-type infections. To ensure that these findings were not phenomena of the Kasumi-3 progenitor cell line, we isolated and infected human CD34+ cells derived from cord blood. This system is established and accepted as a suitable ex vivo model for HCMV latency (32). At 5 dpi, we harvested the cells to determine viral gene transcription and also harvested the supernatant to evaluate the production of extracellular virus. We found that infection of these primary cells with the TB40/EgfpIE2cisΔ mutant virus resulted in a significant increase in IE gene transcript levels compared to those obtained by either wild-type or TB40/EgfpIE2cisΔrep virus infection at 5 dpi (Fig. 5A). Additionally, we were able to quantitate the extracellular viral genomes in the supernatant of TB40/EgfpIE2cisΔ mutant virus-infected CD34+ cells, and the levels were significantly higher than those produced from cells infected with either the wild-type or TB40/EgfpIE2cisΔrep virus (Fig. 5B). Importantly, the phenotypes that we observed in the infected ex vivo CD34+ progenitor cells were consistent with our findings for infected Kasumi-3 cells presented in Fig. 4. Together, these data suggest that the TB40/EgfpIE2cisΔ mutant virus favors a lytic infection rather than a latent infection in the in vitro model system as well as the human umbilical cord-derived CD34+ ex vivo system.
FIG 4.
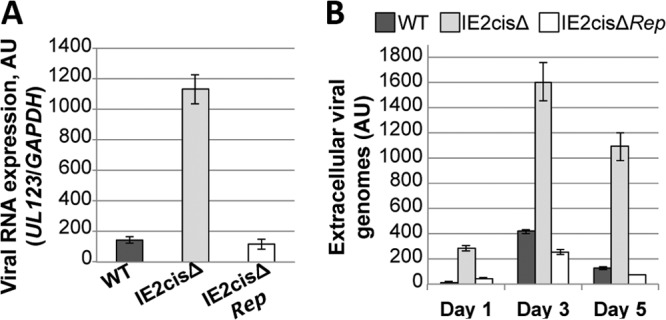
Infection of Kasumi-3 cells with virus lacking the miRNA-binding site in the UL122 (IE2) 3′ UTR favors lytic replication. Kasumi-3 cells were infected with wild-type TB40/Egfp (WT), TB40/EgfpIE2cisΔ (IE2cisΔ), or TB40/EgfpIE2cisΔrep (IE2cisΔRep) virus. (A) Viral gene expression was assessed at 7 dpi by RT-qPCR with primers directed at UL123. All samples were analyzed in triplicate, and levels were normalized to the level of GAPDH gene production. (B) Extracellular virion production was assessed over a 5-day time course. Viral genomes derived from the supernatants of the infected cells were analyzed by qPCR using primers that detect UL123. All samples were analyzed in triplicate. AU, arbitrary units.
FIG 5.
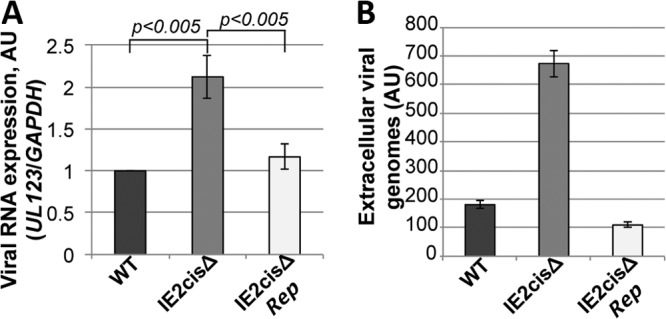
Infection of ex vivo-cultured primary CD34+ cells with a virus that lacks the miRNA seed binding sequence favors lytic gene expression. Primary human CD34+ hematopoietic progenitor cells were isolated from umbilical cord blood by magnetic separation. The cells were then infected with wild-type TB40/Egfp (WT), TB40/EgfpIE2cisΔ (IE2cisΔ), or TB40/EgfpIE2cisΔrep (IE2cisΔRep) viruses at a multiplicity of 2.0 PFU/cell, and then the cells were harvested at 5 dpi for IE gene transcription by RT-qPCR analysis (A) or quantification of extracellular genomes by qPCR (B). Primers directed at UL123 were used to detect IE gene transcripts as well as viral genomes. The levels of the viral transcripts in panel A were normalized to the level of the cellular GAPDH gene. All samples were analyzed in triplicate.
The hsa-miR-200 cluster is expressed in cells that permit HCMV latent infection.
Our findings suggest that the hsa-miR-200 cluster targets the UL122 3′ UTR to aid in HCMV latent infection. Such a function would require the hsa-miR-200 cluster to be appropriately expressed in the cells of the myeloid lineage, specifically, the less differentiated myeloid progenitors that support HCMV latency. Cellular differentiation, in addition to a role in immune insult, plays an important role in viral reactivation (38). Thus, we hypothesized that the hsa-miR-200 cluster is highly expressed in undifferentiated cells, where the virus favors a latent state, and as the cells differentiate, the expression level of this cluster decreases. To test this, we assessed the levels of the hsa-miR-200 cluster in untreated and tetradecanoyl phorbol acetate (TPA)-treated Kasumi-3 cells by RT-qPCR. The treatment of Kasumi-3 cells with TPA differentiates these progenitor cells toward the monocyte/macrophage lineage (39). We found that the hsa-miR-200 cluster was expressed to high levels in untreated Kasumi-3 cells and that upon TPA-induced differentiation, the levels of expression significantly decreased (Fig. 6A). We also determined the expression levels of the hsa-miR-200 cluster in primary CD34+ cells, monocytes, and monocyte-derived macrophages. Consistent with our findings in the Kasumi-3 cells, we found that hsa-miR-200 levels were high in CD34+ cells and monocytes but not in macrophages (Fig. 6B). These results are consistent with our hypothesis that hsa-miR-200 levels are high in cells where the virus favors latency and the levels decrease as the cells differentiate into cells permissive for lytic infection.
FIG 6.
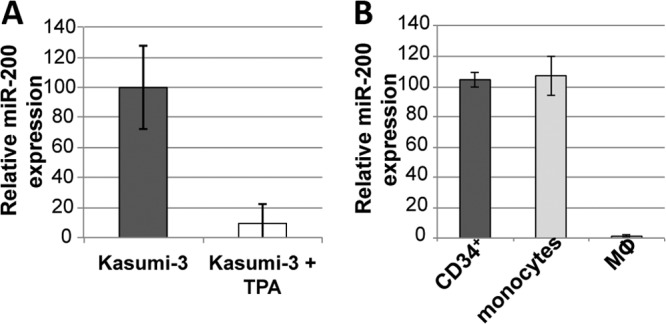
The cellular hsa-miR-200 miRNA family is highly expressed in cells that support HCMV latent infection. hsa-miR-200 levels were detected by qPCR in Kasumi-3 cells (dark gray bar) or TPA-differentiated Kasumi-3 cells (white bar) (A) or in primary human umbilical cord CD34+ cells (dark gray bar), primary human monocytes (light gray bar), or primary human monocyte-derived macrophages (Mϕ; white bar) (B). Samples were analyzed in triplicate, and the levels were normalized to the values for human RNU44.
DISCUSSION
In the present study, we have shown that several of the hsa-miR-200 miRNA family members target the 3′ UTR of UL122 (IE2) to aid in a successful latent infection in vitro. This is one of only a few reports of a cellular miRNA that targets a herpesvirus transcript and the only report of cellular miRNAs regulating HCMV latency. The cellular miR-17 family members have been shown to target the Epstein-Barr virus (EBV) latent transcripts LMP1 and BHRF1 (40), although the functional consequences of this targeting during latency remain unknown. However, since the main targets are the latency transcripts, these interactions would presumably promote a reactivation from latency. Additionally, cellular miR-498 and miR-320d target the Kaposi's sarcoma-associated herpesvirus (KSHV) ORF50/Rta, which promotes the latency-to-lytic replication switch, and overexpression of these miRNAs in BCBL-1 cells, which harbor latent KSHV, results in a decrease of lytic reactivation in the presence of TPA (41). Finally, Ellis-Connell and colleagues showed that hsa-miR-200b and hsa-miR-429, two of the five miRNAs that encompass the hsa-miR-200 family, downregulate cellular ZEB1 and ZEB2 expression, thereby facilitating EBV lytic replication (42). The data that we have presented herein lead us to favor the hypothesis that miRNA targeting of HCMV IE genes is a secondary or backup mechanism (in addition to chromatin remodeling as a means to ensure latency) that the virus has evolved to support viral latency in hematopoietic progenitor cells (Fig. 7).
FIG 7.
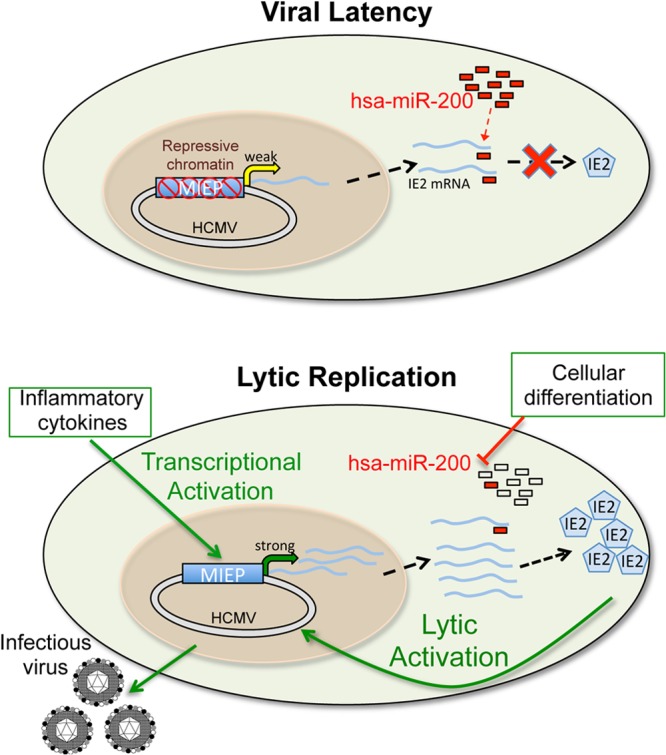
Proposed model of host miRNA control in viral latency. Upon infection of hematopoietic progenitor cells, the HCMV MIEP is marked with repressive chromatin, thereby transcriptionally silencing the promoter. UL122 transcripts generated as a result of low-level basal transcription from this promoter are then repressed by the hsa-miR-200 family of miRNAs to inhibit IE2 translation. Together, these processes ensure viral latency in these cells. However, under conditions during which inflammatory cytokine production is increased, the MIEP is transcriptionally activated and UL122 transcripts accumulate to levels that supersede hsa-miR-200 suppression. Additionally, as the cells differentiate toward mature myeloid cells (e.g., macrophages), the levels of the cellular hsa-miR-200 family decrease such that they are not sufficient for repressing UL122 transcription. As a result, IE2 is translated and lytic reactivation commences.
During herpesvirus latent infection, repressive chromatin modifications are quickly deposited on viral genomes (12, 43, 44). This results in the efficient suppression of viral lytic transcription. Strong repression of the HCMV MIEP, which drives expression of the viral lytic transcriptional activators IE1 and IE2, is necessary to establish latency. If IE1 and IE2 accumulate over a threshold, the viral transcriptional repression is overcome and the switch to lytic replication occurs. The MIEP is a very strong promoter, and thus, low levels of the IE mRNAs are often detected in ex vivo latency model systems (32, 45). We propose that HCMV has evolved a second layer of regulation employing miRNAs that target the IE transcripts to ensure the inhibition of their translation. This model is supported by the general idea that miRNAs function to buffer transcriptional noise, especially in expression of proteins with positive-feedback regulation. Random fluctuations in transcriptional noise can increase the levels of a positive-feedback transcription factor (such as IE2) over a threshold level that results in a loss of regulatory control. These liable systems require additional mechanisms to regulate the oscillation between on and off states. The preexpression of miRNAs would buffer against random inherent noise that would push a biological switch into an all-or-nothing scenario. Reduction of the miRNAs or increased levels of the transcripts serve as the biological switch between the two states (46).
In support of this model, we and others have previously shown that the HCMV miR-UL112-1 targets the 3′ UTR of UL123 (21, 22). Additionally, we observed that infection of Kasumi-3 cells with an UL123 3′ UTR mutant virus (IE1cisΔ) results in IE gene transcription, suggesting that this viral miRNA targets IE1 to aid in the latent infection as well. Utilizing both viral (hcmv-miR-112-1) and cellular (hsa-miR-200) miRNAs, transcripts are mopped up, thereby preventing their translation and ensuring that lytic reactivation does not occur.
How does HCMV overcome miRNA repression to successfully reactivate? HCMV reactivation is thought to involve immune insult and/or cellular differentiation of progenitor cells to more mature cells of the hematopoietic compartment. Once the MIEP is activated, additional sites within the viral genome are activated, eventually leading to lytic reactivation. Our data suggest that the hsa-miR-200 cluster is more highly expressed in cells that favor a latent (CD34+ cells) or persistent (monocytes) infection than in cells that support lytic replication (macrophages). Therefore, as the cells differentiate, the abundance of the transcripts resulting from this strong MIEP outcompete the cellular (and likely the viral) miRNAs, especially as differentiation continues and the levels of hsa-miR-200 decrease. The cellular miRNAs likely cannot keep up with the abundance of transcripts and thus are unable to efficiently repress their translation (Fig. 7). Attempts to deplete the endogenous pool of hsa-miR-200 family members with lentivirus-based miRNA sponge constructs were not successful, as primary cells and undifferentiated Kasumi-3 cells quickly repressed gene expression from transduced vectors.
That HCMV latency is controlled, in part, by miRNA translational repression is not without precedent. Other herpesviruses encode miRNAs expressed during latency, including herpes simplex virus 1 (HSV-1), HSV-2, KSHV, and EBV, which function to promote latent infections (47–58). As mentioned above, cellular miRNAs target both KSHV and EBV transcripts toward regulating latency and/or lytic reactivation. It is not surprising that herpesviruses have evolved mechanisms to suppress IE gene transcription during latency. All herpesviruses transcribe their lytically expressed genes in a cascade, such that the IE genes are made first and then subsequently activate early genes that facilitate DNA replication, which finally leads to the transcription of late genes and the assembly of mature viral particles. For HCMV, we have shown that IE2 alone is in fact sufficient for turning on early gene transcription (59), and thus, regulating translation of this protein during latency would be key for dictating the fate of the infection (i.e., lytic versus latent). Therefore, miRNA repression of HCMV IE genes would serve as a means by which to prevent the kick start of the lytic gene transcriptional cascade that leads to viral reactivation. Thus, miRNAs are an attractive way to ensure the establishment and/or maintenance of a successful latent infection.
ACKNOWLEDGMENTS
We thank T. Shenk and A. Levine for helpful discussions and support, C. Sinzger for the TB40/E BAC construct, Yibin Kang for the miRNA expression constructs, and R. Kalejta and A. Yurochko for technical support.
This study was supported by grants from the National Institute of Allergy and Infectious Diseases (grant no. RAI101080A to E.A.M.), the American Cancer Society (grant no. PF-10-164-01-MPC to C.M.O.), and the Swiss National Science Foundation (grant no. 200021-124936 to J.V.).
Footnotes
Published ahead of print 5 March 2014
REFERENCES
- 1.Britt W. 2008. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr. Top. Microbiol. Immunol. 325:417–470. 10.1007/978-3-540-77349-8_23 [DOI] [PubMed] [Google Scholar]
- 2.Zaia JA. 1990. Epidemiology and pathogenesis of cytomegalovirus disease. Semin. Hematol. 27:5–10 [PubMed] [Google Scholar]
- 3.Bego MG, Keyes LR, Maciejewski J, St Jeor SC. 2011. Human cytomegalovirus latency-associated protein LUNA is expressed during HCMV infections in vivo. Arch. Virol. 156:1847–1851. 10.1007/s00705-011-1027-7 [DOI] [PubMed] [Google Scholar]
- 4.Beisser PS, Laurent L, Virelizier JL, Michelson S. 2001. Human cytomegalovirus chemokine receptor gene US28 is transcribed in latently infected THP-1 monocytes. J. Virol. 75:5949–5957. 10.1128/JVI.75.13.5949-5957.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kondo K, Mocarski ES. 1995. Cytomegalovirus latency and latency-specific transcription in hematopoietic progenitors. Scand. J. Infect. Dis. Suppl. 99:63–67 [PubMed] [Google Scholar]
- 6.Goodrum F, Reeves M, Sinclair J, High K, Shenk T. 2007. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 110:937–945. 10.1182/blood-2007-01-070078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jenkins C, Abendroth A, Slobedman B. 2004. A novel viral transcript with homology to human interleukin-10 is expressed during latent human cytomegalovirus infection. J. Virol. 78:1440–1447. 10.1128/JVI.78.3.1440-1447.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meier JL. 2001. Reactivation of the human cytomegalovirus major immediate-early regulatory region and viral replication in embryonal NTera2 cells: role of trichostatin A, retinoic acid, and deletion of the 21-base-pair repeats and modulator. J. Virol. 75:1581–1593. 10.1128/JVI.75.4.1581-1593.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murphy JC, Fischle W, Verdin E, Sinclair JH. 2002. Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J. 21:1112–1120. 10.1093/emboj/21.5.1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ioudinkova E, Arcangeletti MC, Rynditch A, De Conto F, Motta F, Covan S, Pinardi F, Razin SV, Chezzi C. 2006. Control of human cytomegalovirus gene expression by differential histone modifications during lytic and latent infection of a monocytic cell line. Gene 384:120–128. 10.1016/j.gene.2006.07.021 [DOI] [PubMed] [Google Scholar]
- 11.Lukashchuk V, McFarlane S, Everett RD, Preston CM. 2008. Human cytomegalovirus protein pp71 displaces the chromatin-associated factor ATRX from nuclear domain 10 at early stages of infection. J. Virol. 82:12543–12554. 10.1128/JVI.01215-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reeves MB, MacAry PA, Lehner PJ, Sissons JG, Sinclair JH. 2005. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc. Natl. Acad. Sci. U. S. A. 102:4140–4145. 10.1073/pnas.0408994102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reeves MB, Sinclair JH. 2010. Analysis of latent viral gene expression in natural and experimental latency models of human cytomegalovirus and its correlation with histone modifications at a latent promoter. J. Gen. Virol. 91:599–604. 10.1099/vir.0.015602-0 [DOI] [PubMed] [Google Scholar]
- 14.Saffert R, Penkert R, Kalejta R. 2010. Cellular and viral control over the initial events of human cytomegalovirus experimental latency in CD34+ cells. J. Virol. 84:5594–5604. 10.1128/JVI.00348-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stinski MF, Meier JL. 2007. Immediate-early viral gene regulation and function. In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses. Cambridge University Press, Cambridge, United Kingdom: [PubMed] [Google Scholar]
- 16.Sinclair J. 2008. Human cytomegalovirus: latency and reactivation in the myeloid lineage. J. Clin. Virol. 41:180–185. 10.1016/j.jcv.2007.11.014 [DOI] [PubMed] [Google Scholar]
- 17.Plaisance-Bonstaff K, Renne R. 2011. Viral miRNAs. Methods Mol. Biol. 721:43–66. 10.1007/978-1-61779-037-9_3 [DOI] [PubMed] [Google Scholar]
- 18.Grey F, Nelson J. 2008. Identification and function of human cytomegalovirus microRNAs. J. Clin. Virol. 41:186–191. 10.1016/j.jcv.2007.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pfeffer S, Sewer A, Lagos-Quintana M, Sheridan R, Sander C, Grasser FA, van Dyk LF, Ho CK, Shuman S, Chien M, Russo JJ, Ju J, Randall G, Lindenbach BD, Rice CM, Simon V, Ho DD, Zavolan M, Tuschl T. 2005. Identification of microRNAs of the herpesvirus family. Nat. Methods 2:269–276. 10.1038/nmeth746 [DOI] [PubMed] [Google Scholar]
- 20.Dunn W, Trang P, Zhong Q, Yang E, van Belle C, Liu F. 2005. Human cytomegalovirus expresses novel microRNAs during productive viral infection. Cell. Microbiol. 7:1684–1695. 10.1111/j.1462-5822.2005.00598.x [DOI] [PubMed] [Google Scholar]
- 21.Grey F, Meyers H, White EA, Spector DH, Nelson J. 2007. A human cytomegalovirus-encoded microRNA regulates expression of multiple viral genes involved in replication. PLoS Pathog. 3:e163. 10.1371/journal.ppat.0030163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murphy E, Vanicek J, Robins H, Shenk T, Levine AJ. 2008. Suppression of immediate-early viral gene expression by herpesvirus-coded microRNAs: implications for latency. Proc. Natl. Acad. Sci. U. S. A. 105:5453–5458. 10.1073/pnas.0711910105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee SH, Kalejta RF, Kerry J, Semmes OJ, O'Connor CM, Khan Z, Garcia BA, Shenk T, Murphy E. 2012. BclAF1 restriction factor is neutralized by proteasomal degradation and microRNA repression during human cytomegalovirus infection. Proc. Natl. Acad. Sci. U. S. A. 109:9575–9580. 10.1073/pnas.1207496109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nachmani D, Stern-Ginossar N, Sarid R, Mandelboim O. 2009. Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe 5:376–385. 10.1016/j.chom.2009.03.003 [DOI] [PubMed] [Google Scholar]
- 25.O'Connor CM, Murphy EA. 2012. A myeloid progenitor cell line capable of supporting human cytomegalovirus latency and reactivation resulting in infectious progeny. J. Virol. 86:9854–9865. 10.1128/JVI.01278-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marin RM, Vanicek J. 2012. Optimal use of conservation and accessibility filters in microRNA target prediction. PLoS One 7:e32208. 10.1371/journal.pone.0032208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rehmsmeier M, Steffen P, Hochsmann M, Giegerich R. 2004. Fast and effective prediction of microRNA/target duplexes. RNA 10:1507–1517. 10.1261/rna.5248604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korpal M, Lee ES, Hu G, Kang Y. 2008. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 283:14910–14914. 10.1074/jbc.C800074200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korpal M, Ell BJ, Buffa FM, Ibrahim T, Blanco MA, Celia-Terrassa T, Mercatali L, Khan Z, Goodarzi H, Hua Y, Wei Y, Hu G, Garcia BA, Ragoussis J, Amadori D, Harris AL, Kang Y. 2011. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat. Med. 17:1101–1108. 10.1038/nm.2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Copeland NG, Jenkins NA, Court DL. 2001. Recombineering: a powerful new tool for mouse functional genomics. Nat. Rev. Genet. 2:769–779. 10.1038/35093556 [DOI] [PubMed] [Google Scholar]
- 31.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol. Biol. 634:421–430. 10.1007/978-1-60761-652-8_30 [DOI] [PubMed] [Google Scholar]
- 32.Goodrum FD, Jordan CT, High K, Shenk T. 2002. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: a model for latency. Proc. Natl. Acad. Sci. U. S. A. 99:16255–16260. 10.1073/pnas.252630899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan G, Nogalski MT, Yurochko AD. 2012. Human cytomegalovirus stimulates monocyte-to-macrophage differentiation via the temporal regulation of caspase 3. J. Virol. 86:10714–10723. 10.1128/JVI.07129-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Terhune S, Torigoi E, Moorman N, Silva M, Qian Z, Shenk T, Yu D. 2007. Human cytomegalovirus UL38 protein blocks apoptosis. J. Virol. 81:3109–3123. 10.1128/JVI.02124-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu H, Shen Y, Shenk T. 1995. Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J. Virol. 69:7960–7970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao KQ, Livak KJ, Guegler KJ. 2005. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 33:e179. 10.1093/nar/gni178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nevels M, Paulus C, Shenk T. 2004. Human cytomegalovirus immediate-early 1 protein facilitates viral replication by antagonizing histone deacetylation. Proc. Natl. Acad. Sci. U. S. A. 101:17234–17239. 10.1073/pnas.0407933101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weinshenker BG, Wilton S, Rice GP. 1988. Phorbol ester-induced differentiation permits productive human cytomegalovirus infection in a monocytic cell line. J. Immunol. 140:1625–1631 [PubMed] [Google Scholar]
- 39.Asou H, Suzukawa K, Kita K, Nakase K, Ueda H, Morishita K, Kamada N. 1996. Establishment of an undifferentiated leukemia cell line (Kasumi-3) with t(3;7)(q27;q22) and activation of the EVI1 gene. Jpn. J. Cancer Res. 87:269–274. 10.1111/j.1349-7006.1996.tb00216.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Riley KJ, Rabinowitz GS, Yario TA, Luna JM, Darnell RB, Steitz JA. 2012. EBV and human microRNAs co-target oncogenic and apoptotic viral and human genes during latency. EMBO J. 31:2207–2221. 10.1038/emboj.2012.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yan Q, Li W, Tang Q, Yao S, Lv Z, Feng N, Ma X, Bai Z, Zeng Y, Qin D, Lu C. 2013. Cellular microRNAs 498 and 320d regulate herpes simplex virus 1 induction of Kaposi's sarcoma-associated herpesvirus lytic replication by targeting RTA. PLoS One 8:e55832. 10.1371/journal.pone.0055832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ellis-Connell AL, Iempridee T, Xu I, Mertz JE. 2010. Cellular microRNAs 200b and 429 regulate the Epstein-Barr virus switch between latency and lytic replication. J. Virol. 84:10329–10343. 10.1128/JVI.00923-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Amelio AL, Giordani NV, Kubat NJ, O'Neil JE, Bloom DC. 2006. Deacetylation of the herpes simplex virus type 1 latency-associated transcript (LAT) enhancer and a decrease in LAT abundance precede an increase in ICP0 transcriptional permissiveness at early times postexplant. J. Virol. 80:2063–2068. 10.1128/JVI.80.4.2063-2068.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cliffe AR, Garber DA, Knipe DM. 2009. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J. Virol. 83:8182–8190. 10.1128/JVI.00712-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rossetto CC, Tarrant-Elorza M, Pari GS. 2013. Cis and trans acting factors involved in human cytomegalovirus experimental and natural latent infection of CD14 (+) monocytes and CD34 (+) cells. PLoS Pathog. 9:e1003366. 10.1371/journal.ppat.1003366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herranz H, Cohen SM. 2010. MicroRNAs and gene regulatory networks: managing the impact of noise in biological systems. Genes Dev. 24:1339–1344. 10.1101/gad.1937010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bellare P, Ganem D. 2009. Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: an evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe 6:570–575. 10.1016/j.chom.2009.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cai X, Lu S, Zhang Z, Gonzalez CM, Damania B, Cullen BR. 2005. Kaposi's sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc. Natl. Acad. Sci. U. S. A. 102:5570–5575. 10.1073/pnas.0408192102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Conrad NK. 2009. Posttranscriptional gene regulation in Kaposi's sarcoma-associated herpesvirus. Adv. Appl. Microbiol. 68:241–261. 10.1016/S0065-2164(09)01206-4 [DOI] [PubMed] [Google Scholar]
- 50.Gottwein E, Cai X, Cullen BR. 2006. Expression and function of microRNAs encoded by Kaposi's sarcoma-associated herpesvirus. Cold Spring Harbor Symp. Quant. Biol. 71:357–364. 10.1101/sqb.2006.71.004 [DOI] [PubMed] [Google Scholar]
- 51.Liang D, Gao Y, Lin X, He Z, Zhao Q, Deng Q, Lan K. 2011. A human herpesvirus miRNA attenuates interferon signaling and contributes to maintenance of viral latency by targeting IKKepsilon. Cell Res. 21:793–806. 10.1038/cr.2011.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu CC, Li Z, Chu CY, Feng J, Sun R, Rana TM. 2010. MicroRNAs encoded by Kaposi's sarcoma-associated herpesvirus regulate viral life cycle. EMBO Rep. 11:784–790. 10.1038/embor.2010.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu F, Stedman W, Yousef M, Renne R, Lieberman PM. 2010. Epigenetic regulation of Kaposi's sarcoma-associated herpesvirus latency by virus-encoded microRNAs that target Rta and the cellular Rbl2-DNMT pathway. J. Virol. 84:2697–2706. 10.1128/JVI.01997-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Samols MA, Hu J, Skalsky RL, Renne R. 2005. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:9301–9305. 10.1128/JVI.79.14.9301-9305.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang S, Bertke AS, Patel A, Margolis TP, Krause PR. 2011. Herpes simplex virus 2 microRNA miR-H6 is a novel latency-associated transcript-associated microRNA, but reduction of its expression does not influence the establishment of viral latency or the recurrence phenotype. J. Virol. 85:4501–4509. 10.1128/JVI.01997-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tang S, Patel A, Krause PR. 2009. Novel less-abundant viral microRNAs encoded by herpes simplex virus 2 latency-associated transcript and their roles in regulating ICP34.5 and ICP0 mRNAs. J. Virol. 83:1433–1442. 10.1128/JVI.01723-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. 2008. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 454:780–783. 10.1038/nature07103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang H. 2009. Reversal of HIV-1 latency with anti-microRNA inhibitors. Int. J. Biochem. Cell Biol. 41:451–454. 10.1016/j.biocel.2008.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Murphy EA. 2000. Molecular interactions and functions of human cytomegalovirus IE86 protein. Ph.D. thesis The University of Iowa, Iowa City, IA [Google Scholar]