

ABSTRACT
Exogenous HIV-1 matrix protein p17 (p17) deregulates the function of different cells after its N-terminal loop (AT20) binding to the chemokine receptors CXCR1 and CXCR2. One site within AT20 has been recently found to be the major determinant of viral fitness following transmission of simian immunodeficiency virus (SIV) to the human host. Therefore, we sought to determine whether SIV matrix protein (MA) was already capable of interacting with CXCR1 and CXCR2 and mimic p17 biological activities rather than this being a newly acquired function during host adaptation. We show here that SIV MA binds with the same affinity of p17 to CXCR1 and CXCR2 and displays both p17 proangiogenic on human primary endothelial cells and chemotactic activity on human primary monocytes and B cells. However, SIV MA exhibited a higher degree of plasticity than p17 in the C terminus, a region known to play a role in modulating B cell growth. Indeed, in contrast to p17, SIV MA was found to activate the phosphatidylinositol 3-kinase/Akt signaling pathway and strongly promote B cell proliferation and clonogenic activity. Interestingly, we have recently highlighted the existence of a Ugandan HIV-1 strain-derived p17 variant (S75X) with the same B cell growth-promoting activity of SIV MA. Computational modeling allowed us to hypothesize an altered C terminus/core region interaction behind SIV MA and S75X activity. Our findings suggest the appearance of a structural constraint in the p17 C terminus that controls B cell growth, which may help to elucidate the evolutionary trajectory of HIV-1.
IMPORTANCE The HIV-1 matrix protein p17 (p17) deregulates the biological activities of different cells after binding to the chemokine receptors CXCR1 and CXCR2. The p17 functional domain responsible for receptors interaction includes an amino acid which is considered the major determinant of SIV replication in humans. Therefore, we sought to determine whether SIV matrix protein (SIV MA) already had the ability to bind to both chemokine receptors rather than being a function newly acquired during host adaptation. We show here that SIV MA binds to CXCR1 and CXCR2 and fully mimics the p17 proangiogenic and chemokine activity. However, it differs from p17 in its ability to signal into B cells and promote B cell growth and clonogenicity. Computational analysis suggests that the accumulation of mutations in the C-terminal region may have led to a further SIV MA adaptation to the human host. This finding in turn sheds light on the evolutionary trajectory of HIV-1.
INTRODUCTION
The HIV-1 matrix protein p17 (p17) plays a key role in the virus life cycle (1). It is released in the extracellular space from HIV-1-infected cells and is easily detected in the plasma and tissue specimens of patients (2, 3), including those successfully treated with highly active antiretroviral therapy) (4). Extracellularly, p17 has been found to deregulate the biological activities of many different cells that are directly or indirectly involved in AIDS pathogenesis (3, 5–10). All p17 activities occur after interaction between the functional epitope (AT20) located at the N-terminal region (amino acids 11 to 30) of the viral protein with receptors expressed on different target cells (5). Recent studies have described the capability of p17 to exert chemokine (9) and proangiogenic (3) activities. These activities were mediated by p17 binding to CXCR1 and CXCR2, the physiological receptors for interleukin-8 (IL-8) and, indeed, p17 was found to mimic some of the biological activities of this chemokine (3, 9).
Interestingly, we have recently shown that in B cells a p17 variant derived from a Ugandan HIV-1 strain A1, named S75X, differing from the prototype clade B isolate BH10 p17 (10), triggers an activation of the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway. As a consequence, S75X was found to increase B cell proliferation and clonogenicity on soft agar (10), providing the first evidence on the existence of a p17 variant with oncogenic activity on human B cells. This study also showed the role of the C-terminal region in modulating the p17 signaling pathway and oncogenic activity, thereby highlighting the complexity and consequent implications of p17 binding to and signaling through its receptors. This knowledge will be crucial for understanding the contribution of p17 variants to the development of lymphoma in HIV-1-infected patients.
HIV-1 and HIV-2 are the causes of AIDS. These viruses originated from independent cross-species transmissions of simian immunodeficiency viruses (SIVs) infecting chimpanzees and sooty mangabeys (11). Like other viruses, primate lentiviruses such as SIV also had to adapt to their host in order to replicate efficiently (12, 13). Many cellular factors vary among different species (14), and it is obvious that viruses tend to cross more readily between more closely related host species (15, 16). However, even the genomes of closely related species, such as humans and primates, exhibit multiple differences (17, 18), so that adaptation of SIV proteins to the new host are required. Pioneering studies by Wain et al. (19) identified the viral matrix protein as the modulator of viral fitness following transmission to the new human host. Interestingly, this analysis recognized one site in the matrix protein (amino acid 30) that encodes a Met or Leu in SIV but switched to an Arg or Lys in many current pandemic HIV strains, as the major determinant of SIV replication fitness in humans. The requirement of a viral matrix protein for SIV fitness has been further confirmed by the finding that an HIV-1 strain that had been extensively passaged in chimpanzees underwent a revision of this host-specific signature (20), suggesting that the amino acid at position 30 in the viral matrix protein is under strong host-specific selective pressure (21).
The amino acid at position 30 is included in the p17 functional domain responsible for CXCR1 and CXCR2 interaction (3, 9). Due to the importance that the viral matrix protein has in contributing to SIV adaptation to the human host, we sought to determine whether SIV matrix protein (MA) already could interact with both IL-8 receptors and display functional activities rather than this being a newly acquired function during human host adaptation.
We demonstrate here that SIV MA is per se able to bind to both CXCR1 and CXCR2 and to fully mimic the proangiogenic activity of p17. Moreover, similarly to p17, SIV MA displayed chemotactic activity on human primary monocytes and B cells. On the other hand, SIV MA completely differed from p17 in signaling into B cells and resembled the S75X variant in its ability to promote B cell growth and clonogenicity. Integration of computational modeling and site-specific mutagenesis data allowed us to hypothesize an altered interaction between the C-terminal and core region behind SIV MA and S75X oncogenic activity.
MATERIALS AND METHODS
Cell cultures.
The Jurkat leukemic T-cell line, the lymphoblastoid Raji cell line, and the AIDS-related HBL-1 lymphoma cell line were cultured in RPMI 1640 containing 10% fetal bovine serum (FBS) and 1 mM l-glutamine. Human umbilical vein endothelial cells (HUVECs) were isolated and characterized as previously described (3) and cultured in endothelial growth medium (EGM; Lonza) containing 10% FBS. All experiments were carried out with HUVECs at passages 2 to 6.
Human primary monocyte and B cell isolation and cultures.
Blood was collected from healthy donors, who gave informed consent for this research according to the Declaration of Helsinki. Human monocytes were isolated from whole blood as previously described (7). Primary B lymphocytes were isolated by negative selection from peripheral blood mononuclear cells obtained from healthy donors using the B cell isolation kit II from Miltenyi Biotec according to the manufacturer's instructions. Cells were resuspended in adhesion buffer (phosphate-buffered saline [PBS], 1 mM CaCl2, 1 mM MgCl2, 10% FBS; pH 7.2) or plated in complete medium (RPMI 1640 containing 10% FBS and 1 mM l-glutamine). Cells were pretreated, when indicated, with an isotype-matched monoclonal antibody (MAb; Ctrl MAb), a neutralizing MAb to CXCR1 (MAb330; R&D Systems), and/or a neutralizing MAb to CXCR2 (MAb331; R&D Systems).
Recombinant proteins and antibodies.
Purified endotoxin (lipopolysaccharide)-free recombinant HIV-1 matrix protein p17, S75X, and SIV MA (in their monomeric forms) and glutathione S-transferase (GST) were produced as previously described (10). The absence of endotoxin contamination (<0.25 endotoxin units/ml) in protein preparations was assessed by Limulus amebocyte assay (Associates of Cape Cod). SIV MA, p17, and S75X proteins were labeled with fluorescein isothiocyanate (FITC; SIV MA-FITC) and allophycocyanin (APC; p17-APC and S75X-APC) using the Lightning-Link conjugation system (Innova Biosciences).
Surface plasmon resonance (SPR) binding assay.
The interactions of SIV MA or p17 with CXCR1 and CXCR2 were carried out as previously described (3, 9). Briefly, anti-GST antibodies (GE Healthcare) were immobilized onto a Sensor Chip CM5 using standard amine-coupling chemistry. Recombinant human CXCR1 or CXCR2 with a C-terminal GST tag (Alexa) at 10 μg/ml in 50 mM HEPES (pH 7.0) containing 0.01% cholesteryl hemisuccinate Tris salt (CHS), 0.1% CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate}, and 0.33 mM DOPC-DOPS (synthetic phospholipid blend [dioleoyl], 7:3 [wt/wt]; Avanti Polar Lipids) was then injected over the anti-GST surface, allowing the immobilization of 1,600 and 900 RU (0.025 and 0.014 pmol, respectively). A Sensor Chip coated with anti-GST antibodies was used as a negative control and for blank subtraction for both chemokine receptors. The kinetic parameters of the interactions (association [Kon] and dissociation [Koff] rates and the dissociation constant [Kd, derived from the Koff/Kon ratio]) were determined by injecting increasing concentrations of the different proteins in 10 mM HEPES (pH 7.4) containing 150 mM NaCl, 3 mM EDTA, and 0.005% surfactant P20 (HBS-EP) over the various surfaces and then washed until dissociation. The sensorgram overlays obtained were processed using the nonlinear fitting (single site model) software package BIAevaluation 3.2.
In vitro tube formation assay.
Prechilled 48-well culture plates were coated with 150 μl of basement membrane extract (BME; 10 mg/ml; Cultrex; Trevigen) per well, followed by incubation for 30 min at 37°C. HUVECs were nutrient starved for 24 h in endothelial basal medium containing 0.5% FBS for 24 h before they were harvested and resuspended in EGM. Cells were seeded into a BME-coated plate (5 × 104 per well) and treated or not treated with 10 ng of GST, p17, or SIV MA/ml. Starved HUVECs were cocultured, where indicated, with an isotype-matched MAb, a neutralizing MAb to CXCR1 (MAb330), and/or a neutralizing MAb to CXCR2 (MAb331). Wells were then analyzed over time (from 2 to 24 h) for the formation of tubule structure by examination with a Leica DM IRB microscope. The center of each well was digitally photographed with a Hitachi KP-D50 camera, and capillary-like structures were quantified by analyzing the number of tubes per well formed by HUVECs.
Chemotaxis assays.
Monocyte migration was assessed using 3-μm-pore-size transwells (BD Biosciences), while Jurkat cell and B cell migration was evaluated by using 8-μm-pore-size transwells. Cells were suspended at 2 × 106/ml in adhesion buffer. Cell suspension (100 μl) was added to the top well, and medium containing agonists (600 μl) was added to the bottom well. Chemotaxis was performed for 90 min at 37°C, and then the filters were removed. After fixation with 1.5% paraformaldehyde, migrated cells were counted in five high-power fields by light microscopy at ×10 magnification.
siRNA technique.
Nucleoporation of cells was performed using Amaxa Nucleofector (Lonza). Small interfering RNAs (siRNAs) at 100 nM were added to 3 × 106 cells resuspended in 100 μl of nucleofection buffer. CXCR1 and CXCR2 silencing was performed using four distinct siRNAs targeting four different regions of the CXCR1 and CXCR2 receptors (Dharmacon), respectively. Fluorescein-labeled irrelevant siRNAs (scr siRNAs; Invitrogen) were used as a negative control and to evaluate the efficiency of siRNA nucleoporation by flow cytometry. The efficacy of CXCR1 and CXCR2 siRNA was evaluated by real-time PCR analysis and/or flow cytometry.
Real-time PCR for gene expression analysis.
Total RNA was isolated from cells (106) using an RNeasy minikit (Qiagen). After retrotranscription, 50 ng of cDNA was mixed with sterile water and SYBR green qPCR master mix (Promega) and amplified using the following PCR primers (0.2 μM concentrations of each; Primm): CXCR1, 5′-TGG GAA ATG ACA CAG CAA AA-3′ (forward) and 5′-AGT GTA CGC AGG GTG AAT CC-3′ (reverse); CXCR2, 5′-TTG TTG GCT CTT CTT CAG GG-3′ (forward) and 5′-TGA GGC TTG GAA TGT GAC TG-3′ (reverse); and human β-actin, 5′-GGCACCCAGCACAATGAAG-3′ (forward), and 5′-GCTGATCCACATCTGCTGG-3′ (reverse). Quantification of CXCR1 and CXCR2 cDNA was normalized in each reaction according to the internal β-actin control. The results are expressed as a percentage of the control.
Transfections.
Jurkat cells were plated at 3 × 105/ml and, after 24 h, nucleofected with 2 μg of the endotoxin-free plasmids pEGFP-N3, pEGFP-N3-CXCR1, or pEGFP-N3-CXCR2 (kindly provided by Ingrid U. Schraufstatter, Department of Vascular Biology, University of California–San Diego, La Jolla, CA). The vector pEGFP-N3 expressing a red-shifted variant of wild-type green fluorescent protein (GFP) was obtained from Clontech. After transfection, Jurkat cells were incubated for 24 h at 37°C, and the transfection efficiency was evaluated as enhanced GFP (EGFP) expression by flow cytometry. Cell surface expression of CXCR1 or CXCR2 was evaluated by flow cytometry using a MAb to CXCR1 or CXCR2 and APC-conjugated secondary antibody.
SIV MA binding and internalization into target B cells.
Human primary B cells were activated for 7 days with 1 μg of soluble CD40 ligand (Alexis)/ml, 1 μg of the corresponding enhancer (Enzo Life Sciences)/ml, and 10 ng of IL-4 (R&D Systems)/ml. HBL-1 cells and activated B cells were used to investigate SIV MA binding and internalization. Cells were seeded in complete medium at a final concentration of 4 × 105/ml, and 0.5 μg of each single FITC- or APC-conjugated protein/106 cells was added separately to each culture. CXCR1/2 neutralization assays were performed by pretreating activated B cells with 5 μg/ml of an isotype-matched MAb, an MAb to CXCR1, or an MAb to CXCR2 for 1 h at room temperature, before the addition of labeled proteins. After 48 and 96 h of treatment, cells (2 × 106 per sample) were collected, fixed with 2% of paraformaldehyde in RPMI 1640 containing 10% FBS, and labeled with DRAQ5 (Alexis Biochemicals) for SIV MA-FITC cultures or CyTRAK Orange (eBioscience) nuclear dye for p17-APC and S75X-APC cultures. Cells were run on the ImageStream X cytometer using INSPIRE software (Amnis Corp.), and images were analyzed using IDEAS software (Amnis Corp.). The cytoplasmic localization of the proteins was measured using the internalization algorithm of the IDEAS software, defined as the ratio of the intensity inside the cell to the intensity of the entire cell. The inside of the cell is defined by the erosion mask that fits the cell membrane, allowing the possible identification of membrane localization of the proteins. Cells containing small concentrated fluorescent spots have positive scores, whereas cells showing little and diffuse fluorescence have negative scores. For the analysis of the proteins' nuclear internalization, we used an IDEAS software feature, the similarity score (SS) (22), to compare the similarity of the nuclear (DRAQ5/CyTRAK) and labeled-protein staining patterns. All events showing a positive SS were considered with high similarity between proteins and DRAQ5/CyTRAK, thus indicating a nuclear localization of the proteins. Only viable cells were selected on the basis of morphological features.
Western blot analysis.
Cells were treated for 5 min with recombinant p17, S75X, or SIV MA at a concentration of 0.1 μg/ml and then lysed in 200 μl of 10 mM HEPES (pH 7.9), 10 mM KCl, 1.5 mM MgCl2, 0.5 mM EGTA, 0.5 mM EDTA, and 0.6% NP-40 containing a mixture of protease inhibitors (Complete Mini; Roche) and phosphatase inhibitors (sodium vanadate, phenylarsine oxide (PAO), and sodium fluoride). Equal amounts of total proteins were resolved on an 11% sodium dodecyl sulfate-polyacrylamide gel and then electroblotted onto a nitrocellulose membrane. The blots were incubated overnight at 4°C with MAb to pAkt, MAb to pERK1/2, or rabbit polyclonal antibody to ERK1/2 (Santa Cruz Biotechnology). The antigen-antibody complex was detected by incubation of the membranes for 1 h at room temperature with peroxidase-coupled goat anti-rabbit IgG or goat anti-mouse IgG (Thermo Scientific) and revealed using an enhanced chemiluminescence system.
Soft agar anchorage-independent growth assay.
Raji cell clonogenic activity was evaluated in a soft agar assay as previously described (10). Briefly, Raji cells (20,000/well) were plated in 4 ml of 0.35% agarose–5% charcoal-stripped FBS in phenol red-free RPMI 1640 with a 0.7% agarose base in six-well plates. At 2 days after plating, medium containing the proteins as indicated was added to the top of the layer and replaced every 4 days. After 10 days, 300 ml of MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide; Sigma] was added to each well, and the cells were incubated at 37°C for 4 h. The plates were then placed in 4°C overnight, and colonies >50 mm in diameter were counted.
SIV MA structural modeling.
The structural analysis was based on the following input data: for the p17 model, we used UniProtKB locus GAG_HV1N5 (accession number P12493) and the X-ray structure PDB code 1hiw (23), and for SIV MA, we used UniProtKB locus Q03859_SIVCZ (accession number Q03859) and the X-ray structure PDB code 1ed1 (24). For S75X, we used its amino acid primary sequence (25). To understand and find the local interactions of p17 and SIV MA, both proteins were analyzed using BPlus (26). In addition, we used BRAGI (27) to check for all interactions between different residues of p17. The putative effects resulting from differences in the sequences of p17, SIV MA, and S75X were analyzed using SNAPFUN (28). This software predicts the influence of amino acid exchanges on the secondary structure elements. The input for SNAPFUN was generated using the sequence alignment created by Clustal-Omega (26). From this alignment a list of “mutations” was extracted. For the comparison of p17 and SIV MA, the C-terminal part was not included due to the uncertainty of the alignment. The Clustal-Omega alignment was used to model S75X based on p17 using BRAGI (27) in order to get a raw three-dimensional model of S75X. Since many of these exchanges are on the accessible surface of the protein, the model was refined by hand using the mutagenesis wizard of PYMOL (PyMOL). Most of the modified side chains were arranged to maintain the original network of H bonds and salt bridges.
Statistical analysis.
Data obtained from multiple independent experiments are expressed as the means ± the standard deviations (SD). The data were analyzed for statistical significance using a Student two-tailed t test and one-way or two-way analysis of variance (ANOVA) when appropriate. Bonferroni's post test was used to compare data. Differences were considered significant at P < 0.05. Statistical tests were performed using Prism 5 software (GraphPad).
RESULTS
SPR analyses of the interaction of SIV MA and p17 with CXCR1 and CXCR2.
SPR was exploited to compare the capacity of SIV MA and p17 to interact with the known p17 receptors CXCR1 (9) and CXCR2 (3). As reported in Table 1, SIV MA and p17 interacted with CXCR1 with similar affinities (Kd = 2,210 and 1,538 nM, respectively) that are, however, about 200 times lower than that of IL-8 (29), the physiologic CXCR1 ligand. This difference was mainly due to the very rapid dissociation rate of the two viral proteins compared to IL-8. On the other hand, as already evidenced for p17 (3), the interaction of SIV MA with CXCR2 occurred with higher affinity (Table 1), only 2 to 7 times lower than that of IL-8 (29).
TABLE 1.
Binding parameters of the interactions of SIV MA and p17 with CXCR1 and CXCR2
| Protein | Mean binding ± SDa |
|||||
|---|---|---|---|---|---|---|
| CXCR1 |
CXCR2 |
|||||
| Kon (M−1 s−1) | Koff (s−1) | Kd (nM) | Kon (M−1 s−1) | Koff (s−1) | Kd (nM) | |
| SIV MA | (1.9 ± 0.5) × 105 | (4.2 ± 0.9) × 10−1 | 2,210 ± 723 | (2.0 ± 1.8) × 101 | (9.9 ± 0.05) × 10−6 | 495 ± 174 |
| p17 | (1.3 ± 0.4) × 105 | (2.0 ± 0.5) × 10−1 | 1,538 ± 158 | (8.5 ± 0.1) × 104 | (1.0 ± 0.03) × 10−2 | 118 ± 5.3 |
The association rate (Kon) and the dissociation rate (Koff) are reported. The dissociation constant (Kd) was derived from the Koff/Kon ratio. The results are means of three independent experiments with similar results. The Kd values were also calculated independently from binding kinetics by performing a Scatchard plot analysis of the equilibrium binding data.
SIV MA induces capillary-like structures by binding to CXCR1 and CXCR2.
We recently demonstrated that p17 was able to promote capillary-like structure formation in vitro (3). To determine a possible role of SIV MA in angiogenesis, we investigated the capability of SIV MA to promote capillary-like structures formation in vitro. In the presence of SIV MA, HUVECs formed a consistent network of tube-like structures much earlier than control cultures. Dose-response experiments showed that SIV MA exerted its angiogenic activity at a concentration as low as 2.5 ng/ml, reaching a peak of angiogenic potency at a protein concentration of 10 ng/ml. At this SIV MA concentration, HUVECs formed tube-like structures within 8 h of cell culture (data not shown). As shown in Fig. 1A, the angiogenic activity of SIV MA was similar to that exerted by p17, whereas the irrelevant protein GST did not induce any capillary-like structure formation. Since the angiogenic response of HUVECs to p17 is mediated by both CXCR1 and CXCR2 (3), which are known to coordinate cytoskeletal rearrangement necessary for angiogenic response (30), we determined the involvement of both receptors in SIV MA-induced tube formation. As shown in Fig. 1B, after 8 h of culture, HUVECs formed capillary-like structures in medium containing SIV MA or SIV MA plus the isotype-matched MAb, but not in the presence of the neutralizing MAb to CXCR1 or CXCR2. Blocking of both receptors by using a combination of the two MAbs resulted in an almost complete interference of SIV MA-induced capillary-like structure formation. Our data demonstrate that both CXCR1 and CXCR2 are involved in SIV MA-induced capillary-like structures formation and probably they cooperate in promoting this activity.
FIG 1.

SIV MA tube formation is mediated by CXCR1 and CXCR2. (A) Capillary-like structure formation of HUVECs is shown in response to the indicated treatments. HUVECs were seeded on BME and stimulated at 37°C with PBS (not treated cells [NT]) or 10 ng of GST, p17, or SIV MA/ml. (B) SIV MA-induced capillary-like structure formation is mediated specifically by both CXCR1 and CXCR2. HUVECs were stimulated at 37°C with PBS (NT) or SIV MA (10 ng/ml) after preincubation of viral protein with 2.5 μg/ml of a neutralizing MAb to CXCR1, a neutralizing MAb to CXCR2, a combination of MAb to CXCR1 and CXCR2, or a control isotype-matched MAb (Ctrl MAb). Pictures were taken after 8 h of culture on BME (original magnification, ×10) and are representative of three independent experiments with similar results. Values reported for HUVEC tube formation are means ± the SD of three independent experiments. Statistical analysis was performed by one-way ANOVA, and a Bonferroni's post test was used to compare data (***, P < 0.001).
SIV MA exerts a chemokine-like activity on human monocytes.
We recently demonstrated that p17 exerts IL-8-like chemokine activity on human primary monocytes (9). To determine a possible role of SIV MA as a chemokine-like protein, we performed monocyte migration assay in response to the viral protein. Figure 2A shows that SIV MA was able to induce monocyte migration in a transwell chemotaxis assay. The viral protein exhibited a statistically significant chemoattraction of monocytes compared to control untreated cells (NT). As expected, a strong induction of monocyte migration was obtained using p17 as a reference chemoattractant, whereas migration was not promoted by GST, which was used as an irrelevant protein.
FIG 2.

SIV MA-induced monocyte migration is mediated specifically by CXCR1. (A) Transwell migration assay of monocytes in response to PBS (NT), GST, p17, or SIV MA. Cells were stimulated for 90 min at 37°C with proteins at a concentration of 1 μg/ml. Bars represent the means ± the SD of three independent experiments performed in duplicate. Statistical analysis was performed by one-way ANOVA, and a Bonferroni's post test was used to compare data (***, P < 0.001). (B) Transwell migration assay of monocytes in response to the indicated treatments. Monocytes were stimulated for 90 min at 37°C with PBS (NT) or pretreated for 1 h at 37°C with 50 μg of MAb to CXCR1, MAb to CXCR2, or Ctrl MAb/ml and then stimulated for 90 min at 37°C with SIV MA (1 μg/ml). Bars represent the means ± the SD of three independent experiments performed in duplicate. Statistical analysis was performed by one-way ANOVA, and a Bonferroni's post test was used to compare data (***, P < 0.001). (C) Analysis of CXCR1 gene expression performed by using quantitative real-time PCR. Monocytes were nucleoporated with scr siRNAs used as a negative control or with SMARTpool siRNAs specific for four distinct regions of CXCR1. Analysis of real-time PCR data was performed using the 2−ΔΔCT method and relative quantitation study software. Quantification of CXCR1 mRNA was normalized in each reaction according to the internal β-actin control. Bars represent the means ± the SD of three independent experiments performed in triplicate. (D) Effect of CXCR1 silencing on surface receptor expression. Monocytes nucleofected with scr siRNA or CXCR1 siRNA were incubated with isotype control antibody (solid histogram) or MAb to CXCR1 (open histogram), stained with APC-conjugated secondary antibody, and analyzed by flow cytometry. (E) Transwell migration assay of monocytes nucleoporated with CXCR1 siRNAs or with scr siRNAs in response to the indicated treatments. Bars represent the means ± the SD of three independent experiments performed in duplicate. Statistical analysis was performed by paired two-tailed Student t test (***, P < 0.001). (F) CXCR1 expression in Jurkat cells is sufficient to mediate chemotaxis in response to SIV MA. Transwell migration assay of Jurkat cells transfected with pEGFP-N3 or pEGFP-N3-CXCR1 in response to the indicated treatments. Bars represent the means ± the SD of three independent experiments performed in duplicate. Statistical analysis was performed by one-way ANOVA, and a Bonferroni's post test was used to compare data (**, P < 0.01; ***, P < 0.001).
p17 exerts chemokine activity by interacting with CXCR1 expressed on human monocytes, so we investigated whether monocyte migration triggered by SIV MA could be ascribed to its interaction with CXCR1 and/or CXCR2, since both receptors are capable of mediating cell migration (31). We performed experiments with monocytes pretreated for 1 h at 37°C with neutralizing MAbs to CXCR1 and CXCR2 or with a control isotype-matched MAb. As shown in Fig. 2B, MAb to CXCR1 suppressed SIV MA chemotaxis to control levels. This finding is in agreement with the evidence that CXCR1 is the major factor responsible for the chemotactic response of cells to IL-8 (32). No significant interference with SIV MA activity was observed in monocytes pretreated with the control MAb or with neutralizing MAb to CXCR2. This result demonstrates that the SIV MA chemokine activity is specifically mediated by CXCR1.
To further assess the involvement of CXCR1 in SIV MA-induced cell migration, receptor expression on primary monocytes was suppressed by silencing, using Amaxa nucleofection technology to deliver CXCR1 siRNAs. The efficiency of nucleofection was determined by flow cytometric analysis and showed an average of 80% ± 5% nucleoporated monocytes (data not shown). The effects of siRNA on CXCR1 expression were evaluated by real-time PCR analysis and flow cytometry. As shown in Fig. 2C, ca. 73% inhibition of CXCR1 transcripts was observed in monocytes 24 h after nucleofection with CXCR1 siRNAs compared to monocytes nucleofected with scr siRNA. As expected, the expression of CXCR1 on monocytes nucleofected with CXCR1 siRNA was strongly impaired compared to cells nucleofected with scr siRNA (Fig. 2D). The biologic consequence of CXCR1 silencing was evident in monocytes collected 24 h after nucleofection, which showed a significant inhibition in SIV MA-induced migration of CXCR1-silenced monocytes compared to monocytes nucleofected with scr siRNAs (Fig. 2E).
We also investigated whether the de novo expression of CXCR1 was responsible for SIV MA chemotactic activity. Jurkat cells were used to express CXCR1 because they do not express this receptor but do possess the necessary signaling machinery to support cell migration mediated by CXCR1/2 (31). Cells were nucleofected with pEGFP-N3 or pEGFP-N3 expressing human CXCR1 (pEGFP-N3-CXCR1). The efficiency of nucleofection was determined by flow cytometry and found to be 70% ± 5% in three different experiments (data not shown). Flow cytometric analysis showed that a high percentage (65%) of cells transfected with pEGFP-N3-CXCR1 expressed the receptor on their surfaces compared to cells transfected with pEGFPN3, which were CXCR1 negative (data not shown). The migratory behavior of Jurkat cells expressing GFP or CXCR1 after p17 or SIV MA stimulation was examined by a transwell chemotaxis assay. Figure 2F shows that Jurkat cells nucleofected with the pEGFP-N3 did not migrate in response to GST, IL-8, p17, or SIV MA. Conversely, CXCR1-expressing Jurkat cells were able to migrate in response to IL-8, p17, and SIV MA only. Therefore, our results show that CXCR1 expression in Jurkat cells is sufficient to mediate chemotaxis in response to SIV MA.
SIV MA and p17 exert a chemokine-like activity on human B cells.
B cells do express IL-8 receptors on their surface (33). Therefore, we determined a possible role of p17 and SIV MA viral proteins in B cell migration. Figure 3A and B show that p17 and SIV MA exhibited a statistically significant chemoattraction of B cells at the concentration of 100 ng/ml. As expected, a strong chemoattraction of B cells was obtained using IL-8 (100 ng/ml), a reference chemoattractant.
FIG 3.

p17 and SIV MA induce CXCR2-mediated B cell migration. (A and B) Transwell migration assay of B cells in response to PBS (NT), IL-8, p17, or SIV MA at the indicated concentrations. Bars represent the means ± the SD of three independent experiments performed in duplicate. Statistical analysis was performed by one-way ANOVA, and a Bonferroni's post test was used to compare data (***, P < 0.001). (C and D) Transwell migration assay of B cells in response to the indicated treatments. B cells were stimulated for 90 min at 37°C with PBS (NT) or pretreated for 1 h at 37°C with 50 μg of MAb to CXCR1, MAb to CXCR2, or Ctrl MAb/ml and then stimulated for 90 min at 37°C with p17 or SIV MA (100 ng/ml). Bars represent the means ± the SD of three independent experiments performed in duplicate. Statistical analysis was performed by one-way ANOVA, and a Bonferroni's post-test was used to compare data (*, P < 0.05; **, P < 0.01). (E) Transwell migration assay of B cells nucleoporated with CXCR2 siRNAs or with scr siRNAs in response to the indicated treatments. Bars represent the means ± the SD of three independent experiments performed in duplicate. Statistical analysis was performed by using a paired two-tailed Student t test (**, P < 0.01). (F) Transwell migration assay of Jurkat cells transfected with pEGFP-N3 or pEGFP-N3-CXCR2 in response to the indicated treatments. Bars represent the means ± the SD of three independent experiments performed in duplicate. Statistical analysis was performed by one-way ANOVA, and a Bonferroni's post test was used to compare data (**, P < 0.01; ***, P < 0.001).
The role of CXCR1 and/or CXCR2 in sustaining p17 and SIV MA chemokine activity on B cells was then investigated. As shown in Fig. 3C and D, MAb to CXCR2 significantly inhibited p17 and SIV MA chemotaxis, whereas no significant inhibition of p17 and SIV MA chemotactic activity was observed in B cells pretreated with control MAb or MAb to CXCR1.
To further assess the involvement of CXCR2 in p17- and SIV MA-induced B cell migration, the expression of this receptor was suppressed on primary B cells by silencing, using Amaxa nucleofection technology to deliver CXCR2 siRNAs. The efficiency of nucleofection was determined by flow cytometric analysis and showed an average of 86% ± 4% nucleoporated B cells (data not shown). The biologic consequence of CXCR2 silencing was evident in B cells collected 24 h after nucleofection, which showed a significant inhibition in p17 and SIV MA-induced migration of CXCR2-silenced B cells, compared to B cells nucleofected with scr siRNAs (Fig. 3E).
The involvement of CXCR2 in p17 and SIV MA-triggered B cell migration was also investigated by inducing its de novo expression on Jurkat cells. The efficiency of nucleofection of Jurkat cells with pEGFP-N3 or pEGFP-N3 expressing human CXCR2 (pEGFP-N3-CXCR2) was evaluated by flow cytometry and found to be 67% ± 4% in three different experiments (data not shown). Flow cytometric analysis also showed that 60% of cells transfected with pEGFP-N3-CXCR2 expressed the receptor on their surface compared to cells transfected with pEGFPN3 (data not shown). Transwell chemotaxis assays were performed to analyze the migratory behavior of Jurkat cells expressing GFP or CXCR2 after treatment with p17 or SIV MA (100 ng/ml). Figure 3F shows that CXCR2-expressing Jurkat cells were able to migrate in response to IL-8 (100 ng/ml), p17, and SIV MA compared to Jurkat cells nucleofected with the pEGFP-N3. Therefore, our results show that CXCR2 is sufficient to mediate chemotaxis in response to p17 and SIV MA.
SIV MA internalization.
Compelling evidence has been provided for host-specific adaptation during the emergence of HIV-1 and identifies the viral matrix protein as a modulator of viral fitness following transmission to the new human host (19). Due to its key role in HIV-1 life cycle (34), adaptation of SIV MA to the new host may dispose to an optimal interplay of matrix protein with different cellular molecules to allow proviral integration and viral assembly to cell surface. HIV-1 p17 is a nuclear targeting protein and following exogenous p17/receptor interaction, it is internalized and accumulates into the nuclear compartment (3). Our aim was to investigate whether exogenous SIV MA was able to internalize following receptor interaction and to accumulate into the nucleus of B cells. After a few hours, SIV MA was found to efficiently internalize in HBL-1 (Fig. 4A) and accumulate almost entirely in the nucleus after 2 days of culture, as demonstrated by multispectral imaging flow cytometry (Fig. 4B). SIV MA nuclear localization was found to be significantly higher (P < 0.05) than that displayed by FITC-conjugated p17 or S75X (Fig. 4A).
FIG 4.

SIV MA efficiently internalizes into HBL-1 cells and human primary B cells and rapidly translocates into the nucleus. (A) HBL-1 cells were treated for 48 and 96 h with p17-APC, S75X-APC, and SIV MA-FITC at a final concentration of 0.5 μg/106 cells. A total of 2 × 104 HBL-1 cells were acquired with ImageStream X and analyzed using IDEAS software. Nuclear localization of p17, S75X, and SIV MA was calculated as similarity score between labeled protein and nuclear dye intensities. The graph represents a mean of four independent experiments. After both 48 and 96 h of treatment, the SIV MA nuclear localization was significantly higher than the p17 and S75X nuclear localization (*, P < 0.05). The statistical significance was calculated by using Student two-tailed t test. (B) HBL-1 were cultured for 48 h in the presence of SIV MA-FITC and then labeled with the vital nuclear dye DRAQ5. SIV MA-FITC localized almost completely into the nucleus, as demonstrated by multispectral imaging analysis showing SIV MA-FITC/DRAQ5 colocalization. BF, bright field. The data are representative from one representative experiment. (C) Human primary B cells were pretreated with 5 μg of MAb to CXCR1, MAb to CXCR2, or Ctrl MAb/ml for 1 h at room temperature and then treated for 48 and 96 h with p17-APC and SIV MA-FITC (0.5 μg/106 cells). Cells were acquired with ImageStream X and analyzed with IDEAS software. Nuclear localization of p17 and SIV MA was calculated as the similarity score between labeled protein and nuclear dye intensities. The graph represents the mean of two independent experiments. Internalization and nuclear localization of p17 and SIV MA, after both 48 and 96 h of treatment, was significantly inhibited by the MAb to CXCR2. The statistical significance was calculated by using the Student two-tailed t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
SIV MA, like p17, was shown to be a nuclear targeting protein also in human primary B cells (Fig. 4C). To assess whether the binding of SIV MA and p17 to the B cell surface was specifically mediated by CXCR1 or CXCR2 receptors, we cultured primary human B cells with FITC-labeled SIV MA or APC-labeled p17 protein in the presence of MAb to CXCR1 or to CXCR2. No significant interference with SIV MA internalization and nuclear localization was observed in B cells pretreated with the control MAb or with neutralizing MAb to CXCR1 (Fig. 4C). On the other hand, pretreatment of B cells with the neutralizing anti-CXCR2 MAb strongly limited both SIV MA and p17 internalization and nuclear localization (Fig. 4C). This result demonstrates that SIV MA, like p17, is internalized into B cells after binding to CXCR2 expressed on the B cell surface and acts as a nuclear targeting protein.
p17 and SIV MA show different patterns in modulating the Akt signaling pathway.
The binding of p17 to Raji cells was easily detectable by flow cytometry, and B cells proved very useful in differentiating p17 from the Ugandan-derived p17 variant S75X by marking an opposite effect of the two proteins on cell signaling and B cell proliferation and clonogenicity (10). In particular, the binding of p17 to Raji cells was followed by the activation of ERK1/2 and downmodulation of PI3K/Akt pathways, whereas S75X triggered an activation of both ERK1/2 and PI3K/Akt signaling pathways. Activation of PI3K/Akt pathway by S75X was followed by an increased B cell clonogenic activity in soft agar (10). Therefore, we used Raji cells to investigate whether SIV MA retains the biological properties of p17 or shows those of S75X. First, we explored the capability of SIV MA to differently modulate the phosphorylation status of ERK1/2 and Akt in Raji cells by Western blotting. As shown in Fig. 5A, p17, S75X, and SIV MA induced the activation of ERK1/2, as evidenced by the increase of ERK1/2 phosphorylation. On the contrary, the three proteins showed different phosphorylation patterns with the Akt kinase: p17 inhibited the activation of Akt, as shown by the decreased phosphorylation of the kinase, whereas S75X and SIV MA induced an increase in Akt phosphorylation. These results were also confirmed using primary human B cells. Indeed, p17 was found to inhibit the activation of Akt, whereas S75X and SIV MA increased the phosphorylation status of Akt (Fig. 5B). In conclusion, our data show that SIV MA and p17 have opposite effects on the activation of Akt and that SIV MA has the signal signature characteristic of S75X.
FIG 5.

Effect of p17, S75X, and SIV MA on pERK1/2 and pAkt in Raji and B cells. (A and B) Cells were treated for 5 min with p17, S75X, and SIV MA at a concentration of 0.1 μg/ml. Untreated cells were used as a control. Western blot analysis of Raji lysates shows that p17 inhibited the activation of Akt and induced the activation of ERK1/2, whereas S75X and SIV MA activated both Akt and ERK1/2, as verified by densitometric analysis and plotting of the pAkt/ERK1/2 and pERK1/2/ERK1/2. (Upper panels) Blots from one representative experiment of three with similar results are shown. (Lower panels) Values reported for pERK1/2 and pAkt are the means ± the SD of three independent experiments. Statistical analysis was performed by one-way ANOVA, and a Bonferroni's post test was used to compare data (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
SIV MA and S75X show similar effects on Raji colony formation.
Activation of the Akt signaling pathway is a hallmark of different types of malignancies, and intensive studies of the PI3K/Akt pathway have firmly established a central role for Akt in tumorigenesis and cancer progression (35–37). Activation of the PI3K/Akt signal transduction pathway in B cells and increased B cell clonogenic activity in soft agar are features of the S75X p17 variant. Having shown that SIV MA is capable of activating the PI3K/Akt signaling pathway, we investigated the capacity of SIV MA to promote B cell growth and clonogenicity. As shown in Fig. 6, at concentrations of 0.1 and 0.2 μg/ml, p17 significantly inhibited the colony-forming ability of Raji cells compared to untreated control cultures. On the contrary, SIV MA and S75X, at the same concentrations, induced a significant increase in the number of colonies compared to untreated cells. This result confirms the peculiar antiproliferative activity of p17 but, more interestingly, highlights the capacity of the natural variant S75X and of SIV MA to increase the growth and clonogenicity of Raji cells.
FIG 6.
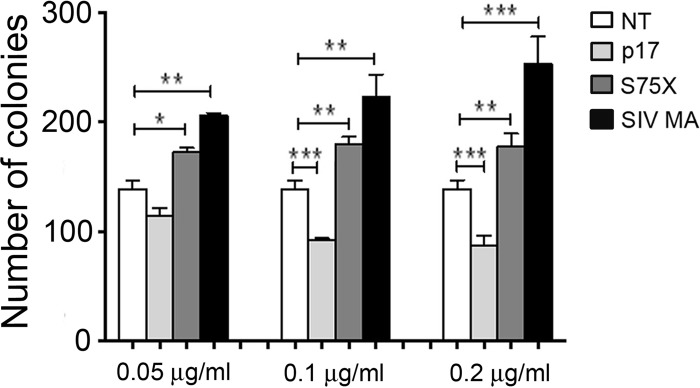
Effects of p17, S75X, and SIV MA on the colony-forming ability of Raji cells. Cells were plated in six-well plates and, after 2 days, the medium was replaced using fresh medium with various concentrations of p17, S75X, or SIV MA: 0.05, 0.1, and 0.2 μg/ml. Cells that were not stimulated (NT) were used as a negative control. Cell growth was analyzed by using MTT. The data represent the average number of colonies ± the SD from three independent experiments performed in triplicate. The statistical significance between control and treated cultures was calculated using two-way ANOVA, and a Bonferroni's post test was used to compare data (*, P < 0.05; **, P < 0.01, ***, P < 0.001).
Computational analysis.
The three-dimensional structure of p17 has been determined by nuclear magnetic resonance (NMR) and X-ray crystallography. Individual folded p17 molecules are composed of five major α-helixes and a highly basic platform consisting of three β strands (23, 38). This partially globular protein presents four helixes centrally organized to form a compact globular domain capped by the β-sheet. The fifth helix (H5) in the C terminus of the protein projects away from the packed bundle of helixes and in the NMR-determined p17 conformation may be partially unfolded (39). Previous results showed that the H5 region of p17 and S75X was involved in modulating the activation of the AKT signaling pathway and in promoting B cell oncogenesis (10). Therefore, we looked for similarity between SIV MA and S75X H5 regions by using computational analysis.
Upon comparing p17 and SIV MA C-terminal region using SNAPFUN (28), we found six exchanges in the SIV MA protein which are non-neutral to the secondary structure. Of these, the most important are D86H, Q117P, and A120P, which break the H5 α-helix fully represented in p17. In total, the exchanges are largely responsible for the different secondary structures, which are seen as an α-helix in p17 and as a β-sheet in SIV MA (Fig. 7A). When the C-terminal regions of p17 and S75X are compared, all exchanges are considered neutral with a high “reliability index”. This suggests that p17 and S75X have the same secondary structure in their C-terminal regions. Even most of the exchanges of the charged amino acids are compensated for by related reciprocal exchanges, e.g., K113Q is compensated for by Q127K. Therefore, it seems unlikely that changes in the secondary structure elements of p17 and S75X could explain the differential behavior in terms of biological activities. Thus, further analyses of the residue interactions and the stabilizing factors of the arrangement of secondary structure elements were performed using BRAGI (27) and HBPLUS (39). Among other bonds, we found an essential H-bond Ile92 to Glu107 in p17 that bridges the end of the fourth α-helix (H4) to H5 (Fig. 7B, left panel). This appears to be the interaction fixating H5 to the compact globular domain of the protein. Of the 10 nonbackbone interactions that we found, this was the only one involving H5. In SIV MA, H5 has pi-pi interactions, e.g., His107 to Arg92 or Trp86 to Leu108 (Fig. 7B, central panel). Interestingly, since Glu107 in p17 is replaced by an Ile in S75X (Fig. 7B, right panel), the stabilizing H-bond is lost and the conformation of H5 is predicted to change, at least in solution. This can explain in part the different biological activities exerted on B cells by SIV MA or S75X compared to those exerted by p17.
FIG 7.

Similarities and differences in the C-terminal regions of p17, SIV MA, and S75X. (A) Three-dimensional alignment of p17 (blue) and SIV MA (red). Both proteins have a nearly identical three-dimensional structure in the N-terminal region down to residue 107. The conformation of the AT20 loops of p17 and SIV MA is preserved for binding to CXCR1 and CXCR2. (B) The α-helix H5 in p17 is bound to the loop following H4 by an H-bond of Ile92 to Glu107 (red), thus stabilizing the conformation between H5 and the rest of the protein (left panel). In the C-terminal end of SIV MA the residue at position 107 is His, and residues 110 to 117 build a β-sheet (middle panel). The model shows that there is no way to keep H5 in place when Glu107 is mutated to Ile, as in S75X (right panel).
DISCUSSION
In this study, we demonstrate that SIV MA, similarly to p17, binds to both CXCR1 and CXCR2. In light of this evidence, we investigated the potential direct effect of SIV MA on previously identified p17 target cells (3, 9). SIV MA showed a potent p17-like proangiogenic activity on human primary endothelial cells. This angiogenic activity was dependent, as already described for p17, on SIV MA interaction with both CXCR1 and CXCR2 expressed on the endothelial cell surface. At the same time, SIV MA showed a chemokine-like activity on human primary monocytes. This was found to be superimposable to that exerted by p17 and was dependent on its interaction with CXCR1. The chemokine activity of SIV MA and p17 was also tested, for the first time, on human primary B cells, since these cells do express both chemokine receptors on their surface (33). Here we show that both proteins were able to chemoattract B cells, and that their activity was mediated by CXCR2. These findings are in agreement with previous reports showing that CXCR1 and CXCR2 are both capable of inducing cell migration (31). Despite the fact that CXCR1 is more involved in sustaining the chemoattaction of different immune cells (32), CXCR2 was found to drive the migratory activity of B cells (33). The diverse doses of protein ligands used in the different assays and the magnitude of the cellular response to SIV MA and p17 are dependent on the affinity of the two matrix proteins to CXCR1 and CXCR2 and on the level of receptor expression at the plasma membrane (40), which differs among the different cells tested.
Similar capability of SIV MA and p17 in using CXCR1 and CXCR2 to perform chemokine and proangiogenic activities has to be ascribed to the finding that the overall network of intramolecular bonds does not change much and the fold of these proteins should be nearly identical. In fact, the X-ray structures (23, 24) confirm that there are minor differences in the structure of SIV MA and p17 up to residue 100 and, as shown in Fig. 7, the functional region responsible for SIV MA and p17 binding to their receptors (AT20 loop) is structurally unchanged. This finding is in line with previous data showing that the functional loop is a structural constraint being an hot spot for viral proteins interaction with several host cell proteins (25, 41–43) and that critical mutations in this highly basic domain do not affect the spatial conformation of the AT20 epitope to an extent that the folded state is destabilized (25).
SIV MA and p17 are known to have interactions with as many as 20 different cellular proteins (44, 45). It is therefore likely that certain sites will be constrained due to these unavoidable vital interactions. In this respect, we previously described the capability of p17 to act as a nuclear targeting protein, being able to reach the nucleus of endothelial cells following receptor-mediated endocytosis (3). The hypothesis that SIV MA is, similarly to p17, capable of interacting with different host proteins and therefore acting as a nuclear targeting protein, was verified in the human B cell model. Here we provide evidence that both SIV MA and p17 are internalized after their binding to CXCR2 expressed on human primary B cells. Based on our evidence, mutation in Gag30 does not involve changes in SIV MA and p17 interaction with their specific receptors or changes in biological activity. Therefore, improvement of viral fitness by Gag30 mutation must reflect the ability of matrix protein to contribute, by interacting with one or more of host proteins or with as-yet-unidentified host factors, to key events in the viral life cycle. It is worth noting that virions carrying mutations in the matrix protein N-terminal basic domain included within the functional AT20 loop were found to harbor not only an early entry defect but also to display a severe impairment in their ability to incorporate envelope (46, 47) and a reduced capacity to integrate their proviral genomes (21).
In contrast, several observations indicate that mutations in the C-terminal region do not affect viral replication and infectivity (47). It is therefore likely that the H5 helix has to be considered evolutionarily relevant, since mutations present in this region may generate proteins with partially unfolded C-terminal regions without resulting in severe fitness consequences. Indeed, recent data point to the H5 region of SIV MA and p17 as highly flexible and with high level of predicted intrinsic disorder (48), which subtends to at least a partially unfolded status of this region (39). The prediction of protein sequence evolution based on structural templates relies heavily on amino acid side chain packing analysis and interpretation of interactions at the atomic level. Interaction between side chains are important for maintaining the stability of the protein, as well as enabling correct folding of the molecule (49, 50). Substitutions that alter the amino acid side chain will have an impact on the surrounding residues. In the case of p17, we found an essential H-bond Ile92 to Glu107 that bridges the end of the fourth α-helix (H4) to H5, thus fixating H5 to the compact globular domain of the protein. Interestingly, since Glu107 in p17 is replaced by an Ile in S75X, the stabilizing H-bond is lost, and the conformation of H5 is predicted to change, at least in solution. In the same region, SIV MA shows at least three critical amino acid mutations that are largely responsible for the different secondary structures, which change from an α-helix in p17 to a β-sheet. This may explain, at least in part, the different biological activities exerted on B cells by SIV MA or S75X compared to those exerted by p17. The role of the C-terminal region in modulating the p17 signaling pathway and biological activity has been recently described. In particular, a C-terminal truncated form of p17 (p17Δ36) induced activation of the PI3K/Akt pathway by maintaining the phosphatase PTEN in an inactive phosphorylated form leading to increased B cell proliferation and clonogenic activity on soft agar. This is opposite to the effects exerted by the entire p17 protein, which displayed decreased B cell proliferation and clonogenic activity by downmodulating the PI3K/Akt pathway and maintaining the phosphatase PTEN in an active but not phosphorylated form (10). This finding allowed us to hypothesize that a N-terminal epitope of p17 may be responsible for an activation signal on Akt signaling pathway opportunely balanced by a second inhibitory signal promoted by the C-terminal region (10). Therefore, losing its helical character, as in SIV MA, or changing the H5 helix conformation because of amino acid mutations, as in S75X, is likely to be responsible for differences in B cell signaling and growth-modulating activity between SIV MA, S75X, and p17. Our hypothesis is in line with the evidence that many crucial biological functions are performed by proteins that lack ordered and/or secondary structure (51–53). It is worth noting that the highly dynamic nature, correlated with the high propensity of HIV-1 and SIV matrix proteins for intrinsic disorders in the H5 region, is a characteristic feature of HIV-1 and its closest relatives, SIV and HIV-2 (48).
Retroviruses are programmed to integrate within cells and, consequently, to live together with the infected host. Indeed, genome sequencing reveals that 8% of the human genome consists of human endogenous retroviruses (54, 55). In this respect, it is interesting to consider that pandemic plagues are ultimately a brutal manifestation of an evolutionary mechanism resulting in changes to the species gene pool through a protracted series of invasions with repeated large-scale attrition and that the exogenous forebears of our human endogenous retroviruses once behaved as highly contagious infections, following transmission pathways akin to that of HIV-1 (56). HIV-1 has a highly diverse viral population both within an infected individual and across populations of hosts (57). Despite HIV-1 unusually high mutability, we might expect that there are significant constraints on how the sequence can change and still code for a replication-competent virus. The analysis of fitness costs of evolving viruses indicates that evolutionary constraints restrict viral change (58–60). The absence of constraints in H5 does not limit the number and location of mutations that can be accepted per replication cycle (61). This is in agreement with several phylogenetic studies tracing the degree of HIV-1 evolution which showed that protein structure constrains evolutionary change in HIV-1 and that the surface of a protein is more likely to accept replacements than the compact core (62, 63). Based on the change of the H5 region from a β-sheet in SIV MA to an α-helix in p17, we can hypothesize that there has been a functional change to a dramatic change in protein function. Both SIV and HIV-1 may cause lymphoma in their respective hosts (64, 65). If the finding of p17 variants capable of sustaining in vitro B cell proliferation and enhancing clonogenicity (10) is validated by in vivo studies, showing the presence in HIV-1 seropositive patients of p17 variants at work in the process of lymphomagenesis, this may suggest that sequence evolutions from SIV MA to p17 could be a means of maintaining optimal structural and functional integrity to deliver inhibitory signals to B cells. In this respect, small structural rearrangements, or a combination of coevolution and structural changes, due to a few mutations in the H5 α-helix and/or scattered through the entire protein allowed S75X to acquire biological activities that belong to the ancestral SIV MA protein. It is worth noting that S75X is a p17 protein derived from a Ugandan clade A1 virus, which is phylogenetically distinct from the p17 derived from B clade viruses.
In conclusion, p17 exhibits a lower degree of plasticity in the H5 α-helix than SIV MA and S75X that allows the formation of distinct ligand-receptor interactions capable of selectively deactivate Akt signaling and control B cell proliferation and clonogenic efficiency by maintaining PTEN in an active form. This finding suggests that accumulation of the necessary mutations may have led to a further p17 adaptation to its host, by the insertion of key amino acid residues in H5 required to specify its fold and activity. Although at a very initial step, we may be in front of the appearance of a structural constrain in the H5 region of p17, which may offer the possibility of understanding the evolutionary trajectory of HIV-1. This link between H5 sequence/structure and protein biological activity points to the possibility that HIV-1 is going to evolve by selecting for changes that preserve and strengthen the relationship with its human host.
ACKNOWLEDGMENTS
We thank Genoveffa Franchini for helpful suggestions.
This study was supported by the Istituto Superiore di Sanità (AIDS grant 40G.16).
The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Published ahead of print 12 March 2014
REFERENCES
- 1.Fiorentini S, Giagulli C, Caccuri F, Magiera A, Caruso A. 2010. HIV-1 matrix protein p17: a candidate antigen for therapeutic vaccines against AIDS. Pharmacol. Ther. 128:433–444. 10.1016/j.pharmthera.2010.08.005 [DOI] [PubMed] [Google Scholar]
- 2.Fiorentini S, Riboldi E, Facchetti F, Avolio M, Fabbri M, Tosti G, Becker PD, Guzman CA, Sozzani S, Caruso A. 2008. HIV-1 matrix protein p17 induces human plasmacytoid dendritic cells to acquire a migratory immature cell phenotype. Proc. Natl. Acad. Sci. U. S. A. 105:3867–3872. 10.1073/pnas.0800370105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caccuri F, Giagulli C, Bugatti A, Benetti A, Alessandri G, Ribatti D, Marsico S, Apostoli P, Slevin MA, Rusnati M, Guzman CA, Fiorentini S, Caruso A. 2012. HIV-1 matrix protein p17 promotes angiogenesis via chemokine receptors CXCR1 and CXCR2. Proc. Natl. Acad. Sci. U. S. A. 109:14580–14585. 10.1073/pnas.1206605109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Popovic M, Tenner-Racz K, Pelser C, Stellbrink HJ, van Lunzen J, Lewis G, Kalyanaraman VS, Gallo RC, Racz P. 2005. Persistence of structural proteins and glycoproteins in lymph nodes of patients under highly active antiretroviral therapy. Proc. Natl. Acad. Sci. U. S. A. 102:14807–14812. 10.1073/pnas.0506857102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Francesco MA, Baronio M, Fiorentini S, Signorini C, Bonfanti C, Poiesi C, Popovic M, Grassi M, Garrafa E, Bozzo L, Lewis GK, Licenziati S, Gallo RC, Caruso A. 2002. HIV-1 matrix protein p17 increases the production of proinflammatory cytokines and counteracts IL-4 activity by binding to a cellular receptor. Proc. Natl. Acad. Sci. U. S. A. 99:9972–9977. 10.1073/pnas.142274699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vitale M, Caruso A, De Francesco MA, Rodella L, Bozzo L, Garrafa E, Grassi M, Gobbi G, Cacchioli A, Fiorentini S. 2003. HIV-1 matrix protein p17 enhances the proliferative activity of natural killer cells and increases their ability to secrete proinflammatory cytokines. Br. J. Haematol. 120:337–343. 10.1046/j.1365-2141.2003.04053.x [DOI] [PubMed] [Google Scholar]
- 7.Marini E, Tiberio L, Caracciolo S, Tosti G, Guzman CA, Schiaffonati L, Fiorentini S, Caruso A. 2008. HIV-1 matrix protein p17 binds to monocytes and selectively stimulates MCP-1 secretion: role of transcriptional factor AP-1. Cell Microbiol. 10:655–666. 10.1111/j.1462-5822.2007.01073.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Avolio M, Caracciolo S, Tosti G, Vollero L, Fiorentini S, Caruso A. 2008. HIV-1 matrix protein p17 prevents loss of CD28 expression during IL-2-induced maturation of naive CD8+ T cells. Viral Immunol. 21:189–202. 10.1089/vim.2007.0095 [DOI] [PubMed] [Google Scholar]
- 9.Giagulli C, Magiera AK, Bugatti A, Caccuri F, Marsico S, Rusnati M, Vermi W, Fiorentini S, Caruso A. 2012. HIV-1 matrix protein p17 binds to the IL-8 receptor CXCR1 and shows IL-8-like chemokine activity on monocytes through Rho/ROCK activation. Blood 119:2274–2283. 10.1182/blood-2011-06-364083 [DOI] [PubMed] [Google Scholar]
- 10.Giagulli C, Marsico S, Magiera AK, Bruno R, Caccuri F, Barone I, Fiorentini S, ò S, Caruso A. 2011. Opposite effects of HIV-1 p17 variants on PTEN activation and cell growth in B cells. PLoS One 6:e17831. 10.1371/journal.pone.0017831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharp PM, Bailes E, Chaudhuri RR, Rodenburg CM, Santiago MO, Hahn BH. 2001. The origins of acquired immune deficiency syndrome viruses: where and when? Philos. Trans. R. Soc. Lond. B Biol. Sci. 356:867–876. 10.1098/rstb.2001.0863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirchhoff F. 2010. Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe 8:55–67. 10.1016/j.chom.2010.06.004 [DOI] [PubMed] [Google Scholar]
- 13.Bushman FD, Malani N, Fernandes J, D'Orso I, Cagney G, Diamond TL, Zhou H, Hazuda DJ, Espeseth AS, König R, Bandyopadhyay S, Ideker T, Goff SP, Krogan NJ, Frankel AD, Young JA, Chanda SK. 2009. Host cell factors in HIV replication: meta-analysis of genome-wide studies. PLoS Pathog. 5:e1000437. 10.1371/journal.ppat.1000437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ortiz M, Bleiber G, Martinez R, Kaessmann H, Telenti A. 2006. Patterns of evolution of host proteins involved in retroviral pathogenesis. Retrovirology 3:11–18. 10.1186/1742-4690-3-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Longdon B, Hadfield JD, Webster CL, Obbard DJ, Jiggins FM. 2011. Host phylogeny determines viral persistence and replication in novel hosts. PLoS Pathog. 7:e1002260. 10.1371/journal.ppat.1002260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Streicker DG, Turmelle AS, Vonhof MJ, Kuzmin IV, McCracken GF, Rupprecht CE. 2010. Host phylogeny constrains cross-species emergence and establishment of rabies virus in bats. Science 329:676–679. 10.1126/science.1188836 [DOI] [PubMed] [Google Scholar]
- 17.The Chimpanzee Sequencing and Analysis Consortium. 2005. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437:69–87. 10.1038/nature04072 [DOI] [PubMed] [Google Scholar]
- 18.McLean CY, Reno PL, Pollen AA, Bassan AI, Capellini TD, Guenther C, Indjeian VB, Lim X, Menke DB. 2011. Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature 471:216–219. 10.1038/nature09774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wain LV, Bailes E, Bibollet-Ruche F, Decker JM, Keele BF, Van Heuverswyn F, Li Y, Takehisa J, Ngole EM, Shaw GM, Peeters M, Hahn BH, Sharp PM. 2007. Adaptation of HIV-1 to its human host. Mol. Biol. Evol. 24:1853–1860. 10.1093/molbev/msm110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mwaengo DM, Novembre FJ. 1998. Molecular cloning and characterization of viruses isolated from chimpanzees with pathogenic human immunodeficiency virus type 1 infections. J. Virol. 72:8976–8987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bibollet-Ruche F, Heigele A, Keele BF, Easlick JL, Decker JM, Takehisa J, Learn G, Sharp PM, Hahn BH, Kirchhoff F. 2012. Efficient SIVcpz replication in human lymphoid tissue requires viral matrix protein adaptation. J. Clin. Invest. 122:1644–1652. 10.1172/JCI61429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ackerman ME, Moldt B, Wyatt RT, Dugast AS, McAndrew E, Tsoukas S, Jost S, Berger CT, Sciaranghella G, Liu Q, Irvine DJ, Burton DR, Alter G. 2011. A robust, high-throughput assay to determine the phagocytic activity of clinical antibody samples. J. Immunol. Methods 366:8–19. 10.1016/j.jim.2010.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hill CP, Worthylake D, Bancroft DP, Christensen AM, Sundquist WI. 1996. Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc. Natl. Acad. Sci. U. S. A. 93:3099–3104. 10.1073/pnas.93.7.3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rao Z, Belyaev AS, Fry E, Roy P, Jones IM, Stuart DI. 1995. Crystal structure of SIV matrix antigen and implications for virus assembly. Nature 378:743–747. 10.1038/378743a0 [DOI] [PubMed] [Google Scholar]
- 25.Fiorentini S, Marsico S, Becker PD, Iaria ML, Bruno R, Guzmán CA, Caruso A. 2008. Synthetic peptide AT20 coupled to KLH elicits antibodies against a conserved conformational epitope from a major functional area of the HIV-1 matrix protein p17. Vaccine 26:4758–4765. 10.1016/j.vaccine.2008.06.082 [DOI] [PubMed] [Google Scholar]
- 26.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reichelt J, Dieterich G, Kvesic M, Schomburg D, Heinz DW. 2005. BRAGI: linking and visualization of database information in a 3D viewer and modeling tool. Bioinformatics 21:1291–1293. 10.1093/bioinformatics/bti138 [DOI] [PubMed] [Google Scholar]
- 28.Bromberg Y, Rost B. 2007. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 35:3823–3835. 10.1093/nar/gkm238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee J, Horuk R, Rice GC, Bennett GL, Camerato T, Wood WI. 1992. Characterization of two high-affinity human interleukin-8 receptors. J. Biol. Chem. 267:16283–16287 [PubMed] [Google Scholar]
- 30.Brat DJ, Bellail AC, Van Meir EG. 2005. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro-oncol. 7:122–133. 10.1215/S1152851704001061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loetscher P, Seitz M, Clark-Lewis I, Baggiolini M, Moser B. 1994. Both interleukin-8 receptors independently mediate chemotaxis. Jurkat cells transfected with IL-8R1 or IL-8R2 migrate in response to IL-8, GRO alpha and NAP-2. FEBS Lett. 341:187–192 [DOI] [PubMed] [Google Scholar]
- 32.Hammond ME, Lapointe GR, Feucht PH, Hilt S, Gallegos CA, Gordon CA, Giedlin MA, Mullenbach G, Tekamp-Olson P. 1995. IL-8 induces neutrophil chemotaxis predominantly via type I IL-8 receptors. J. Immunol. 155:1428–1433 [PubMed] [Google Scholar]
- 33.Jinquan T, Møller B, Storgaard M, Mukaida N, Bonde J, Grunnet N, Black FT, Larsen CG, Matsushima K, Thestrup-Pedersen K. 1997. Chemotaxis and IL-8 receptor expression in B cells from normal and HIV-infected subjects. J. Immunol. 158:475–484 [PubMed] [Google Scholar]
- 34.Fiorentini S, Marini E, Caracciolo S, Caruso A. 2006. Functions of the HIV-1 matrix protein p17. New Microbiol. 29:1–10 [PubMed] [Google Scholar]
- 35.Jiang BH, Liu LZ. 2008. PI3K/PTEN signaling in tumorigenesis and angiogenesis. Biochim. Biophys. Acta 1784:150–158. 10.1016/j.bbapap.2007.09.008 [DOI] [PubMed] [Google Scholar]
- 36.Li L, Ittmann MM, Ayala G, Tsai MJ, Amato RJ, Wheeler TM, Miles BJ, Kadmon D, Thompson TC. 2005. The emerging role of the PI3-K-Akt pathway in prostate cancer progression. Prostate Cancer Prostatic Dis. 8:108–118. 10.1038/sj.pcan.4500776 [DOI] [PubMed] [Google Scholar]
- 37.McCubrey JA, Steelman LS, Abrams SL, Lee JT, Chang F, Bertrand FE, Navolanic PM, Terrian DM, Franklin RA, D'Assoro AB, Salisbury JL, Mazzarino MC, Stivala F, Libra M. 2006. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv. Enzyme Regul. 46:249–279. 10.1016/j.advenzreg.2006.01.004 [DOI] [PubMed] [Google Scholar]
- 38.Massiah MA, Starich MR, Paschall C, Summers MF, Christensen AM, Sundquist WI. 1994. Three-dimensional structure of the human immunodeficiency virus type 1 matrix protein. J. Mol. Biol. 244:198–223. 10.1006/jmbi.1994.1719 [DOI] [PubMed] [Google Scholar]
- 39.Verli H, Calazans A, Brindeiro R, Tanuri A, Guimarães JA. 2007. Molecular dynamics analysis of HIV-1 matrix protein: clarifying differences between crystallographic and solution structures. J. Mol. Graph. Model. 26:62–68. 10.1016/j.jmgm.2006.09.009 [DOI] [PubMed] [Google Scholar]
- 40.Feniger-Barish R, Belkin D, Zaslaver A, Gal S, Dori M, Ran M, Ben-Baruch A. 2000. GCP-2-induced internalization of IL-8 receptors: hierarchical relationships between GCP-2 and other ELR(+)-CXC chemokines and mechanisms regulating CXCR2 internalization and recycling. Blood 95:1551–1559 [PubMed] [Google Scholar]
- 41.McDonald IK, Thornton JM. 1994. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 238:777–793. 10.1006/jmbi.1994.1334 [DOI] [PubMed] [Google Scholar]
- 42.Chukkapalli V, Oh SJ, Ono A. 2010. Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc. Natl. Acad. Sci. U. S. A. 107:1600–1605. 10.1073/pnas.0908661107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bugatti A, Giagulli C, Urbinati C, Caccuri F, Chiodelli P, Oreste P, Fiorentini S, Orro A, Milanesi L, d'Ursi P, Caruso A, Rusnati M. 2013. Molecular interaction studies of HIV-1 matrix protein p17 and heparin: identification of the heparin-binding motif of p17 as a target for the development of multitarget antagonists. J. Biol. Chem. 288:1150–1161. 10.1074/jbc.M112.400077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pinney JW, Dickerson JE, Fu W, Sanders-Beer BE, Ptak RG, Robertson DL. 2009. HIV-host interactions: a map of viral perturbation of the host system. AIDS 23:549–554. 10.1097/QAD.0b013e328325a495 [DOI] [PubMed] [Google Scholar]
- 45.Jäger S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, Shales M, Mercenne G, Pache L, Li K, Hernandez H, Jang GM, Roth SL, Akiva E, Marlett J, Stephens M, D'Orso I, Fernandes J, Fahey M, Mahon C, O'Donoghue AJ, Todorovic A, Morris JH, Maltby DA, Alber T, Cagney G, Bushman FD, Young JA, Chanda SK, Sundquist WI, Kortemme T, Hernandez RD, Craik CS, Burlingame A, Sali A, Frankel AD, Krogan NJ. 2011. Global landscape of HIV-human protein complexes. Nature 481:365–370. 10.1038/nature10719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Casella CR, Raffini LJ, Panganiban AT. 1997. Pleiotropic mutations in the HIV-1 matrix protein that affect diverse steps in replication. Virology 228:294–306. 10.1006/viro.1996.8355 [DOI] [PubMed] [Google Scholar]
- 47.Bhatia AK, Campbell N, Panganiban A, Ratner L. 2007. Characterization of replication defects induced by mutations in the basic domain and C terminus of HIV-1 matrix. Virology 369:47–54. 10.1016/j.virol.2007.06.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goh GK, Dunker AK, Uversky VN. 2008. A comparative analysis of viral matrix proteins using disorder predictors. Virol. J. 5:126. 10.1186/1743-422X-5-126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dahiyat BI, Mayo SL. 1997. Probing the role of packing specificity in protein design. Proc. Natl. Acad. Sci. U. S. A. 94:10172–10177. 10.1073/pnas.94.19.10172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sandberg WS, Terwilliger TC. 1989. Influence of interior packing and hydrophobicity on the stability of a protein. Science 245:54–57. 10.1126/science.2787053 [DOI] [PubMed] [Google Scholar]
- 51.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradović Z. 2002. Intrinsic disorder and protein function. Biochemistry 41:6573–6582. 10.1021/bi012159+ [DOI] [PubMed] [Google Scholar]
- 52.Uversky VN, Oldfield CJ, Dunker AK. 2005. Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit. 18:343–384. 10.1002/jmr.747 [DOI] [PubMed] [Google Scholar]
- 53.Dyson HJ, Wright PE. 2005. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell. Biol. 6:197–208. 10.1038/nrm1589 [DOI] [PubMed] [Google Scholar]
- 54.Bannert N, Kurth R, Medstrand P, van de Lagemaat LN, Mager DL. 2002. Retroelement distributions in the human genome: variations associated with age and proximity to genes. Genome Res. 12:1483–1495. 10.1101/gr.388902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bannert N, Kurth R. 2004. Retroelements and the human genome: new perspectives on an old relation. Proc. Natl. Acad. Sci. U. S. A. 101:S14572–S14579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ryan FP. 2004. Human endogenous retroviruses in health and disease: a symbiotic perspective. J. R. Soc. Med. 97:560–565. 10.1258/jrsm.97.12.560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rambaut A, Posada D, Crandall KA, Holmes EC. 2004. The causes and consequences of HIV evolution. Nat. Rev. Genet. 5:52–61. 10.1038/nrg1246 [DOI] [PubMed] [Google Scholar]
- 58.Peyerl FW, Bazick HS, Newberg MH, Barouch DH, Sodroski J, Letvin NL. 2004. Fitness costs limit viral escape from cytotoxic T lymphocytes at a structurally constrained epitope. J. Virol. 78:13901–13910. 10.1128/JVI.78.24.13901-13910.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kent SJ, Fernandez CS, Dale CJ, Davenport MP. 2005. Reversion of immune escape HIV variants upon transmission: insights into effective viral immunity. Trends Microbiol. 13:243–246. 10.1016/j.tim.2005.03.011 [DOI] [PubMed] [Google Scholar]
- 60.Crawford H, Prado JG, Leslie A, Hué S, Honeyborne I, Reddy S, van der Stok M, Mncube Z, Brander C, Rousseau C, Mullins JI, Kaslow R, Goepfert P, Allen S, Hunter E, Mulenga J, Kiepiela P, Walker BD, Goulder PJ. 2007. Compensatory mutation partially restores fitness and delays reversion of escape mutation within the immunodominant HLA-B*5703-restricted Gag epitope in chronic human immunodeficiency virus type 1 infection. J. Virol. 81:8346–8351. 10.1128/JVI.00465-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holmes EC. 2003. Error thresholds and the constraints to RNA virus evolution. Trends Microbiol. 11:543–546. 10.1016/j.tim.2003.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Overington J, Donnelly D, Johnson MS, Sali A, Blundell TL. 1992. Environment-specific amino acid substitution tables: tertiary templates and prediction of protein folds. Protein Sci. 1:216–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gong S, Blundell TL. 2008. Discarding functional residues from the substitution table improves predictions of active sites within three-dimensional structures. PLoS Comput. Biol. 4:e1000179. 10.1371/journal.pcbi.1000179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Feichtinger H, Putkonen P, Parravicini C, Li SL, Kaaya EE, Böttiger D, Biberfeld G, Biberfeld P. 1990. Malignant lymphomas in cynomolgus monkeys infected with simian immunodeficiency virus. Am. J. Pathol. 137:1311–1315 [PMC free article] [PubMed] [Google Scholar]
- 65.Gaidano G, Carbone A, Dalla-Favera R. 1998. Pathogenesis of AIDS-related lymphomas: molecular and histogenetic heterogeneity. Am. J. Pathol. 152:623–630 [PMC free article] [PubMed] [Google Scholar]